
A Novel and Cross-Species Active Mammalian INDY (NaCT) Inhibitor Ameliorates Hepatic Steatosis in Mice with Diet-Induced Obesity

 , , and
, , and
Abstract
:1. Introduction
2. Results
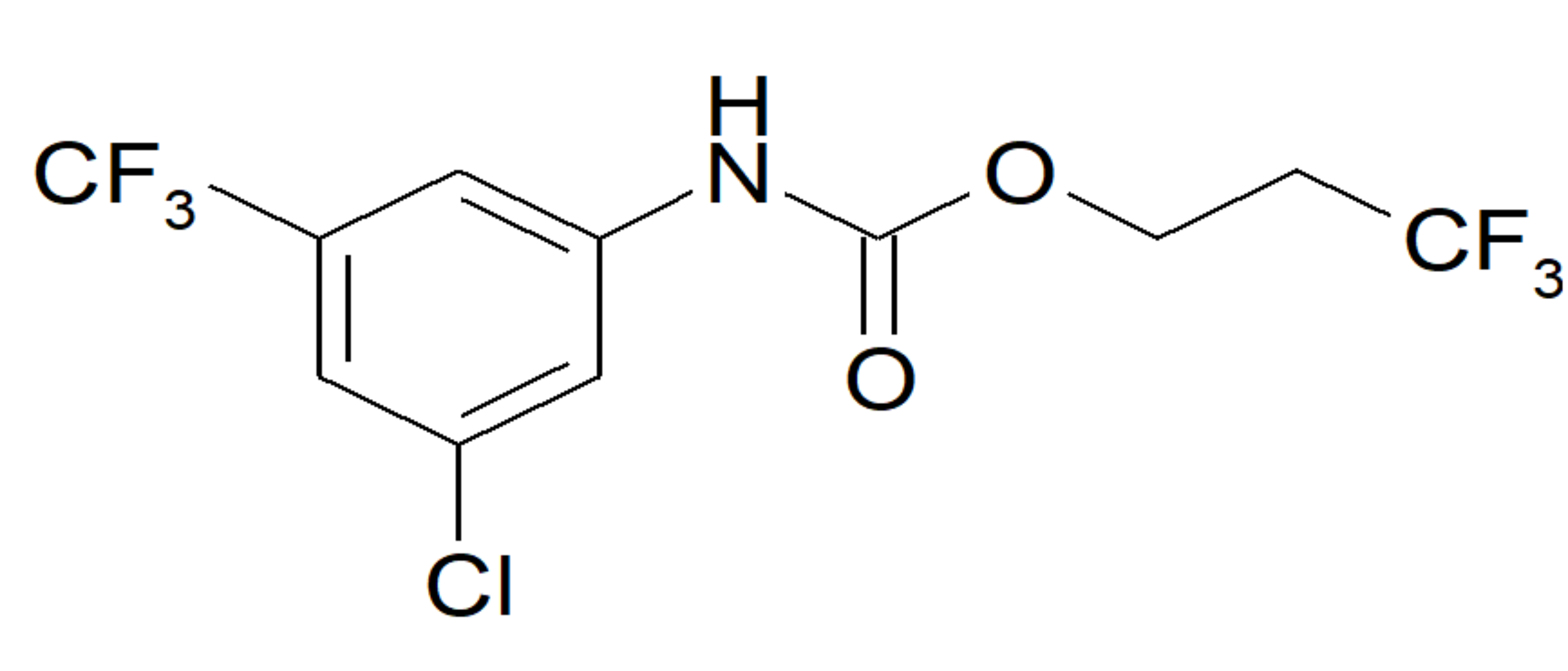
2.1. Selection of the Inhibitor
2.2. In Vitro Activity and Selectivity of ETG-5773
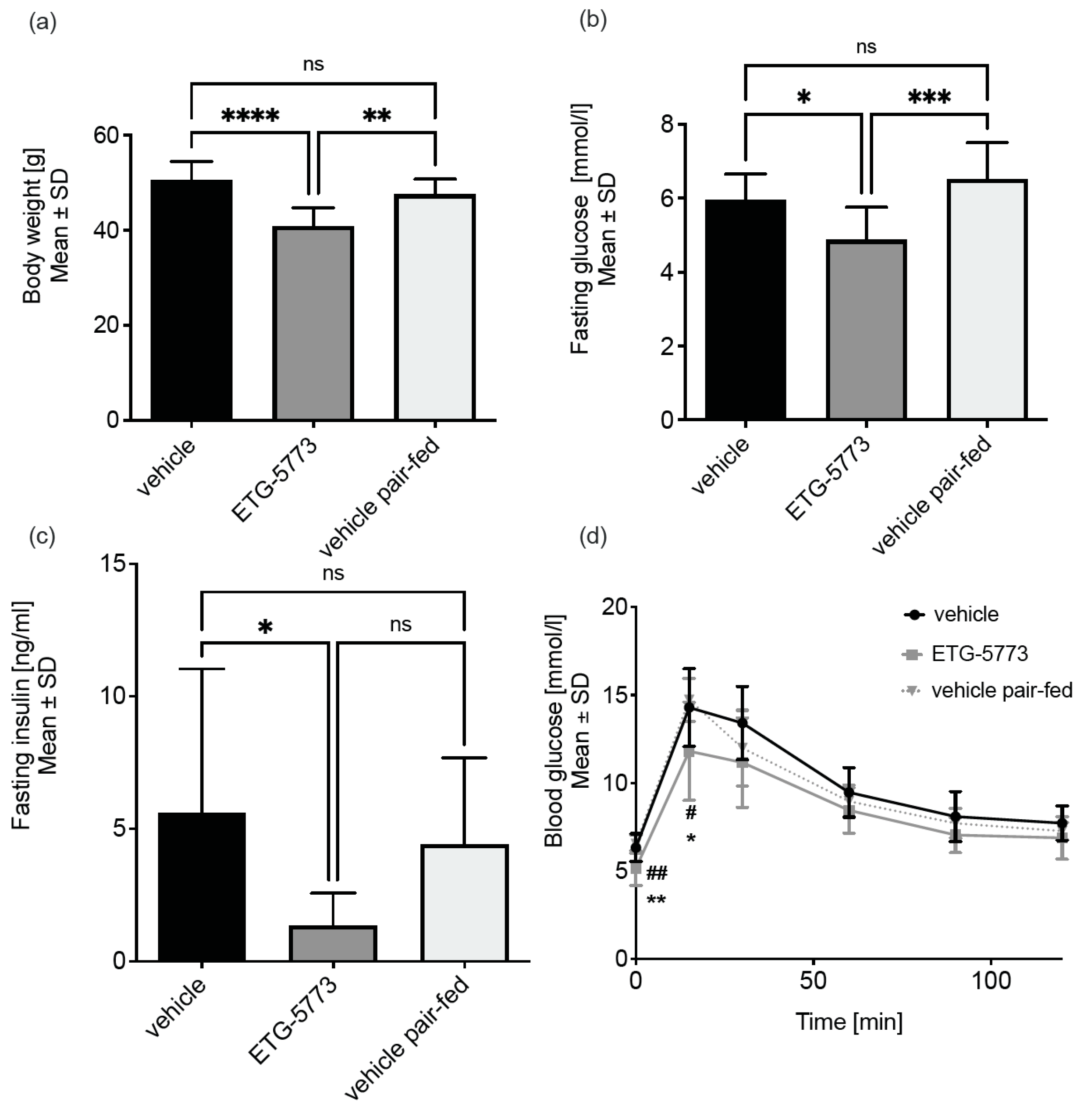
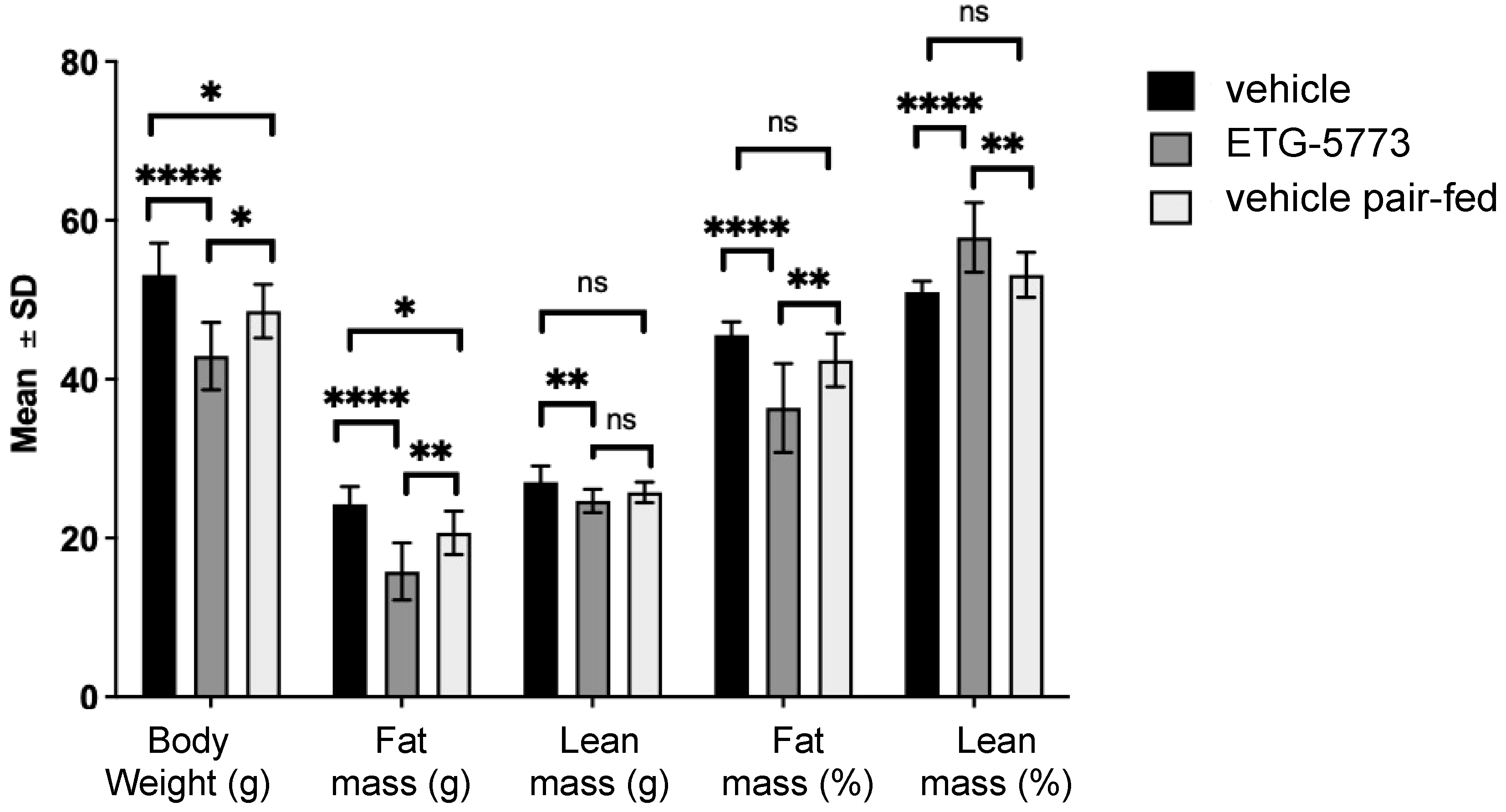
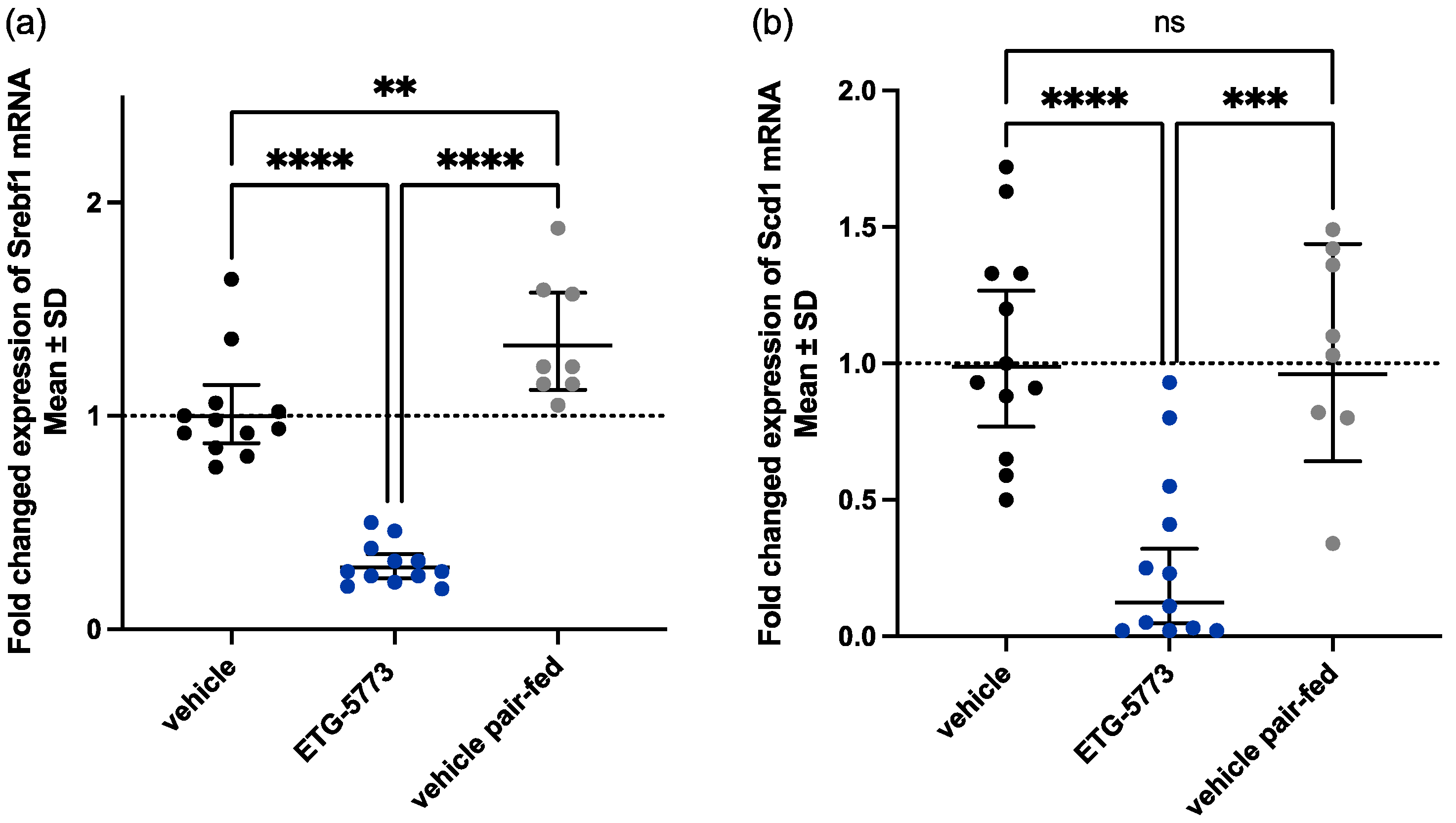
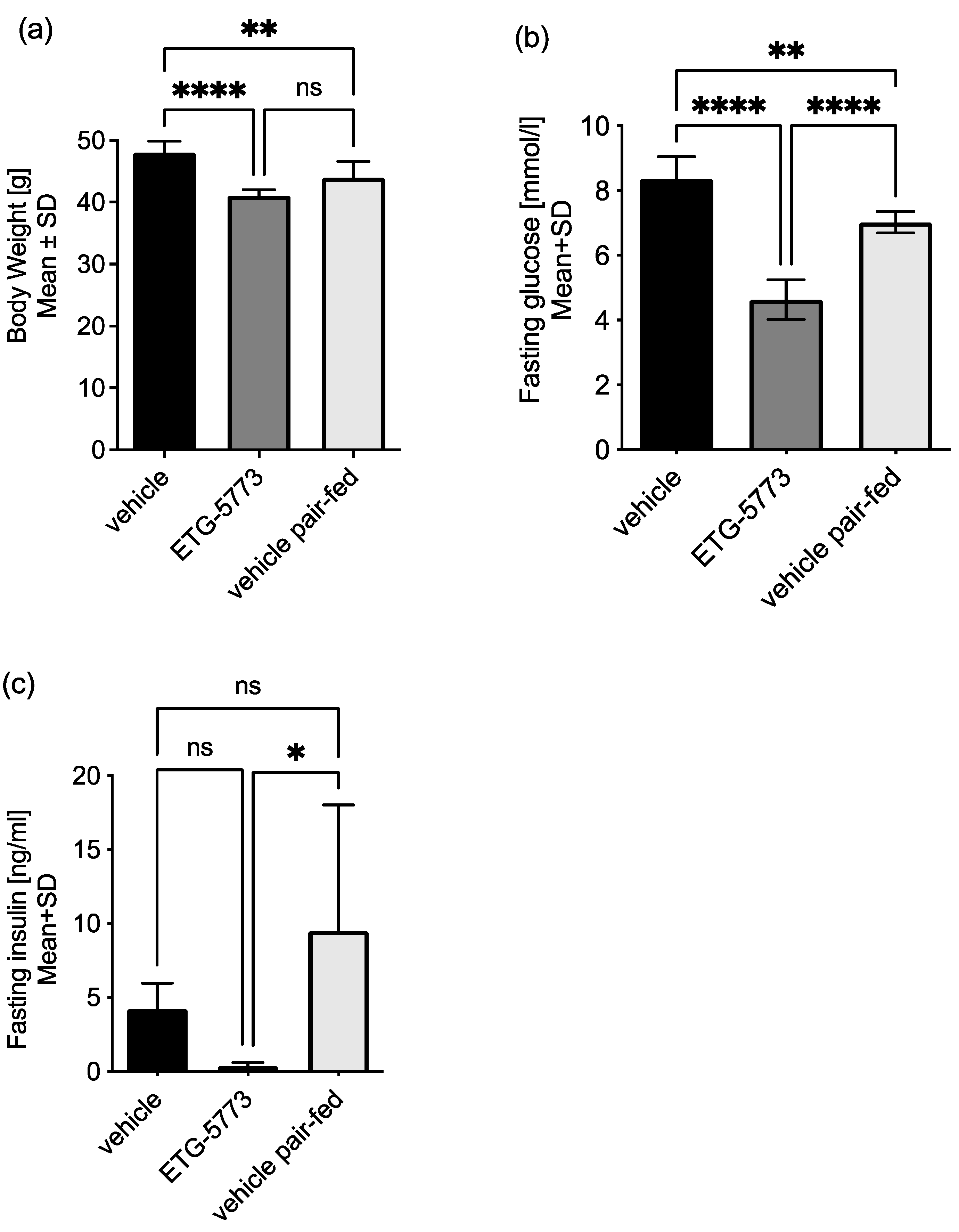
2.3. In Vivo Investigation of ETG-5773 in a Mouse Model of Diet-Induced Obesity
3. Discussion
4. Materials and Methods
4.1. Screening and Selection of the NaCT Inhibitor
4.2. Cell Culture
4.3. Citrate and Succinate Uptake Assays
4.4. Fatty Acid Synthesis (FAS) and Viability Assay
4.5. Animals
4.6. Animal Treatment
4.7. Body Composition Analysis
4.8. Lipogenesis Gene Expression Analysis
4.9. Beta-Hydroxybutyrate ELISA
4.10. AMPK Analysis
4.11. Plasma Lipid Analysis
4.12. Compounds and Cell Lines
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Sánchez, N.; Bugianesi, E.; Gish, R.G.; Lammert, F.; Tilg, H.; Nguyen, M.H.; Sarin, S.K.; Fabrellas, N.; Zelber-Sagi, S.; Fan, J.G.; et al. Global multi-stakeholder endorsement of the MAFLD definition. Lancet Gastroenterol. Hepatol. 2022, 7, 388–390. [Google Scholar] [CrossRef]
- Ferraioli, G.; Soares Monteiro, L.B. Ultrasound-based techniques for the diagnosis of liver steatosis. World J. Gastroenterol. 2019, 25, 6053–6062. [Google Scholar] [CrossRef]
- Koch, L.K.; Yeh, M.M. Nonalcoholic fatty liver disease (NAFLD): Diagnosis, pitfalls, and staging. Ann. Diagn. Pathol. 2018, 37, 83–90. [Google Scholar] [CrossRef]
