
Altered TDP-43 Structure and Function: Key Insights into Aberrant RNA, Mitochondrial, and Cellular and Systemic Metabolism in Amyotrophic Lateral Sclerosis



Abstract
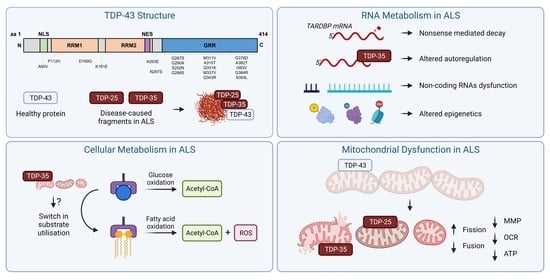
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Normal Physiological TDP-43 Structure and Function
2.1. The N-Terminal Domain
2.2. RNA Recognition Motifs
2.3. Nuclear Localisation and Export Signals
2.4. Glycine-Rich C-Terminal Domain
3. RNA Instability and Metabolism
3.1. Autoregulation of TDP-43
3.2. Phosphorylation and Autophagy of TDP-43
3.3. Alternative Splicing of RNA Targets
3.4. TDP-43 Alters Non-Coding RNA Biogenesis and Function
3.5. TDP-43 and Epigenetic Regulation
4. Oxidative Stress and Stress Granule Formation
5. Mitochondrial Dysfunction
5.1. Mitochondrial Structure
5.2. OXPHOS Complex Impairment
5.3. Disrupted Mitochondrial Pathways and Functions
5.4. Mitochondrial Dynamics
6. Mitochondria, Energy Metabolism, and TDP-43 in ALS
6.1. Mouse Models
6.2. Human Studies
7. Discussion and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mitchell, J.D.; Borasio, G.D. Amyotrophic lateral sclerosis. Lancet 2007, 369, 2031–2041. [Google Scholar] [CrossRef]
- Zufiria, M.; Gil-Bea, F.J.; Fernandez-Torron, R.; Poza, J.J.; Muñoz-Blanco, J.L.; Rojas-Garcia, R.; Riancho, J.; de Munain, A.L. ALS: A bucket of genes, environment, metabolism and unknown ingredients. Prog. Neurobiol. 2016, 142, 104–129. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Leigh, P.; Whitwell, H.; Garofalo, O.; Buller, J.; Swash, M.; Martin, J.; Gallo, J.-M.; Weller, R.; Anderton, B. Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis: Morphology, distribution, and specificity. Brain 1991, 114, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Cykowski, M.D.; Takei, H.; Schulz, P.E.; Appel, S.H.; Powell, S.Z. TDP-43 pathology in the basal forebrain and hypothalamus of patients with amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2014, 2, 171. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, F.E. TDP-43: Gumming up neurons through protein–protein and protein–RNA interactions. Trends Biochem. Sci. 2012, 37, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.-L. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef] [PubMed]
- Lattante, S.; Rouleau, G.A.; Kabashi, E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: Summary and update. Hum. Mutat. 2013, 34, 812–826. [Google Scholar] [CrossRef]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Velde, C.V.; Bouchard, J.-P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Daoud, H.; Valdmanis, P.N.; Kabashi, E.; Dion, P.; Dupre, N.; Camu, W.; Meininger, V.; Rouleau, G.A. Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. J. Med. Genet. 2009, 46, 112–114. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Wobst, H.J.; Delsing, L.; Brandon, N.J.; Moss, S.J. Truncation of the TAR DNA-binding protein 43 is not a prerequisite for cytoplasmic relocalization, and is suppressed by caspase inhibition and by introduction of the A90V sequence variant. PLoS ONE 2017, 12, e0177181. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Mazzini, L.; D’Alfonso, S.; Corrado, L.; Canosa, A.; Moglia, C.; Manera, U.; Bersano, E.; Brunetti, M.; Barberis, M. The multistep hypothesis of ALS revisited: The role of genetic mutations. Neurology 2018, 91, e635–e642. [Google Scholar] [CrossRef]
- Kuo, P.-H.; Chiang, C.-H.; Wang, Y.-T.; Doudeva, L.G.; Yuan, H.S. The crystal structure of TDP-43 RRM1-DNA complex reveals the specific recognition for UG-and TG-rich nucleic acids. Nucleic Acids Res. 2014, 42, 4712–4722. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.-H.; Doudeva, L.G.; Wang, Y.-T.; Shen, C.-K.J.; Yuan, H.S. Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res. 2009, 37, 1799–1808. [Google Scholar] [CrossRef]
- Buratti, E. TDP-43 post-translational modifications in health and disease. Expert Opin. Ther. Targets 2018, 22, 279–293. [Google Scholar] [CrossRef]
- Costessi, L.; Porro, F.; Iaconcig, A.; Muro, A.F. TDP-43 regulates β-adducin (Add2) transcript stability. RNA Biol. 2014, 11, 1280–1290. [Google Scholar] [CrossRef] [PubMed]
- Avendaño-Vázquez, S.E.; Dhir, A.; Bembich, S.; Buratti, E.; Proudfoot, N.; Baralle, F.E. Autoregulation of TDP-43 mRNA levels involves interplay between transcription, splicing, and alternative polyA site selection. Genes Dev. 2012, 26, 1679–1684. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, F.E. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front. Biosci. 2008, 13, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Freibaum, B.D.; Chitta, R.K.; High, A.A.; Taylor, J.P. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J. Proteome Res. 2010, 9, 1104–1120. [Google Scholar] [CrossRef] [PubMed]
- De Conti, L.; Akinyi, M.V.; Mendoza-Maldonado, R.; Romano, M.; Baralle, M.; Buratti, E. TDP-43 affects splicing profiles and isoform production of genes involved in the apoptotic and mitotic cellular pathways. Nucleic Acids Res. 2015, 43, 8990–9005. [Google Scholar] [CrossRef] [PubMed]
- Alami, N.H.; Smith, R.B.; Carrasco, M.A.; Williams, L.A.; Winborn, C.S.; Han, S.S.; Kiskinis, E.; Winborn, B.; Freibaum, B.D.; Kanagaraj, A. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 2014, 81, 536–543. [Google Scholar] [CrossRef]
- Kawahara, Y.; Mieda-Sato, A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 3347–3352. [Google Scholar] [CrossRef] [PubMed]
- Pacetti, M.; De Conti, L.; Marasco, L.E.; Romano, M.; Rashid, M.M.; Nubiè, M.; Baralle, F.E.; Baralle, M. Physiological tissue-specific and age-related reduction of mouse TDP-43 levels is regulated by epigenetic modifications. Dis. Models Mech. 2022, 15, dmm049032. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.R.; Prudencio, M.; Koike, Y.; Vatsavayai, S.C.; Kim, G.; Harbinski, F.; Briner, A.; Rodriguez, C.M.; Guo, C.; Akiyama, T. TDP-43 represses cryptic exon inclusion in the FTD–ALS gene UNC13A. Nature 2022, 603, 124–130. [Google Scholar] [CrossRef]
- Buratti, E.; Dörk, T.; Zuccato, E.; Pagani, F.; Romano, M.; Baralle, F.E. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001, 20, 1774–1784. [Google Scholar] [CrossRef] [PubMed]
- Shiina, Y.; Arima, K.; Tabunoki, H.; Satoh, J.-I. TDP-43 dimerizes in human cells in culture. Cell. Mol. Neurobiol. 2010, 30, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Caulfield, T.; Xu, Y.-F.; Gendron, T.F.; Hubbard, J.; Stetler, C.; Sasaguri, H.; Whitelaw, E.C.; Cai, S.; Lee, W.C. The dual functions of the extreme N-terminus of TDP-43 in regulating its biological activity and inclusion formation. Hum. Mol. Genet. 2013, 22, 3112–3122. [Google Scholar] [CrossRef] [PubMed]
- Afroz, T.; Hock, E.-M.; Ernst, P.; Foglieni, C.; Jambeau, M.; Gilhespy, L.A.; Laferriere, F.; Maniecka, Z.; Plückthun, A.; Mittl, P. Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 2017, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.-L.; Xue, W.; Hong, J.-Y.; Zhang, J.-T.; Li, M.-J.; Yu, S.-N.; He, J.-H.; Hu, H.-Y. The N-terminal dimerization is required for TDP-43 splicing activity. Sci. Rep. 2017, 7, 6196. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Conicella, A.E.; Schmidt, H.B.; Martin, E.W.; Rhoads, S.N.; Reeb, A.N.; Nourse, A.; Ramirez Montero, D.; Ryan, V.H.; Rohatgi, R. A single N-terminal phosphomimic disrupts TDP-43 polymerization, phase separation, and RNA splicing. EMBO J. 2018, 37, e97452. [Google Scholar] [CrossRef]
- Vivoli-Vega, M.; Guri, P.; Chiti, F.; Bemporad, F. Insight into the folding and dimerization mechanisms of the N-terminal domain from human TDP-43. Int. J. Mol. Sci. 2020, 21, 6259. [Google Scholar] [CrossRef] [PubMed]
- Tziortzouda, P.; Van Den Bosch, L.; Hirth, F. Triad of TDP43 control in neurodegeneration: Autoregulation, localization and aggregation. Nat. Rev. Neurosci. 2021, 22, 197–208. [Google Scholar] [CrossRef]
