Abstract
Activating KRAS mutations occur in about 30% of pulmonary adenocarcinoma (AC) cases and the discovery of specific inhibitors of G12C-mutated KRAS has considerably improved the prognosis for a subgroup of about 14% of non-small cell lung cancer (NSCLC) patients. However, even in patients with a KRAS G12C mutation, the overall response rate only reaches about 40% and mutations other than G12C still cannot be targeted. Despite the fact that one-carbon metabolism (1CM) and epigenetic regulation are known to be dysregulated by aberrant KRAS activity, we still lack evidence that co-treatment with drugs that regulate these factors might ameliorate response rates and patient prognosis. In this study, we show a direct dependency of Methylenetetrahydrofolate dehydrogenase 2 (MTHFD2) and Enhancer of Zeste Homolog 2 (EZH2) expression on mutationally activated KRAS and their prognostic relevance in KRAS-mutated AC. We show that aberrant KRAS activity generates a vulnerability of AC cancer cell lines to both MTHFD2 and EZH2 inhibitors. Importantly, co-inhibition of both factors was synergistically effective and comparable to KRASG12C inhibition alone, paving the way for their use in a therapeutic approach for NSCLC cancer patients.
1. Introduction
Lung cancer is one of the most aggressive and deadliest cancer types [1] and causes about 1.4 × 106 global deaths every year [2]. Pulmonary adenocarcinoma (AC) is the main subgroup of non-small cell lung cancer, accounting for nearly 40% of all cases [2,3]. On the molecular level, AC can be further subdivided, and activating mutations of KRAS define the largest molecular subgroup (~30%) [4,5]. For a long time, KRAS mutations were considered to be undruggable, but, eventually, the KRAS G12C (KRASG12C) specific inhibitor AMG510 (Sotorasib) was FDA-approved for second-line treatment of AC patients. However, mutations other than G12C are still not targetable and the overall response rate for Sotorasib among KRASG12C cases remains below its expected level, with about 60% of patients evading response [6,7,8,9].
KRAS mutations play a vital role in controlling tumor metabolism, e.g., by stimulating glucose uptake [10,11,12], and an increased need for energy and elevated aerobic glycolysis are closely associated with chemoresistance, tumor progression, and metastasis of malignant tumors [13,14,15,16]. We and others have recently shown that KRAS mutations are connected to an enhanced dependency on one-carbon metabolism (1CM) in non-small cell lung cancer (NSCLC) and colorectal cancer [17,18,19]. One-carbon metabolism includes the methionine and folate cycles and is essential for the maintenance of cellular homeostasis. Integrating the cell’s nutritional status 1CM catabolizes different carbon sources to derive one-carbon (methyl) units. In cancer cells, the high proliferation rate requires these one-carbon units for nucleotide synthesis, methylation, and reductive metabolism [20]. Furthermore, we have shown that Methylenetetrahydrofolate dehydrogenase 2 (MTHFD2) is essential for the survival of NSCLC cell lines and is a prognostic factor in AC [18]. MTHFD2 is one of the key enzymes in 1CM and is strongly expressed in embryonic development but it is almost absent in most healthy adult tissues, making it a promising potential therapeutic target for cancer treatment [21]. High levels of MTHFD2 have been associated with tumor recurrence and worse prognosis in multiple solid and liquid malignancies and participate in resistance against gemcitabine and pemetrexed [18,19,21,22,23,24,25,26,27,28].
Enhancer of Zeste Homolog 2 (EZH2) is a member of the polycomb repressive complex 2 (PRC2), which plays an important role in maintaining cellular identity by regulating the transcription of genes through deposition of the H3K27me3 repressive mark. EZH2 is upregulated in multiple malignancies, including NSCLC [29,30,31,32,33,34,35]; therefore, several attempts have been made to inhibit EZH2 as a clinical treatment approach. The specific EZH2 inhibitors GSK126 and EPZ-6438 have yet not reached clinical stages [36,37]; however, tazemetostat was granted accelerated approval at the beginning of 2020 by USFDA for Follicular Lymphoma and has since been tested in multiple solid tumors [38,39,40]. Recent findings connected EZH2 expression to KRAS mutations and metabolism. Compared to a moderate expression in KRAS wild-type (KRASWT) cell lines, expression of EZH2 is increased in cells with an activating KRAS mutation [35]. EZH2 has also been described to facilitate metabolic reprogramming in glioblastomas with a substantial increase in glycolytic metabolism [41].