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565. [Google Scholar] [CrossRef]
- Bellentani, S. The epidemiology of non-alcoholic fatty liver disease. Liver Int. 2017, 37 (Suppl. 1), 81–84. [Google Scholar] [CrossRef]
- Haufe, S.; Engeli, S.; Kast, P.; Böhnke, J.; Utz, W.; Haas, V.; Hermsdorf, M.; Mähler, A.; Wiesner, S.; Birkenfeld, A.L.; et al. Randomized comparison of reduced fat and reduced carbohydrate hypocaloric diets on intrahepatic fat in overweight and obese human subjects. Hepatology 2011, 53, 1504–1514. [Google Scholar] [CrossRef]
- Haufe, S.; Haas, V.; Utz, W.; Birkenfeld, A.L.; Jeran, S.; Böhnke, J.; Mähler, A.; Luft, F.C.; Schulz-Menger, J.; Boschmann, M.; et al. Long-lasting improvements in liver fat and metabolism despite body weight regain after dietary weight loss. Diabetes Care 2013, 36, 3786–3792. [Google Scholar] [CrossRef]
- Rogina, B.; Reenan, R.A.; Nilsen, S.P.; Helfand, S.L. Extended life-span conferred by cotransporter gene mutations in Drosophila. Science 2000, 290, 2137–2140. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Zhuang, L.; Maddox, D.M.; Smith, S.B.; Ganapathy, V. Structure, function, and expression pattern of a novel sodium-coupled citrate transporter (NaCT) cloned from mammalian brain. J. Biol. Chem. 2002, 277, 39469–39476. [Google Scholar] [CrossRef] [PubMed]