- Mompeán, M.; Romano, V.; Pantoja-Uceda, D.; Stuani, C.; Baralle, F.E.; Buratti, E.; Laurents, D.V. Point mutations in the N-terminal domain of transactive response DNA-binding protein 43 kDa (TDP-43) compromise its stability, dimerization, and functions. J. Biol. Chem. 2017, 292, 11992–12006. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, F.E. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator ofCFTR Exon 9. J. Biol. Chem. 2001, 276, 36337–36343. [Google Scholar] [CrossRef] [PubMed]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; König, J.; Hortobágyi, T.; Nishimura, A.L.; Župunski, V. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Lukavsky, P.J.; Daujotyte, D.; Tollervey, J.R.; Ule, J.; Stuani, C.; Buratti, E.; Baralle, F.E.; Damberger, F.F.; Allain, F.H. Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 2013, 20, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Mackness, B.C.; Tran, M.T.; McClain, S.P.; Matthews, C.R.; Zitzewitz, J.A. Folding of the RNA recognition motif (RRM) domains of the amyotrophic lateral sclerosis (ALS)-linked protein TDP-43 reveals an intermediate state. J. Biol. Chem. 2014, 289, 8264–8276. [Google Scholar] [CrossRef] [PubMed]
- Bose, J.K.; Wang, I.-F.; Hung, L.; Tarn, W.-Y.; Shen, C.-K.J. TDP-43 Overexpression Enhances Exon 7 Inclusion during the Survival of Motor Neuron Pre-mRNA Splicing. J. Biol. Chem. 2008, 283, 28852–28859. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-J.; Topp, S.D.; Hui, H.S.; Zacco, E.; Katarya, M.; McLoughlin, C.; King, A.; Smith, B.N.; Troakes, C.; Pastore, A. RRM adjacent TARDBP mutations disrupt RNA binding and enhance TDP-43 proteinopathy. Brain 2019, 142, 3753–3770. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-C.; Lin, K.-F.; He, R.-Y.; Tu, P.-H.; Koubek, J.; Hsu, Y.-C.; Huang, J.J.-T. Inhibition of TDP-43 aggregation by nucleic acid binding. PLoS ONE 2013, 8, e64002. [Google Scholar] [CrossRef]
- Liu, W.; Li, C.; Shan, J.; Wang, Y.; Chen, G. Insights into the aggregation mechanism of RNA recognition motif domains in TDP-43: A theoretical exploration. R. Soc. Open Sci. 2021, 8, 210160. [Google Scholar] [CrossRef] [PubMed]
- Shodai, A.; Morimura, T.; Ido, A.; Uchida, T.; Ayaki, T.; Takahashi, R.; Kitazawa, S.; Suzuki, S.; Shirouzu, M.; Kigawa, T. Aberrant assembly of RNA recognition motif 1 links to pathogenic conversion of TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 2013, 288, 14886–14905. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-K.; Chiang, M.-H.; Toh, E.K.-W.; Chang, C.-F.; Huang, T.-H. Molecular mechanism of oxidation-induced TDP-43 RRM1 aggregation and loss of function. FEBS Lett. 2013, 587, 575–582. [Google Scholar] [CrossRef]
- Colombrita, C.; Zennaro, E.; Fallini, C.; Weber, M.; Sommacal, A.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 2009, 111, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Ayala, Y.M.; Zago, P.; D’Ambrogio, A.; Xu, Y.-F.; Petrucelli, L.; Buratti, E.; Baralle, F.E. Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 2008, 121, 3778–3785. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Qiao, T.; Yu, J.; Wang, H.; Guo, Y.; Salameh, J.; Metterville, J.; Parsi, S.; Yusuf, I.; Brown, R.H. Low-level overexpression of wild type TDP-43 causes late-onset, progressive neurodegeneration and paralysis in mice. PLoS ONE 2022, 17, e0255710. [Google Scholar] [CrossRef]
- Winton, M.J.; Igaz, L.M.; Wong, M.M.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M.-Y. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 2008, 283, 13302–13309. [Google Scholar] [CrossRef]
- Stade, K.; Ford, C.S.; Guthrie, C.; Weis, K. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell 1997, 90, 1041–1050. [Google Scholar] [CrossRef]
- Pinarbasi, E.S.; Cağatay, T.; Fung, H.Y.J.; Li, Y.C.; Chook, Y.M.; Thomas, P.J. Active nuclear import and passive nuclear export are the primary determinants of TDP-43 localization. Sci. Rep. 2018, 8, 7083. [Google Scholar] [CrossRef]
- Buratti, E.; Brindisi, A.; Giombi, M.; Tisminetzky, S.; Ayala, Y.M.; Baralle, F.E. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: An important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem. 2005, 280, 37572–37584. [Google Scholar] [CrossRef]
- Suk, T.R.; Rousseaux, M.W. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 45. [Google Scholar] [CrossRef]
- Ayala, Y.M.; De Conti, L.; Avendaño-Vázquez, S.E.; Dhir, A.; Romano, M.; D’ambrogio, A.; Tollervey, J.; Ule, J.; Baralle, M.; Buratti, E. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011, 30, 277–288. [Google Scholar] [CrossRef]
- Mercado, P.A.; Ayala, Y.M.; Romano, M.; Buratti, E.; Baralle, F.E. Depletion of TDP 43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene. Nucleic Acids Res. 2005, 33, 6000–6010. [Google Scholar] [CrossRef] [PubMed]
- Udan, M.; Baloh, R.H. Implications of the prion-related Q/N domains in TDP-43 and FUS. Prion 2011, 5, 1–5. [Google Scholar] [CrossRef]
- Sun, Y.; Chakrabartty, A. Phase to phase with TDP-43. Biochemistry 2017, 56, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Monahan, Z.; Shewmaker, F.; Pandey, U.B. Stress granules at the intersection of autophagy and ALS. Brain Res. 2016, 1649, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, Y.; Ito, D.; Yagi, T.; Nihei, Y.; Tsunoda, Y.; Suzuki, N. Characterization of alternative isoforms and inclusion body of the TAR DNA-binding protein-43. J. Biol. Chem. 2010, 285, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.; Li, Y.; Duan, W.; Guo, Y.; Jiang, H.; Li, W.; Li, C. Full-length TDP-43 and its C-terminal fragments activate mitophagy in NSC34 cell line. Neurosci. Lett. 2012, 530, 144–149. [Google Scholar] [CrossRef]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.-J.; Vanderwyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; Sherman, M. Tar DNA binding protein-43 (TDP-43) associates with stress granules: Analysis of cultured cells and pathological brain tissue. PLoS ONE 2010, 5, e13250. [Google Scholar] [CrossRef] [PubMed]
- Igaz, L.M.; Kwong, L.K.; Xu, Y.; Truax, A.C.; Uryu, K.; Neumann, M.; Clark, C.M.; Elman, L.B.; Miller, B.L.; Grossman, M. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am. J. Pathol. 2008, 173, 182–194. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Xu, Y.-F.; Cook, C.; Gendron, T.F.; Roettges, P.; Link, C.D.; Lin, W.-L.; Tong, J.; Castanedes-Casey, M.; Ash, P. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 7607–7612. [Google Scholar] [CrossRef] [PubMed]
- Che, M.-X.; Jiang, L.-L.; Li, H.-Y.; Jiang, Y.-J.; Hu, H.-Y. TDP-35 sequesters TDP-43 into cytoplasmic inclusions through binding with RNA. FEBS Lett. 2015, 589, 1920–1928. [Google Scholar] [CrossRef] [PubMed]
- Che, M.X.; Jiang, Y.J.; Xie, Y.Y.; Jiang, L.L.; Hu, H.Y. Aggregation of the 35-kDa fragment of TDP-43 causes formation of cytoplasmic inclusions and alteration of RNA processing. FASEB J. 2011, 25, 2344–2353. [Google Scholar] [CrossRef]
- Xiao, S.; Sanelli, T.; Chiang, H.; Sun, Y.; Chakrabartty, A.; Keith, J.; Rogaeva, E.; Zinman, L.; Robertson, J. Low molecular weight species of TDP-43 generated by abnormal splicing form inclusions in amyotrophic lateral sclerosis and result in motor neuron death. Acta Neuropathol. 2015, 130, 49–61. [Google Scholar] [CrossRef]
- Tsuji, H.; Arai, T.; Kametani, F.; Nonaka, T.; Yamashita, M.; Suzukake, M.; Hosokawa, M.; Yoshida, M.; Hatsuta, H.; Takao, M. Molecular analysis and biochemical classification of TDP-43 proteinopathy. Brain 2012, 135, 3380–3391. [Google Scholar] [CrossRef]
- Siomi, H.; Dreyfuss, G. RNA-binding proteins as regulators of gene expression. Curr. Opin. Genet. Dev. 1997, 7, 345–353. [Google Scholar] [CrossRef]
- Corley, M.; Burns, M.C.; Yeo, G.W. How RNA-binding proteins interact with RNA: Molecules and mechanisms. Mol. Cell 2020, 78, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Barmada, S.J. Linking RNA dysfunction and neurodegeneration in amyotrophic lateral sclerosis. Neurotherapeutics 2015, 12, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Sephton, C.F.; Cenik, C.; Kucukural, A.; Dammer, E.B.; Cenik, B.; Han, Y.; Dewey, C.M.; Roth, F.P.; Herz, J.; Peng, J. Identification of Neuronal RNA Targets of TDP-43-containing Ribonucleoprotein Complexes. J. Biol. Chem. 2011, 286, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Godena, V.K.; Romano, G.; Romano, M.; Appocher, C.; Klima, R.; Buratti, E.; Baralle, F.E.; Feiguin, F. TDP-43 regulates Drosophila neuromuscular junctions growth by modulating Futsch/MAP1B levels and synaptic microtubules organization. PLoS ONE 2011, 6, e17808. [Google Scholar] [CrossRef]
- Kraemer, B.C.; Schuck, T.; Wheeler, J.M.; Robinson, L.C.; Trojanowski, J.Q.; Lee, V.M.; Schellenberg, G.D. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 2010, 119, 409–419. [Google Scholar] [CrossRef]
- Swarup, V.; Phaneuf, D.; Bareil, C.; Robertson, J.; Rouleau, G.A.; Kriz, J.; Julien, J.-P. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain 2011, 134, 2610–2626. [Google Scholar] [CrossRef]
- Sephton, C.F.; Good, S.K.; Atkin, S.; Dewey, C.M.; Mayer, P.; Herz, J.; Yu, G. TDP-43 is a developmentally regulated protein essential for early embryonic development. J. Biol. Chem. 2010, 285, 6826–6834. [Google Scholar] [CrossRef] [PubMed]
- Bembich, S.; Herzog, J.S.; De Conti, L.; Stuani, C.; Avendano-Vazquez, S.E.; Buratti, E.; Baralle, M.; Baralle, F.E. Predominance of spliceosomal complex formation over polyadenylation site selection in TDP-43 autoregulation. Nucleic Acids Res. 2014, 42, 3362–3371. [Google Scholar] [CrossRef][Green Version]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.-C.; Sun, E.; Wancewicz, E.; Mazur, C. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef]
- Sugai, A.; Kato, T.; Koyama, A.; Koike, Y.; Konno, T.; Ishihara, T.; Onodera, O. Non-genetically modified models exhibit TARDBP mRNA increase due to perturbed TDP-43 autoregulation. Neurobiol. Dis. 2019, 130, 104534. [Google Scholar] [CrossRef] [PubMed]
- Koyama, A.; Sugai, A.; Kato, T.; Ishihara, T.; Shiga, A.; Toyoshima, Y.; Koyama, M.; Konno, T.; Hirokawa, S.; Yokoseki, A. Increased cytoplasmic TARDBP mRNA in affected spinal motor neurons in ALS caused by abnormal autoregulation of TDP-43. Nucleic Acids Res. 2016, 44, 5820–5836. [Google Scholar] [CrossRef]
- D’Alton, S.; Altshuler, M.; Lewis, J. Studies of alternative isoforms provide insight into TDP-43 autoregulation and pathogenesis. RNA 2015, 21, 1419–1432. [Google Scholar] [CrossRef]
- D’Ambrogio, A.; Buratti, E.; Stuani, C.; Guarnaccia, C.; Romano, M.; Ayala, Y.M.; Baralle, F.E. Functional mapping of the interaction between TDP-43 and hnRNP A2 in vivo. Nucleic Acids Res. 2009, 37, 4116–4126. [Google Scholar] [CrossRef]
- Xu, Y.-F.; Zhang, Y.-J.; Lin, W.-L.; Cao, X.; Stetler, C.; Dickson, D.W.; Lewis, J.; Petrucelli, L. Expression of mutant TDP-43 induces neuronal dysfunction in transgenic mice. Mol. Neurodegener. 2011, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Melamed, Z.e.; López-Erauskin, J.; Baughn, M.W.; Zhang, O.; Drenner, K.; Sun, Y.; Freyermuth, F.; McMahon, M.A.; Beccari, M.S.; Artates, J.W. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 2019, 22, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.-L.; Wilkins, O.G.; Keuss, M.J.; Hill, S.E.; Zanovello, M.; Lee, W.C.; Bampton, A.; Lee, F.C.; Masino, L.; Qi, Y.A. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 2022, 603, 131–137. [Google Scholar] [CrossRef]
- Liu, E.Y.; Russ, J.; Cali, C.P.; Phan, J.M.; Amlie-Wolf, A.; Lee, E.B. Loss of nuclear TDP-43 is associated with decondensation of LINE retrotransposons. Cell Rep. 2019, 27, 1409–1421. e1406. [Google Scholar] [CrossRef]
- Ryan, V.H.; Perdikari, T.M.; Naik, M.T.; Saueressig, C.F.; Lins, J.; Dignon, G.L.; Mittal, J.; Hart, A.C.; Fawzi, N.L. Tyrosine phosphorylation regulates hnRNPA2 granule protein partitioning and reduces neurodegeneration. EMBO J. 2021, 40, e105001. [Google Scholar] [CrossRef]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Habelhah, H.; Shah, K.; Huang, L.; Ostareck-Lederer, A.; Burlingame, A.; Shokat, K.M.; Hentze, M.W.; Ronai, Z.E. ERK phosphorylation drives cytoplasmic accumulation of hnRNP-K and inhibition of mRNA translation. Nat. Cell Biol. 2001, 3, 325–330. [Google Scholar] [CrossRef]
- Hasegawa, M.; Arai, T.; Nonaka, T.; Kametani, F.; Yoshida, M.; Hashizume, Y.; Beach, T.G.; Buratti, E.; Baralle, F.; Morita, M. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2008, 64, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Brady, O.A.; Meng, P.; Zheng, Y.; Mao, Y.; Hu, F. Regulation of TDP-43 aggregation by phosphorylation andp62/SQSTM1. J. Neurochem. 2011, 116, 248–259. [Google Scholar] [CrossRef]
- Kametani, F.; Nonaka, T.; Suzuki, T.; Arai, T.; Dohmae, N.; Akiyama, H.; Hasegawa, M. Identification of casein kinase-1 phosphorylation sites on TDP-43. Biochem. Biophys. Res. Commun. 2009, 382, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, T.; Suzuki, G.; Tanaka, Y.; Kametani, F.; Hirai, S.; Okado, H.; Miyashita, T.; Saitoe, M.; Akiyama, H.; Masai, H. Phosphorylation of TAR DNA-binding protein of 43 kDa (TDP-43) by truncated casein kinase 1δ triggers mislocalization and accumulation of TDP-43. J. Biol. Chem. 2016, 291, 5473–5483. [Google Scholar] [CrossRef]
- Choksi, D.K.; Roy, B.; Chatterjee, S.; Yusuff, T.; Bakhoum, M.F.; Sengupta, U.; Ambegaokar, S.; Kayed, R.; Jackson, G.R. TDP-43 Phosphorylation by casein kinase Iε promotes oligomerization and enhances toxicity in vivo. Hum. Mol. Genet. 2014, 23, 1025–1035. [Google Scholar] [CrossRef]
- Deng, X.; Sun, X.; Yue, W.; Duan, Y.; Hu, R.; Zhang, K.; Ni, J.; Cui, J.; Wang, Q.; Chen, Y. CHMP2B regulates TDP-43 phosphorylation and cytotoxicity independent of autophagy via CK1. J. Cell Biol. 2021, 221, e202103033. [Google Scholar] [CrossRef]
- Salado, I.G.; Redondo, M.; Bello, M.L.; Perez, C.n.; Liachko, N.F.; Kraemer, B.C.; Miguel, L.; Lecourtois, M.; Gil, C.; Martinez, A. Protein kinase CK-1 inhibitors as new potential drugs for amyotrophic lateral sclerosis. J. Med. Chem. 2014, 57, 2755–2772. [Google Scholar] [CrossRef] [PubMed]
- Alquezar, C.; Salado, I.G.; De la Encarnación, A.; Pérez, D.I.; Moreno, F.; Gil, C.; de Munain, A.L.; Martínez, A.; Martín-Requero, Á. Targeting TDP-43 phosphorylation by Casein Kinase-1δ inhibitors: A novel strategy for the treatment of frontotemporal dementia. Mol. Neurodegener. 2016, 11, 36. [Google Scholar] [CrossRef]
- Arnold, E.S.; Ling, S.-C.; Huelga, S.C.; Lagier-Tourenne, C.; Polymenidou, M.; Ditsworth, D.; Kordasiewicz, H.B.; McAlonis-Downes, M.; Platoshyn, O.; Parone, P.A. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc. Natl. Acad. Sci. USA 2013, 110, E736–E745. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Yalamanchili, H.K.; Park, J.; De Maio, A.; Lu, H.-C.; Wan, Y.-W.; White, J.J.; Bondar, V.V.; Sayegh, L.S.; Liu, X. Extensive cryptic splicing upon loss of RBM17 and TDP43 in neurodegeneration models. Hum. Mol. Genet. 2016, 25, 5083–5093. [Google Scholar] [CrossRef]
- Hu, F.; Padukkavidana, T.; Vægter, C.B.; Brady, O.A.; Zheng, Y.; Mackenzie, I.R.; Feldman, H.H.; Nykjaer, A.; Strittmatter, S.M. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 2010, 68, 654–667. [Google Scholar] [CrossRef]
- Mohagheghi, F.; Prudencio, M.; Stuani, C.; Cook, C.; Jansen-West, K.; Dickson, D.W.; Petrucelli, L.; Buratti, E. TDP-43 functions within a network of hnRNP proteins to inhibit the production of a truncated human SORT1 receptor. Hum. Mol. Genet. 2016, 25, 534–545. [Google Scholar] [CrossRef]