- Birkenfeld, A.L.; Lee, H.Y.; Guebre-Egziabher, F.; Alves, T.C.; Jurczak, M.J.; Jornayvaz, F.R.; Zhang, D.; Hsiao, J.J.; Martin-Montalvo, A.; Fischer-Rosinsky, A.; et al. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. 2011, 14, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Willmes, D.M.; Daniels, M.; Kurzbach, A.; Lieske, S.; Bechmann, N.; Schumann, T.; Henke, C.; El-Agroudy, N.N.; Da Costa Goncalves, A.C.; Peitzsch, M.; et al. The longevity gene mIndy (I’m Not Dead, Yet) affects blood pressure through sympathoadrenal mechanisms. JCI Insight 2021, 6, e136083. [Google Scholar] [CrossRef]
- Pesta, D.H.; Perry, R.J.; Guebre-Egziabher, F.; Zhang, D.; Jurczak, M.; Fischer-Rosinsky, A.; Daniels, M.A.; Willmes, D.M.; Bhanot, S.; Bornstein, S.R.; et al. Prevention of diet-induced hepatic steatosis and hepatic insulin resistance by second generation antisense oligonucleotides targeted to the longevity gene mIndy (Slc13a5). Aging 2015, 7, 1086–1093. [Google Scholar] [CrossRef]
- Brachs, S.; Winkel, A.F.; Tang, H.; Birkenfeld, A.L.; Brunner, B.; Jahn-Hofmann, K.; Margerie, D.; Ruetten, H.; Schmoll, D.; Spranger, J. Inhibition of citrate cotransporter Slc13a5/mINDY by RNAi improves hepatic insulin sensitivity and prevents diet-induced non-alcoholic fatty liver disease in mice. Mol. Metab. 2016, 5, 1072–1082. [Google Scholar] [CrossRef]
- Li, L.; Li, H.; Garzel, B.; Yang, H.; Sueyoshi, T.; Li, Q.; Shu, Y.; Zhang, J.; Hu, B.; Heyward, S.; et al. SLC13A5 is a novel transcriptional target of the pregnane X receptor and sensitizes drug-induced steatosis in human liver. Mol. Pharmacol. 2015, 87, 674–682. [Google Scholar] [CrossRef]