- Colombrita, C.; Onesto, E.; Buratti, E.; de la Grange, P.; Gumina, V.; Baralle, F.E.; Silani, V.; Ratti, A. From transcriptomic to protein level changes in TDP-43 and FUS loss-of-function cell models. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2015, 1849, 1398–1410. [Google Scholar] [CrossRef]
- Gumina, V.; Colombrita, C.; Fallini, C.; Bossolasco, P.; Maraschi, A.M.; Landers, J.E.; Silani, V.; Ratti, A. TDP-43 and NOVA-1 RNA-binding proteins as competitive splicing regulators of the schizophrenia-associated TNIK gene. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2019, 1862, 194413. [Google Scholar] [CrossRef]
- Klim, J.R.; Williams, L.A.; Limone, F.; Guerra San Juan, I.; Davis-Dusenbery, B.N.; Mordes, D.A.; Burberry, A.; Steinbaugh, M.J.; Gamage, K.K.; Kirchner, R. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 2019, 22, 167–179. [Google Scholar] [CrossRef] [PubMed]
- San Juan, I.G.; Nash, L.A.; Smith, K.S.; Leyton-Jaimes, M.F.; Qian, M.; Klim, J.R.; Limone, F.; Dorr, A.B.; Couto, A.; Pintacuda, G. Loss of mouse Stmn2 function causes motor neuropathy. Neuron 2022, 110, 1671–1688. e1676. [Google Scholar] [CrossRef] [PubMed]
- Torres, P.; Ramírez-Núñez, O.; Romero-Guevara, R.; Barés, G.; Granado-Serrano, A.B.; Ayala, V.; Boada, J.; Fontdevila, L.; Povedano, M.; Sanchís, D. Cryptic exon splicing function of TARDBP interacts with autophagy in nervous tissue. Autophagy 2018, 14, 1398–1403. [Google Scholar] [CrossRef] [PubMed]
- Shiga, A.; Ishihara, T.; Miyashita, A.; Kuwabara, M.; Kato, T.; Watanabe, N.; Yamahira, A.; Kondo, C.; Yokoseki, A.; Takahashi, M. Alteration of POLDIP3 splicing associated with loss of function of TDP-43 in tissues affected with ALS. PLoS ONE 2012, 7, e43120. [Google Scholar]
- Deshaies, J.-E.; Shkreta, L.; Moszczynski, A.J.; Sidibé, H.; Semmler, S.; Fouillen, A.; Bennett, E.R.; Bekenstein, U.; Destroismaisons, L.; Toutant, J. TDP-43 regulates the alternative splicing of hnRNP A1 to yield an aggregation-prone variant in amyotrophic lateral sclerosis. Brain 2018, 141, 1320–1333. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Oiwa, K.; Murata, Y.; Komine, O.; Sobue, A.; Endo, F.; Takahashi, E.; Yamanaka, K. ALS-linked TDP-43M337V knock-in mice exhibit splicing deregulation without neurodegeneration. Mol. Brain 2020, 13, 8. [Google Scholar] [CrossRef]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef] [PubMed]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C. Molecular biology: The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, A.F.; Lee, E.S. Non-coding RNA: What is functional and what is junk? Front. Genet. 2015, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Barry, G. Integrating the roles of long and small non-coding RNA in brain function and disease. Mol. Psychiatry 2014, 19, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.; Allis, C.D. RNA meets chromatin. Genes Dev. 2005, 19, 1635–1655. [Google Scholar] [CrossRef] [PubMed]
- Brockdorff, N. Noncoding RNA and Polycomb recruitment. RNA 2013, 19, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Nagai, K.; Oubridge, C.; Kuglstatter, A.; Menichelli, E.; Isel, C.; Jovine, L. Structure, function and evolution of the signal recognition particle. EMBO J. 2003, 22, 3479–3485. [Google Scholar] [CrossRef] [PubMed]
- Malecová, B.; Morris, K.V. Transcriptional gene silencing mediated by non-coding RNAs. Curr. Opin. Mol. Ther. 2010, 12, 214. [Google Scholar]
- Varela, M.A.; Roberts, T.C.; Wood, M.J. Epigenetics and ncRNAs in brain function and disease: Mechanisms and prospects for therapy. Neurotherapeutics 2013, 10, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, I.A.; Mehler, M.F. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat. Rev. Neurosci. 2012, 13, 528–541. [Google Scholar] [CrossRef]
- Gagliardi, S.; Milani, P.; Sardone, V.; Pansarasa, O.; Cereda, C. From Transcriptome to Noncoding RNAs: Implications in ALS Mechanism. Neurol. Res. Int. 2012, 2012, 278725. [Google Scholar] [CrossRef]
- Joilin, G.; Gray, E.; Thompson, A.G.; Bobeva, Y.; Talbot, K.; Weishaupt, J.; Ludolph, A.; Malaspina, A.; Leigh, P.N.; Newbury, S.F.; et al. Identification of a potential non-coding RNA biomarker signature for amyotrophic lateral sclerosis. Brain Commun. 2020, 2, fcaa053. [Google Scholar] [CrossRef] [PubMed]
- Chekulaeva, M.; Filipowicz, W. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr. Opin. Cell Biol. 2009, 21, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Benhamed, M.; Servet, C.; Latrasse, D.; Zhang, W.; Delarue, M.; Zhou, D.-X. Histone acetyltransferase GCN5 interferes with the miRNA pathway in Arabidopsis. Cell Res. 2009, 19, 899–909. [Google Scholar] [CrossRef]
- Benigni, M.; Ricci, C.; Jones, A.R.; Giannini, F.; Al-Chalabi, A.; Battistini, S. Identification of miRNAs as potential biomarkers in cerebrospinal fluid from amyotrophic lateral sclerosis patients. Neuromolecular Med. 2016, 18, 551–560. [Google Scholar] [CrossRef]
- Waller, R.; Wyles, M.; Heath, P.R.; Kazoka, M.; Wollff, H.; Shaw, P.J.; Kirby, J. Small RNA sequencing of sporadic amyotrophic lateral sclerosis cerebrospinal fluid reveals differentially expressed miRNAs related to neural and glial activity. Front. Neurosci. 2018, 11, 731. [Google Scholar] [CrossRef]
- Banack, S.A.; Dunlop, R.A.; Cox, P.A. An miRNA fingerprint using neural-enriched extracellular vesicles from blood plasma: Towards a biomarker for amyotrophic lateral sclerosis/motor neuron disease. Open Biol. 2020, 10, 200116. [Google Scholar] [CrossRef]
- Takahashi, I.; Hama, Y.; Matsushima, M.; Hirotani, M.; Kano, T.; Hohzen, H.; Yabe, I.; Utsumi, J.; Sasaki, H. Identification of plasma microRNAs as a biomarker of sporadic amyotrophic lateral sclerosis. Mol. Brain 2015, 8, 67. [Google Scholar] [CrossRef]
- Figueroa-Romero, C.; Hur, J.; Lunn, J.S.; Paez-Colasante, X.; Bender, D.E.; Yung, R.; Sakowski, S.A.; Feldman, E.L. Expression of microRNAs in human post-mortem amyotrophic lateral sclerosis spinal cords provides insight into disease mechanisms. Mol. Cell. Neurosci. 2016, 71, 34–45. [Google Scholar] [CrossRef]
- Paez-Colasante, X.; Figueroa-Romero, C.; Rumora, A.E.; Hur, J.; Mendelson, F.E.; Hayes, J.M.; Backus, C.; Taubman, G.F.; Heinicke, L.; Walter, N.G. Cytoplasmic TDP43 binds microRNAs: New disease targets in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2020, 14, 117. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Yan, P.; Luo, S.; Lu, J.Y.; Shen, X. Cis-and trans-acting lncRNAs in pluripotency and reprogramming. Curr. Opin. Genet. Dev. 2017, 46, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, M.; Kitagawa, K.; Kotake, Y.; Niida, H.; Ohhata, T. Cell cycle regulation by long non-coding RNAs. Cell. Mol. Life Sci. 2013, 70, 4785–4794. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Tan, J.T.; Shelkovnikova, T.A. Stress granules regulate stress-induced paraspeckle assembly. J. Cell Biol. 2019, 218, 4127–4140. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Coller, J. RNA in unexpected places: Long non-coding RNA functions in diverse cellular contexts. Nat. Rev. Mol. Cell Biol. 2013, 14, 699–712. [Google Scholar] [CrossRef]
- Watanabe, T.; Cheng, E.-c.; Zhong, M.; Lin, H. Retrotransposons and pseudogenes regulate mRNAs and lncRNAs via the piRNA pathway in the germline. Genome Res. 2015, 25, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.-C.; Liang, T.Y.; Mazur, C. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef]
- Beaulieu, Y.B.; Kleinman, C.L.; Landry-Voyer, A.-M.; Majewski, J.; Bachand, F. Polyadenylation-dependent control of long noncoding RNA expression by the poly (A)-binding protein nuclear 1. PLoS Genet. 2012, 8, e1003078. [Google Scholar] [CrossRef]
- Nishimoto, Y.; Nakagawa, S.; Hirose, T.; Okano, H.J.; Takao, M.; Shibata, S.; Suyama, S.; Kuwako, K.-i.; Imai, T.; Murayama, S. The long non-coding RNA nuclear-enriched abundant transcript 1_2 induces paraspeckle formation in the motor neuron during the early phase of amyotrophic lateral sclerosis. Mol. Brain 2013, 6, 31. [Google Scholar] [CrossRef]
- Modic, M.; Grosch, M.; Rot, G.; Schirge, S.; Lepko, T.; Yamazaki, T.; Lee, F.C.; Rusha, E.; Shaposhnikov, D.; Palo, M. Cross-regulation between TDP-43 and paraspeckles promotes pluripotency-differentiation transition. Mol. Cell 2019, 74, 951–965. e913. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Duan, Y.; Duan, G.; Wang, Q.; Zhang, K.; Deng, X.; Qian, B.; Gu, J.; Ma, Z.; Zhang, S. Stress induces dynamic, cytotoxicity-antagonizing TDP-43 nuclear bodies via paraspeckle LncRNA NEAT1-mediated liquid-liquid phase separation. Mol. Cell 2020, 79, 443–458.e447. [Google Scholar] [CrossRef] [PubMed]
- Matsukawa, K.; Kukharsky, M.S.; Park, S.-K.; Park, S.; Watanabe, N.; Iwatsubo, T.; Hashimoto, T.; Liebman, S.W.; Shelkovnikova, T.A. Long non-coding RNA NEAT1_1 ameliorates TDP-43 toxicity in in vivo models of TDP-43 proteinopathy. RNA Biol. 2021, 18, 1546–1554. [Google Scholar] [CrossRef]
- Naganuma, T.; Hirose, T. Paraspeckle formation during the biogenesis of long non-coding RNAs. RNA Biol. 2013, 10, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.M.; Kabotyanski, E.B.; Reineke, L.C.; Shao, J.; Xiong, F.; Lee, J.-H.; Dubrulle, J.; Johnson, H.; Stossi, F.; Tsoi, P.S. The SINEB1 element in the long non-coding RNA Malat1 is necessary for TDP-43 proteostasis. Nucleic Acids Res. 2020, 48, 2621–2642. [Google Scholar] [CrossRef]
- Liu, X.; Li, D.; Zhang, W.; Guo, M.; Zhan, Q. Long non-coding RNA gadd7 interacts with TDP-43 and regulates Cdk6 mRNA decay. EMBO J. 2012, 31, 4415–4427. [Google Scholar] [CrossRef]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [PubMed]
- Illingworth, R.S.; Bird, A.P. CpG islands—‘A rough guide’. FEBS Lett. 2009, 583, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.V.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Romero, C.; Hur, J.; Bender, D.E.; Delaney, C.E.; Cataldo, M.D.; Smith, A.L.; Yung, R.; Ruden, D.M.; Callaghan, B.C.; Feldman, E.L. Identification of epigenetically altered genes in sporadic amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e52672. [Google Scholar] [CrossRef]
- Paez-Colasante, X.; Figueroa-Romero, C.; Sakowski, S.A.; Goutman, S.A.; Feldman, E.L. Amyotrophic lateral sclerosis: Mechanisms and therapeutics in the epigenomic era. Nat. Rev. Neurol. 2015, 11, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Berson, A.; Sartoris, A.; Nativio, R.; Van Deerlin, V.; Toledo, J.B.; Porta, S.; Liu, S.; Chung, C.-Y.; Garcia, B.A.; Lee, V.M.-Y. TDP-43 promotes neurodegeneration by impairing chromatin remodeling. Curr. Biol. 2017, 27, 3579–3590.e3576. [Google Scholar] [CrossRef]
- Masala, A.; Sanna, S.; Esposito, S.; Rassu, M.; Galioto, M.; Zinellu, A.; Carru, C.; Carrì, M.T.; Iaccarino, C.; Crosio, C. Epigenetic changes associated with the expression of amyotrophic lateral sclerosis (ALS) causing genes. Neuroscience 2018, 390, 1–11. [Google Scholar] [CrossRef]
- Sanna, S.; Esposito, S.; Masala, A.; Sini, P.; Nieddu, G.; Galioto, M.; Fais, M.; Iaccarino, C.; Cestra, G.; Crosio, C. HDAC1 inhibition ameliorates TDP-43-induced cell death in vitro and in vivo. Cell Death Dis. 2020, 11, 369. [Google Scholar] [CrossRef]
- Wang, P.; Wander, C.M.; Yuan, C.-X.; Bereman, M.S.; Cohen, T.J. Acetylation-induced TDP-43 pathology is suppressed by an HSF1-dependent chaperone program. Nat. Commun. 2017, 8, 82. [Google Scholar] [CrossRef]
- Appleby-Mallinder, C.; Schaber, E.; Kirby, J.; Shaw, P.; Cooper-Knock, J.; Heath, P.; Highley, J. TDP43 proteinopathy is associated with aberrant DNA methylation in human amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 2021, 47, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Zhang, M.; Bruni, A.C.; Maletta, R.G.; Colao, R.; Fratta, P.; Polke, J.M.; Sweeney, M.G.; Mudanohwo, E.; Nacmias, B. The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathol. 2015, 129, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Tremolizzo, L.; Messina, P.; Conti, E.; Sala, G.; Cecchi, M.; Airoldi, L.; Pastorelli, R.; Pupillo, E.; Bandettini Di Poggio, M.; Filosto, M. Whole-blood global DNA methylation is increased in amyotrophic lateral sclerosis independently of age of onset. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 98–105. [Google Scholar] [CrossRef]
- Hop, P.J.; Zwamborn, R.A.; Hannon, E.; Shireby, G.L.; Nabais, M.F.; Walker, E.M.; van Rheenen, W.; van Vugt, J.J.; Dekker, A.M.; Westeneng, H.-J. Genome-wide study of DNA methylation shows alterations in metabolic, inflammatory, and cholesterol pathways in ALS. Sci. Transl. Med. 2022, 14, eabj0264. [Google Scholar] [CrossRef] [PubMed]
- Hop, P.J.; Zwamborn, R.A.; Hannon, E.; Shireby, G.L.; Nabais, M.F.; Walker, E.M.; van Rheenen, W.; van Vugt, J.J.; Dekker, A.M.; Westeneng, H.-J. Genome-wide study of DNA methylation in amyotrophic lateral sclerosis identifies differentially methylated loci and implicates metabolic, inflammatory and cholesterol pathways. medRxiv 2021. [Google Scholar] [CrossRef]
- Morahan, J.M.; Yu, B.; Trent, R.J.; Pamphlett, R. A genome-wide analysis of brain DNA methylation identifies new candidate genes for sporadic amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2009, 10, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Gertz, B.; Chestnut, B.A.; Martin, L.J. Mitochondrial DNMT3A and DNA methylation in skeletal muscle and CNS of transgenic mouse models of ALS. Front. Cell. Neurosci. 2013, 7, 279. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Hadad, Y.; Altarescu, G.; Eldar-Geva, T.; Levi-Lahad, E.; Zhang, M.; Rogaeva, E.; Gotkine, M.; Bartok, O.; Ashwal-Fluss, R.; Kadener, S. Marked differences in C9orf72 methylation status and isoform expression between C9/ALS human embryonic and induced pluripotent stem cells. Stem Cell Rep. 2016, 7, 927–940. [Google Scholar] [CrossRef][Green Version]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown Jr, R.H.; Beal, M.F. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Kirkinezos, I.G.; Bacman, S.R.; Hernandez, D.; Oca-Cossio, J.; Arias, L.J.; Perez-Pinzon, M.A.; Bradley, W.G.; Moraes, C.T. Cytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A-SOD1 mice. J. Neurosci. 2005, 25, 164–172. [Google Scholar] [CrossRef]
- Li, Y.R.; King, O.D.; Shorter, J.; Gitler, A.D. Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 2013, 201, 361–372. [Google Scholar] [CrossRef]
- Protter, D.S.; Parker, R. Principles and properties of stress granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef]
- Orrù, S.; Coni, P.; Floris, A.; Littera, R.; Carcassi, C.; Sogos, V.; Brancia, C. Reduced stress granule formation and cell death in fibroblasts with the A382T mutation of TARDBP gene: Evidence for loss of TDP-43 nuclear function. Hum. Mol. Genet. 2016, 25, 4473–4483. [Google Scholar] [CrossRef]
- McDonald, K.K.; Aulas, A.; Destroismaisons, L.; Pickles, S.; Beleac, E.; Camu, W.; Rouleau, G.A.; Vande Velde, C. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet. 2011, 20, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Stallings, N.R.; Puttaparthi, K.; Dowling, K.J.; Luther, C.M.; Burns, D.K.; Davis, K.; Elliott, J.L. TDP-43, an ALS linked protein, regulates fat deposition and glucose homeostasis. PLoS ONE 2013, 8, e71793. [Google Scholar] [CrossRef]
- Harrison, A.F.; Shorter, J. RNA-binding proteins with prion-like domains in health and disease. Biochem. J. 2017, 474, 1417–1438. [Google Scholar] [CrossRef]
- Coyne, A.N.; Siddegowda, B.B.; Estes, P.S.; Johannesmeyer, J.; Kovalik, T.; Daniel, S.G.; Pearson, A.; Bowser, R.; Zarnescu, D.C. Futsch/MAP1B mRNA is a translational target of TDP-43 and is neuroprotective in a Drosophila model of amyotrophic lateral sclerosis. J. Neurosci. 2014, 34, 15962–15974. [Google Scholar] [CrossRef]