- von Loeffelholz, C.; Lieske, S.; Neuschäfer-Rube, F.; Willmes, D.M.; Raschzok, N.; Sauer, I.M.; König, J.; Fromm, M.F.; Horn, P.; Chatzigeorgiou, A.; et al. The human longevity gene homolog INDY and interleukin-6 interact in hepatic lipid metabolism. Hepatology 2017, 66, 616–630. [Google Scholar] [CrossRef]
- Willmes, D.M.; Kurzbach, A.; Henke, C.; Schumann, T.; Zahn, G.; Heifetz, A.; Jordan, J.; Helfand, S.L.; Birkenfeld, A.L. The longevity gene INDY (I’m Not Dead Yet) in metabolic control: Potential as pharmacological target. Pharmacol. Ther. 2018, 185, 1–11. [Google Scholar] [CrossRef]
- Sun, J.; Aluvila, S.; Kotaria, R.; Mayor, J.A.; Walters, D.E.; Kaplan, R.S. Mitochondrial and Plasma Membrane Citrate Transporters: Discovery of Selective Inhibitors and Application to Structure/Function Analysis. Mol. Cell. Pharmacol. 2010, 2, 101–110. [Google Scholar]
- Pajor, A.M.; Randolph, K.M. Inhibition of the Na+/dicarboxylate cotransporter by anthranilic acid derivatives. Mol. Pharmacol. 2007, 72, 1330–1336. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, K.; Kopel, J.J.; Sivaprakasam, S.; Jaramillo-Martinez, V.; Sutton, R.B.; Urbatsch, I.L.; Ganapathy, V. Functional analysis of a species-specific inhibitor selective for human Na+-coupled citrate transporter (NaCT/SLC13A5/mINDY). Biochem. J. 2020, 477, 4149–4165. [Google Scholar] [CrossRef] [PubMed]
- Huard, K.; Brown, J.; Jones, J.C.; Cabral, S.; Futatsugi, K.; Gorgoglione, M.; Lanba, A.; Vera, N.B.; Zhu, Y.; Yan, Q.; et al. Discovery and characterization of novel inhibitors of the sodium-coupled citrate transporter (NaCT or SLC13A5). Sci. Rep. 2015, 5, 17391. [Google Scholar] [CrossRef] [PubMed]