- Bentmann, E.; Neumann, M.; Tahirovic, S.; Rodde, R.; Dormann, D.; Haass, C. Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 2012, 287, 23079–23094. [Google Scholar] [CrossRef]
- Finelli, M.J.; Liu, K.X.; Wu, Y.; Oliver, P.L.; Davies, K.E. Oxr1 improves pathogenic cellular features of ALS-associated FUS and TDP-43 mutations. Hum. Mol. Genet. 2015, 24, 3529–3544. [Google Scholar] [CrossRef]
- Fernandes, N.; Nero, L.; Lyons, S.M.; Ivanov, P.; Mittelmeier, T.M.; Bolger, T.A.; Buchan, J.R. Stress granule assembly can facilitate but is not required for TDP-43 cytoplasmic aggregation. Biomolecules 2020, 10, 1367. [Google Scholar] [CrossRef]
- Protasoni, M.; Zeviani, M. Mitochondrial structure and bioenergetics in normal and disease conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; Da Rocha, E.L. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, L.; Lin, W.-L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Duan, W.; Guo, Y.; Jiang, H.; Li, Z.; Huang, J.; Hong, K.; Li, C. Mitochondrial dysfunction in human TDP-43 transfected NSC34 cell lines and the protective effect of dimethoxy curcumin. Brain Res. Bull. 2012, 89, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-F.; Gendron, T.F.; Zhang, Y.-J.; Lin, W.-L.; D’Alton, S.; Sheng, H.; Casey, M.C.; Tong, J.; Knight, J.; Yu, X. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci. 2010, 30, 10851–10859. [Google Scholar] [CrossRef]
- Magrane, J.; Cortez, C.; Gan, W.-B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.K.; Spiller, K.J.; Ge, G.; Zheng, A.; Xu, Y.; Zhou, M.; Tripathy, K.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M.-Y. Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol. 2015, 130, 643–660. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Chiang, P.-M.; Price, D.L.; Wong, P.C. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc. Natl. Acad. Sci. USA 2010, 107, 16325–16330. [Google Scholar] [CrossRef] [PubMed]
- Mori, F.; Tanji, K.; Zhang, H.-X.; Nishihira, Y.; Tan, C.-F.; Takahashi, H.; Wakabayashi, K. Maturation process of TDP-43-positive neuronal cytoplasmic inclusions in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol. 2008, 116, 193–203. [Google Scholar] [CrossRef]
- Gautam, M.; Xie, E.F.; Kocak, N.; Ozdinler, P.H. Mitoautophagy: A unique self-destructive path mitochondria of upper motor neurons with TDP-43 pathology take, very early in ALS. Front. Cell. Neurosci. 2019, 13, 489. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D. TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell 2020, 183, 636–649. e618. [Google Scholar] [CrossRef] [PubMed]
- Peggion, C.; Massimino, M.L.; Bonadio, R.S.; Lia, F.; Lopreiato, R.; Cagnin, S.; Calì, T.; Bertoli, A. Regulation of Endoplasmic Reticulum–Mitochondria Tethering and Ca2+ Fluxes by TDP-43 via GSK3β. Int. J. Mol. Sci. 2021, 22, 11853. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.C.; Phung, T.H.; Higgins, N.R.; Greenslade, J.E.; Prado, M.A.; Finley, D.; Karbowski, M.; Polster, B.M.; Monteiro, M.J. ALS/FTD mutations in UBQLN2 are linked to mitochondrial dysfunction through loss-of-function in mitochondrial protein import. Hum. Mol. Genet. 2021, 30, 1230–1246. [Google Scholar] [CrossRef] [PubMed]
- Salvatori, I.; Ferri, A.; Scaricamazza, S.; Giovannelli, I.; Serrano, A.; Rossi, S.; D’Ambrosi, N.; Cozzolino, M.; Giulio, A.D.; Moreno, S. Differential toxicity of TAR DNA-binding protein 43 isoforms depends on their submitochondrial localization in neuronal cells. J. Neurochem. 2018, 146, 585–597. [Google Scholar] [CrossRef]
- Onesto, E.; Colombrita, C.; Gumina, V.; Borghi, M.O.; Dusi, S.; Doretti, A.; Fagiolari, G.; Invernizzi, F.; Moggio, M.; Tiranti, V. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 2016, 4, 47. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, H.; Peixoto, P.; Konrad, C.; Palomo, G.; Bredvik, K.; Gerges, M.; Valsecchi, F.; Petrucelli, L.; Ravits, J.M.; Starkov, A. Mutant TDP-43 does not impair mitochondrial bioenergetics in vitro and in vivo. Mol. Neurodegener. 2017, 12, 37. [Google Scholar] [CrossRef]
- Debska-Vielhaber, G.; Miller, I.; Peeva, V.; Zuschratter, W.; Walczak, J.; Schreiber, S.; Petri, S.; Machts, J.; Vogt, S.; Szczepanowska, J. Impairment of mitochondrial oxidative phosphorylation in skin fibroblasts of SALS and FALS patients is rescued by in vitro treatment with ROS scavengers. Exp. Neurol. 2021, 339, 113620. [Google Scholar] [CrossRef]
- Hollensworth, S.B.; Shen, C.-C.; Sim, J.E.; Spitz, D.R.; Wilson, G.L.; LeDoux, S.P. Glial cell type-specific responses to menadione-induced oxidative stress. Free. Radic. Biol. Med. 2000, 28, 1161–1174. [Google Scholar] [CrossRef]
- Voets, A.; Huigsloot, M.; Lindsey, P.; Leenders, A.; Koopman, W.; Willems, P.; Rodenburg, R.; Smeitink, J.; Smeets, H. Transcriptional changes in OXPHOS complex I deficiency are related to anti-oxidant pathways and could explain the disturbed calcium homeostasis. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2012, 1822, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Lucini, C.B.; Braun, R.J. Mitochondrion-dependent cell death in TDP-43 proteinopathies. Biomedicines 2021, 9, 376. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Baldie, G.; Periz, G.; Wang, J. RNA-processing protein TDP-43 regulates FOXO-dependent protein quality control in stress response. PLoS Genet. 2014, 10, e1004693. [Google Scholar] [CrossRef] [PubMed]
- White, M.A.; Kim, E.; Duffy, A.; Adalbert, R.; Phillips, B.U.; Peters, O.M.; Stephenson, J.; Yang, S.; Massenzio, F.; Lin, Z. TDP-43 gains function due to perturbed autoregulation in a Tardbp knock-in mouse model of ALS-FTD. Nat. Neurosci. 2018, 21, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Craigen, W.J.; Riley, D.J. Nek1 regulates cell death and mitochondrial membrane permeability through phosphorylation of VDAC1. Cell Cycle 2009, 8, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.r.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Rostovtseva, T.K.; Bezrukov, S.M. VDAC regulation: Role of cytosolic proteins and mitochondrial lipids. J. Bioenerg. Biomembr. 2008, 40, 163–170. [Google Scholar] [CrossRef]
- Ni, H.-M.; Williams, J.A.; Ding, W.-X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Detmer, S.A.; Chan, D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Mozdy, A.; McCaffery, J.; Shaw, J. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J. Cell Biol. 2000, 151, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, L.; Liu, J.; Xie, F.; Su, B.; Wang, X. Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants 2017, 6, 25. [Google Scholar] [CrossRef]
- Perfettini, J.-L.; Roumier, T.; Kroemer, G. Mitochondrial fusion and fission in the control of apoptosis. Trends Cell Biol. 2005, 15, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-S.; Lin, S.-C. AMPK promotes autophagy by facilitating mitochondrial fission. Cell Metab. 2016, 23, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Prudent, J.; McBride, H.M. Mitochondrial dynamics: ER actin tightens the Drp1 noose. Curr. Biol. 2016, 26, R207–R209. [Google Scholar] [CrossRef] [PubMed]
- Basu, H.; Pekkurnaz, G.; Falk, J.; Wei, W.; Chin, M.; Steen, J.; Schwarz, T.L. FHL2 anchors mitochondria to actin and adapts mitochondrial dynamics to glucose supply. J. Cell Biol. 2021, 220, e201912077. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Torres, M.; Sha, H.; Halbrook, C.J.; Van den Bergh, F.; Reinert, R.B.; Yamada, T.; Wang, S.; Luo, Y.; Hunter, A.H. Endoplasmic reticulum–associated degradation regulates mitochondrial dynamics in brown adipocytes. Science 2020, 368, 54–60. [Google Scholar] [CrossRef]
- Choi, S.Y.; Lee, J.-H.; Chung, A.-Y.; Jo, Y.; Shin, J.-h.; Park, H.-C.; Kim, H.; Lopez-Gonzalez, R.; Ryu, J.R.; Sun, W. Prevention of mitochondrial impairment by inhibition of protein phosphatase 1 activity in amyotrophic lateral sclerosis. Cell Death Dis. 2020, 11, 888. [Google Scholar] [CrossRef]
- Joshi, A.U.; Saw, N.L.; Vogel, H.; Cunnigham, A.D.; Shamloo, M.; Mochly-Rosen, D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 2018, 10, e8166. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. The link between oxidative stress, redox status, bioenergetics and mitochondria in the pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352. [Google Scholar] [CrossRef]
- Hyder, F.; Rothman, D.L.; Bennett, M.R. Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc. Natl. Acad. Sci. USA 2013, 110, 3549–3554. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, B.M.; Klein, S.; Peters, E.J.; Schmidt, B.F.; Wolfe, R.R. Effect of elevated free fatty acids on glucose oxidation in normal humans. Metabolism 1988, 37, 323–329. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Heath, P.R.; Holden, H.; Kasher, P.; Kirby, J.; Shaw, P.J. Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J. Neurosci. 2007, 27, 9201–9219. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, J.J.; Park, N.Y.; Dubey, S.K.; Kim, T.; Ruan, K.; Lim, S.B.; Park, S.-H.; Ha, S.; Kovlyagina, I. Multi-omic analysis of selectively vulnerable motor neuron subtypes implicates altered lipid metabolism in ALS. Nat. Neurosci. 2021, 24, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Plaitakis, A.; Caroscio, J.T. Abnormal glutamate metabolism in amyotrophic lateral sclerosis. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1987, 22, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Ioannides, Z.A.; Ngo, S.T.; Henderson, R.D.; McCombe, P.A.; Steyn, F.J. Altered metabolic homeostasis in amyotrophic lateral sclerosis: Mechanisms of energy imbalance and contribution to disease progression. Neurodegener. Dis. 2016, 16, 382–397. [Google Scholar] [CrossRef] [PubMed]
- Steyn, F.J.; Ioannides, Z.A.; Van Eijk, R.P.; Heggie, S.; Thorpe, K.A.; Ceslis, A.; Heshmat, S.; Henders, A.K.; Wray, N.R.; van den Berg, L.H. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1016–1023. [Google Scholar] [CrossRef]
- Steyn, F.J.; Li, R.; Kirk, S.E.; Tefera, T.W.; Xie, T.Y.; Tracey, T.J.; Kelk, D.; Wimberger, E.; Garton, F.C.; Roberts, L. Altered skeletal muscle glucose–fatty acid flux in amyotrophic lateral sclerosis. Brain Commun. 2020, 2, fcaa154. [Google Scholar] [CrossRef] [PubMed]
- Tefera, T.W.; Steyn, F.J.; Ngo, S.T.; Borges, K. CNS glucose metabolism in Amyotrophic Lateral Sclerosis: A therapeutic target? Cell Biosci. 2021, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Pradat, P.-F.; Ludolph, A.C.; Loeffler, J.-P. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. 2011, 10, 75–82. [Google Scholar] [CrossRef]
- Noori, A.; Mezlini, A.M.; Hyman, B.T.; Serrano-Pozo, A.; Das, S. Systematic review and meta-analysis of human transcriptomics reveals neuroinflammation, deficient energy metabolism, and proteostasis failure across neurodegeneration. Neurobiol. Dis. 2021, 149, 105225. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.G.; Pedersen, W.A.; Camandola, S.; Rothstein, J.D.; Mattson, M.P. Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress–induced death of motor neurons in amyotrophic lateral sclerosis. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2002, 52, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Scaricamazza, S.; Salvatori, I.; Amadio, S.; Nesci, V.; Torcinaro, A.; Giacovazzo, G.; Primiano, A.; Gloriani, M.; Candelise, N.; Pieroni, L. Repurposing of Trimetazidine for Amyotrophic Lateral Sclerosis: A study in SOD1G93A mice. Br. J. Pharmacol. 2022, 179, 1732–1752. [Google Scholar] [CrossRef] [PubMed]
- Scaricamazza, S.; Salvatori, I.; Giacovazzo, G.; Loeffler, J.P.; Renè, F.; Rosina, M.; Quessada, C.; Proietti, D.; Heil, C.; Rossi, S. Skeletal-muscle metabolic reprogramming in ALS-SOD1G93A mice predates disease onset and is a promising therapeutic target. Iscience 2020, 23, 101087. [Google Scholar] [CrossRef] [PubMed]
- Pecqueur, C.; Bui, T.; Gelly, C.; Hauchard, J.; Barbot, C.; Bouillaud, F.; Ricquier, D.; Miroux, B.; Thompson, C.B. Uncoupling protein-2 controls proliferation by promoting fatty acid oxidation and limiting glycolysis-derived pyruvate utilization. FASEB J. 2008, 22, 9–18. [Google Scholar] [CrossRef]
- Chiang, P.-M.; Ling, J.; Jeong, Y.H.; Price, D.L.; Aja, S.M.; Wong, P.C. Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 16320–16324. [Google Scholar] [CrossRef]
- Yang, C.; Wang, H.; Qiao, T.; Yang, B.; Aliaga, L.; Qiu, L.; Tan, W.; Salameh, J.; McKenna-Yasek, D.M.; Smith, T. Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2014, 111, E1121–E1129. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, M.A.; Panahi, M.; Yadav, S.; Hennings, L.; Kiaei, M. Premature death of TDP-43 (A 315 T) transgenic mice due to gastrointestinal complications prior to development of full neurological symptoms of amyotrophic lateral sclerosis. Int. J. Exp. Pathol. 2013, 94, 56–64. [Google Scholar] [CrossRef]
- Stribl, C.; Samara, A.; Trümbach, D.; Peis, R.; Neumann, M.; Fuchs, H.; Gailus-Durner, V.; de Angelis, M.H.; Rathkolb, B.; Wolf, E. Mitochondrial dysfunction and decrease in body weight of a transgenic knock-in mouse model for TDP-43. J. Biol. Chem. 2014, 289, 10769–10784. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, Y.; Katsuno, M.; Niwa, J.-i.; Takagi, S.; Ishigaki, S.; Ikenaka, K.; Kawai, K.; Watanabe, H.; Yamanaka, K.; Takahashi, R. Loss of TDP-43 causes age-dependent progressive motor neuron degeneration. Brain 2013, 136, 1371–1382. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-L.; Wu, L.-S.; Lee, M.; Chang, C.-W.; Cheng, W.-C.; Fang, Y.-S.; Chen, Y.-R.; Cheng, P.-L.; Shen, C.-K.J. A robust TDP-43 knock-in mouse model of ALS. Acta Neuropathol. Commun. 2020, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Cocozza, G.; Garofalo, S.; Morotti, M.; Chece, G.; Grimaldi, A.; Lecce, M.; Scavizzi, F.; Menghini, R.; Casagrande, V.; Federici, M. The feeding behaviour of Amyotrophic Lateral Sclerosis mouse models is modulated by the Ca2+-activated KCa3.1 channels. Br. J. Pharmacol. 2021, 178, 4891–4906. [Google Scholar] [CrossRef]
- Fontanesi, L.; Bertolini, F. The TBC1D1 gene: Structure, function, and association with obesity and related traits. Vitam. Horm. 2013, 91, 77–95. [Google Scholar] [PubMed]
- Watkins, J.A.; Alix, J.J.; Shaw, P.J.; Mead, R.J. Extensive phenotypic characterisation of a human TDP-43Q331K transgenic mouse model of amyotrophic lateral sclerosis (ALS). Sci. Rep. 2021, 11, 16659. [Google Scholar] [CrossRef] [PubMed]
- Badu-Mensah, A.; Guo, X.; McAleer, C.W.; Rumsey, J.W.; Hickman, J.J. Functional skeletal muscle model derived from SOD1-mutant ALS patient iPSCs recapitulates hallmarks of disease progression. Sci. Rep. 2020, 10, 14302. [Google Scholar] [CrossRef]
- Suzuki, M.; Mikami, H.; Watanabe, T.; Yamano, T.; Yamazaki, T.; Nomura, M.; Yasui, K.; Ishikawa, H.; Ono, S. Increased expression of TDP-43 in the skin of amyotrophic lateral sclerosis. Acta Neurol. Scand. 2010, 122, 367–372. [Google Scholar] [CrossRef]
- Gerou, M.; Hall, B.; Woof, R.; Allsop, J.; Kolb, S.J.; Meyer, K.; Shaw, P.J.; Allen, S.P. Amyotrophic lateral sclerosis alters the metabolic aging profile in patient derived fibroblasts. Neurobiol. Aging 2021, 105, 64–77. [Google Scholar] [CrossRef]