- Huard, K.; Gosset, J.R.; Montgomery, J.I.; Gilbert, A.; Hayward, M.M.; Magee, T.V.; Cabral, S.; Uccello, D.P.; Bahnck, K.; Brown, J.; et al. Optimization of a Dicarboxylic Series for in Vivo Inhibition of Citrate Transport by the Solute Carrier 13 (SLC13) Family. J. Med. Chem. 2016, 59, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.O.; Popov, Y.; Schuppan, D. Optimized Mouse Models for Liver Fibrosis. Methods Mol. Biol. 2017, 1559, 279–296. [Google Scholar] [CrossRef]
- Hardie, D.G. The AMP-activated protein kinase pathway--new players upstream and downstream. J. Cell Sci. 2004, 117, 5479–5487. [Google Scholar] [CrossRef]
- von Loeffelholz, C.; Coldewey, S.M.; Birkenfeld, A.L. A Narrative Review on the Role of AMPK on De Novo Lipogenesis in Non-Alcoholic Fatty Liver Disease: Evidence from Human Studies. Cells 2021, 10, 1822. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; DeBose-Boyd, R.A. Regulation of cholesterol and fatty acid synthesis. Cold Spring Harb. Perspect. Biol. 2011, 3, a004754. [Google Scholar] [CrossRef]
- Ntambi, J.M.; Miyazaki, M.; Stoehr, J.P.; Lan, H.; Kendziorski, C.M.; Yandell, B.S.; Song, Y.; Cohen, P.; Friedman, J.M.; Attie, A.D. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc. Natl. Acad. Sci. USA 2002, 99, 11482–11486. [Google Scholar] [CrossRef]
- Rives, M.L.; Shaw, M.; Zhu, B.; Hinke, S.A.; Wickenden, A.D. State-Dependent Allosteric Inhibition of the Human SLC13A5 Citrate Transporter by Hydroxysuccinic Acids, PF-06649298 and PF-06761281. Mol. Pharmacol. 2016, 90, 766–774. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. β-Hydroxybutyrate: A Signaling Metabolite. Annu. Rev. Nutr. 2017, 37, 51–76. [Google Scholar] [CrossRef] [PubMed]
- Kopel, J.; Higuchi, K.; Ristic, B.; Sato, T.; Ramachandran, S.; Ganapathy, V. The Hepatic Plasma Membrane Citrate Transporter NaCT (SLC13A5) as a Molecular Target for Metformin. Sci. Rep. 2020, 10, 8536. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Niu, Q.; Guo, Y.; Zhuang, Y.; Li, X.; Liu, J.; Li, N.; Li, Z.; Huang, F.; Qiu, Z. Regulation on Citrate Influx and Metabolism through Inhibiting SLC13A5 and ACLY: A Novel Mechanism Mediating the Therapeutic Effects of Curcumin on NAFLD. J. Agric. Food Chem. 2021, 69, 8714–8725. [Google Scholar] [CrossRef] [PubMed]
- Kopel, J.J.; Bhutia, Y.D.; Sivaprakasam, S.; Ganapathy, V. Consequences of NaCT/SLC13A5/mINDY deficiency: Good versus evil, separated only by the blood-brain barrier. Biochem. J. 2021, 478, 463–486. [Google Scholar] [CrossRef]
- Yang, Q.Z.; Spelbrink, E.M.; Nye, K.L.; Hsu, E.R.; Porter, B.E. Epilepsy and EEG Phenotype of SLC13A5 Citrate Transporter Disorder. Child Neurol. Open 2020, 7. [Google Scholar] [CrossRef]
- Fan, S.Z.; Sung, C.W.; Tsai, Y.H.; Yeh, S.R.; Lin, W.S.; Wang, P.Y. Nervous System Deletion of Mammalian INDY in Mice Mimics Dietary Restriction-Induced Memory Enhancement. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2021, 76, 50–56. [Google Scholar] [CrossRef]