- Raman, R.; Allen, S.P.; Goodall, E.F.; Kramer, S.; Ponger, L.L.; Heath, P.R.; Milo, M.; Hollinger, H.C.; Walsh, T.; Highley, J.R. Gene expression signatures in motor neurone disease fibroblasts reveal dysregulation of metabolism, hypoxia-response and RNA processing functions. Neuropathol. Appl. Neurobiol. 2015, 41, 201–226. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Konrad, C.; Sandhu, D.; Roychoudhury, D.; Schwartz, B.I.; Cheng, R.R.; Bredvik, K.; Kawamata, H.; Calder, E.L.; Studer, L. Accelerated transsulfuration metabolically defines a discrete subclass of amyotrophic lateral sclerosis patients. Neurobiol. Dis. 2020, 144, 105025. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Mora, M.I.; Garrabou, G.; Barcos, T.; Garcia-Garcia, F.; Grillo-Risco, R.; Peruga, E.; Gort, L.; Borrego-Écija, S.; Sanchez-Valle, R.; Canto-Santos, J. Bioenergetic and Autophagic Characterization of Skin Fibroblasts from C9orf72 Patients. Antioxidants 2022, 11, 1129. [Google Scholar] [CrossRef]
- Thau, N.; Knippenberg, S.; Körner, S.; Rath, K.J.; Dengler, R.; Petri, S. Decreased mRNA expression of PGC-1α and PGC-1α-regulated factors in the SOD1G93A ALS mouse model and in human sporadic ALS. J. Neuropathol. Exp. Neurol. 2012, 71, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Bouteloup, C.; Desport, J.-C.; Clavelou, P.; Guy, N.; Derumeaux-Burel, H.; Ferrier, A.; Couratier, P. Hypermetabolism in ALS patients: An early and persistent phenomenon. J. Neurol. 2009, 256, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Funalot, B.; Desport, J.-C.; Sturtz, F.; Camu, W.; Couratier, P. High metabolic level in patients with familial amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2009, 10, 113–117. [Google Scholar] [CrossRef]
- Nakken, O.; Meyer, H.E.; Stigum, H.; Holmøy, T. High BMI is associated with low ALS risk: A population-based study. Neurology 2019, 93, e424–e432. [Google Scholar] [CrossRef]
- Dorst, J.; Kühnlein, P.; Hendrich, C.; Kassubek, J.; Sperfeld, A.; Ludolph, A.C. Patients with elevated triglyceride and cholesterol serum levels have a prolonged survival in amyotrophic lateral sclerosis. J. Neurol. 2011, 258, 613–617. [Google Scholar] [CrossRef]
- Jawaid, A.; Murthy, S.B.; Wilson, A.M.; Qureshi, S.U.; Amro, M.J.; Wheaton, M.; Simpson, E.; Harati, Y.; Strutt, A.M.; York, M.K. A decrease in body mass index is associated with faster progression of motor symptoms and shorter survival in ALS. Amyotroph. Lateral Scler. 2010, 11, 542–548. [Google Scholar] [CrossRef]
- Shimizu, T.; Nagaoka, U.; Nakayama, Y.; Kawata, A.; Kugimoto, C.; Kuroiwa, Y.; Kawai, M.; Shimohata, T.; Nishizawa, M.; Mihara, B. Reduction rate of body mass index predicts prognosis for survival in amyotrophic lateral sclerosis: A multicenter study in Japan. Amyotroph. Lateral Scler. 2012, 13, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Moura, M.C.; Novaes, M.R.C.G.; Eduardo, E.J.; Zago, Y.S.; Freitas, R.D.N.B.; Casulari, L.A. Prognostic factors in amyotrophic lateral sclerosis: A population-based study. PLoS ONE 2015, 10, e0141500. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Hatazawa, J.; Brooks, R.A.; Di Chiro, G. Lowered cerebral glucose utilization in amyotrophic lateral sclerosis. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1987, 22, 580–586. [Google Scholar] [CrossRef]
- Van Weehaeghe, D.; Ceccarini, J.; Willekens, S.M.; De Vocht, J.; Van Damme, P.; Van Laere, K. Is there a glucose metabolic signature of spreading TDP-43 pathology in amyotrophic lateral sclerosis? Q. J. Nucl. Med. Mol. Imaging 2017, 64, 96–104. [Google Scholar] [CrossRef]
- Ho, W.Y.; Chang, J.-C.; Lim, K.; Cazenave-Gassiot, A.; Nguyen, A.T.; Foo, J.C.; Muralidharan, S.; Viera-Ortiz, A.; Ong, S.J.; Hor, J.H. TDP-43 mediates SREBF2-regulated gene expression required for oligodendrocyte myelination. J. Cell Biol. 2021, 220, e201910213. [Google Scholar] [CrossRef] [PubMed]
- Broeck, L.V.; Naval-Sánchez, M.; Adachi, Y.; Diaper, D.; Dourlen, P.; Chapuis, J.; Kleinberger, G.; Gistelinck, M.; Van Broeckhoven, C.; Lambert, J.-C. TDP-43 loss-of-function causes neuronal loss due to defective steroid receptor-mediated gene program switching in Drosophila. Cell Rep. 2013, 3, 160–172. [Google Scholar]
- Bigini, P.; Steffensen, K.R.; Ferrario, A.; Diomede, L.; Ferrara, G.; Barbera, S.; Salzano, S.; Fumagalli, E.; Ghezzi, P.; Mennini, T. Neuropathologic and biochemical changes during disease progression in liver X receptor β−/− mice, a model of adult neuron disease. J. Neuropathol. Exp. Neurol. 2010, 69, 593–605. [Google Scholar] [CrossRef]
- Sallam, T.; Jones, M.C.; Gilliland, T.; Zhang, L.; Wu, X.; Eskin, A.; Sandhu, J.; Casero, D.; Vallim, T.Q.; Hong, C. Feedback modulation of cholesterol metabolism by the lipid-responsive non-coding RNA LeXis. Nature 2016, 534, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Andrés-Benito, P.; Gelpi, E.; Jové, M.; Mota-Martorell, N.; Obis, È.; Portero-Otin, M.; Povedano, M.; Pujol, A.; Pamplona, R.; Ferrer, I. Lipid alterations in human frontal cortex in ALS-FTLD-TDP43 proteinopathy spectrum are partly related to peroxisome impairment. Neuropathol. Appl. Neurobiol. 2021, 47, 544–563. [Google Scholar] [CrossRef] [PubMed]
- Esteban-García, N.; Fernández-Beltrán, L.C.; Godoy-Corchuelo, J.M.; Ayala, J.L.; Matias-Guiu, J.A.; Corrochano, S. Body Complexion and Circulating Lipids in the Risk of TDP-43 Related Disorders. Front. Aging Neurosci. 2022, 14, 838141. [Google Scholar] [CrossRef] [PubMed]
- Moujalled, D.; James, J.L.; Yang, S.; Zhang, K.; Duncan, C.; Moujalled, D.M.; Parker, S.J.; Caragounis, A.; Lidgerwood, G.; Turner, B.J. Phosphorylation of hnRNP K by cyclin-dependent kinase 2 controls cytosolic accumulation of TDP-43. Hum. Mol. Genet. 2015, 24, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Sol, J.; Jové, M.; Povedano, M.; Sproviero, W.; Domínguez, R.; Piñol-Ripoll, G.; Romero-Guevara, R.; Hye, A.; Al-Chalabi, A.; Torres, P. Lipidomic traits of plasma and cerebrospinal fluid in amyotrophic lateral sclerosis correlate with disease progression. Brain Commun. 2021, 3, fcab143. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Veyrat-Durebex, C.; Bocca, C.; Patin, F.; Vourc’h, P.; Kouassi Nzoughet, J.; Lenaers, G.; Andres, C.R.; Simard, G.; Corcia, P. Lipidomics reveals cerebrospinal-fluid signatures of ALS. Sci. Rep. 2017, 7, 17652. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, X.; Li, X.; Wang, H.; Wang, T. Elevated cerebrospinal fluid homocysteine is associated with blood-brain barrier disruption in amyotrophic lateral sclerosis patients. Neurol. Sci. 2020, 41, 1865–1872. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Khalik, J.; Yutuc, E.; Crick, P.J.; Gustafsson, J.-Å.; Warner, M.; Roman, G.; Talbot, K.; Gray, E.; Griffiths, W.J.; Turner, M.R. Defective cholesterol metabolism in amyotrophic lateral sclerosis. J. Lipid Res. 2017, 58, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.; Larkin, J.R.; Claridge, T.D.; Talbot, K.; Sibson, N.R.; Turner, M.R. The longitudinal cerebrospinal fluid metabolomic profile of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 456–463. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, L.; Ngo, S.T. Altered TDP-43 Structure and Function: Key Insights into Aberrant RNA, Mitochondrial, and Cellular and Systemic Metabolism in Amyotrophic Lateral Sclerosis. Metabolites 2022, 12, 709. https://doi.org/10.3390/metabo12080709
Jiang L, Ngo ST. Altered TDP-43 Structure and Function: Key Insights into Aberrant RNA, Mitochondrial, and Cellular and Systemic Metabolism in Amyotrophic Lateral Sclerosis. Metabolites. 2022; 12(8):709. https://doi.org/10.3390/metabo12080709
Chicago/Turabian StyleJiang, Leanne, and Shyuan T. Ngo. 2022. "Altered TDP-43 Structure and Function: Key Insights into Aberrant RNA, Mitochondrial, and Cellular and Systemic Metabolism in Amyotrophic Lateral Sclerosis" Metabolites 12, no. 8: 709. https://doi.org/10.3390/metabo12080709
APA StyleJiang, L., & Ngo, S. T. (2022). Altered TDP-43 Structure and Function: Key Insights into Aberrant RNA, Mitochondrial, and Cellular and Systemic Metabolism in Amyotrophic Lateral Sclerosis. Metabolites, 12(8), 709. https://doi.org/10.3390/metabo12080709