- Gopal, E.; Miyauchi, S.; Martin, P.M.; Ananth, S.; Srinivas, S.R.; Smith, S.B.; Prasad, P.D.; Ganapathy, V. Expression and functional features of NaCT, a sodium-coupled citrate transporter, in human and rat livers and cell lines. Am. J. Physiol.-Gastrointest. Liver Physiol. 2007, 292, G402–G408. [Google Scholar] [CrossRef]
- Kaufhold, M.; Schulz, K.; Breljak, D.; Gupta, S.; Henjakovic, M.; Krick, W.; Hagos, Y.; Sabolic, I.; Burckhardt, B.C.; Burckhardt, G. Differential interaction of dicarboxylates with human sodium-dicarboxylate cotransporter 3 and organic anion transporters 1 and 3. Am. J. Physiol. Ren. Physiol. 2011, 301, F1026–F1034. [Google Scholar] [CrossRef]
- Lang, P.; Hasselwander, S.; Li, H.; Xia, N. Effects of different diets used in diet-induced obesity models on insulin resistance and vascular dysfunction in C57BL/6 mice. Sci. Rep. 2019, 9, 19556. [Google Scholar] [CrossRef]
- Li, J.; Wu, H.; Liu, Y.; Yang, L. High fat diet induced obesity model using four strains of mice: Kunming, C57BL/6, BALB/c and ICR. Exp. Anim. 2020, 69, 326–335. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [14C] Substrate Uptake | Fatty Acid Synthesis | Cytotoxicity | |||
|---|---|---|---|---|---|
| Cell line | HepG2 | HEK293 | HEK293 | HepG2 | HepG2 |
| Species | Human | Mouse | Human | Human | Human |
| Substrate | Citrate | Citrate | Succinate | Citrate | - |
| Transporter | NaCT | Nact | NaDC3 | NaCT | - |
| IC50 | 160 nM | 180 nM | >20 µM | 1 µM | >50 µM |
| IC50 Measurements | ETG-5773 | PF-06649298 | BI01383298 |
|---|---|---|---|
| HepG2 citrate uptake (human) | 160 nM | 50 µM | 78 nM |
| HepG2 fatty acid synthesis | 1 µM | 39 µM | 30 nM |
| HEK293 citrate uptake (mouse) | 180 nM | 6.6 µM | >50 µM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zahn, G.; Willmes, D.M.; El-Agroudy, N.N.; Yarnold, C.; Jarjes-Pike, R.; Schaertl, S.; Schreiter, K.; Gehrmann, W.; Wong, A.K.C.; Zordan, T.; et al. A Novel and Cross-Species Active Mammalian INDY (NaCT) Inhibitor Ameliorates Hepatic Steatosis in Mice with Diet-Induced Obesity. Metabolites 2022, 12, 732. https://doi.org/10.3390/metabo12080732
Zahn G, Willmes DM, El-Agroudy NN, Yarnold C, Jarjes-Pike R, Schaertl S, Schreiter K, Gehrmann W, Wong AKC, Zordan T, et al. A Novel and Cross-Species Active Mammalian INDY (NaCT) Inhibitor Ameliorates Hepatic Steatosis in Mice with Diet-Induced Obesity. Metabolites. 2022; 12(8):732. https://doi.org/10.3390/metabo12080732
Chicago/Turabian StyleZahn, Grit, Diana M. Willmes, Nermeen N. El-Agroudy, Christopher Yarnold, Richard Jarjes-Pike, Sabine Schaertl, Kay Schreiter, Wiebke Gehrmann, Andrea Kuan Cie Wong, Tommaso Zordan, and et al. 2022. "A Novel and Cross-Species Active Mammalian INDY (NaCT) Inhibitor Ameliorates Hepatic Steatosis in Mice with Diet-Induced Obesity" Metabolites 12, no. 8: 732. https://doi.org/10.3390/metabo12080732
APA StyleZahn, G., Willmes, D. M., El-Agroudy, N. N., Yarnold, C., Jarjes-Pike, R., Schaertl, S., Schreiter, K., Gehrmann, W., Wong, A. K. C., Zordan, T., König, J., Jordan, J., & Birkenfeld, A. L. (2022). A Novel and Cross-Species Active Mammalian INDY (NaCT) Inhibitor Ameliorates Hepatic Steatosis in Mice with Diet-Induced Obesity. Metabolites, 12(8), 732. https://doi.org/10.3390/metabo12080732