A better understanding of the metabolic and epigenetic network regulated by aberrant KRAS in AC is therefore of primary importance. In the present study, we investigated the expression of MTHFD2 and EZH2 as dependent on KRAS mutational status in a cohort of primary AC patient samples. Our results highlight a functional connection between mutated KRAS, EZH2, and MTHFD2 and reveal a vulnerability of KRAS-mutated AC cancer cell lines to both MTHFD2 and EZH2 inhibitors. Importantly, our study shows that co-inhibition of both factors was synergistically effective in addition to KRASG12C inhibition alone, providing evidence that their use in a therapeutic approach for NSCLC cancer patients may increase overall response rate outcomes in KRASG12C cases.
2. Results
2.1. EZH2 and MTHFD2 Expression Correlate with KRAS Mutation Status and Clinicopathologic Characteristics in AC Patients
KRAS mutations have been connected to metabolism and epigenetic regulation in several cancer types [35,42,43]. Therefore, we investigated a cohort of 109 AC patients for activating KRAS mutations and protein expression of MTHFD2 and EZH2. The clinical characteristics of the patients are summarized in (Table 1 and Supplementary Table S1). All patients underwent surgical tumor resection without prior chemotherapy. Male patients (56%) were slightly more numerous than females and the median age at the time of diagnosis was 67 years (range 34–85 years). Most patients demonstrated a moderately differentiated disease, the frequency of T1–2 stage was 82.6%, and the majority were lymph node-negative (60.2%). The median follow-up time was 23 months and 48% of the patients deceased during the follow-up time.

Table 1.
Clinical data summary.
Among 62 patients with a clinical follow-up, we examined the mutational status of exon 2 of the KRAS gene. Eighteen samples were KRAS-mutated (G12C n = 15, G12V n = 3) and 44 were found to be KRASWT. The MTHFD2 statuses of the investigated samples were published previously [18] and can be found in Supplementary Table S1. Comparing MTHFD2 expression in KRASMUT and KRASWT samples, we observed a significant correlation between high expression of MTHFD2 and KRASMUT tumors (p = 0.0066) (Figure 1a). Furthermore, overall survival (OS) was significantly influenced by MTHFD2 expression in KRASMUT (p = 0.0178, Figure 1b) but not KRASWT cases (p = 0.2906, Figure 1c).
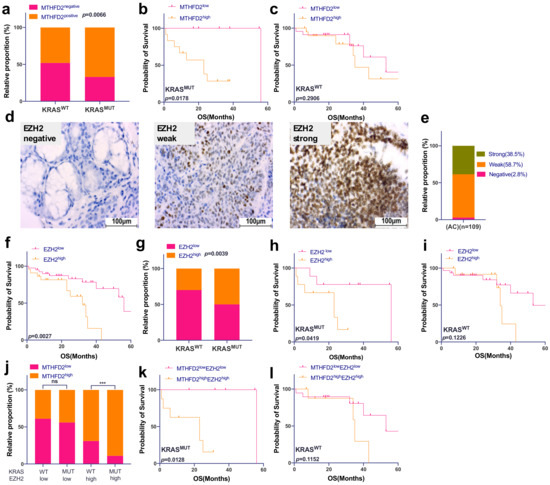
Figure 1.
EZH2 and MTHFD2 expression correlate with KRAS mutation status and patient survival in AC patients. (a) Correlation between MTHFD2 protein expression and KRAS gene mutation in AC samples (p = 0.0066). (b) Kaplan–Meier survival analysis of AC according to MTHFD2 protein expression in KRASMUT samples (p = 0.0178) and (c) KRASWT samples (p = 0.2906). (d) Human AC tissues were immunohistochemically stained for EZH2. All images were captured at 40× magnification. (e) Prevalence of EZH2 protein expression in AC patients. (f) Kaplan–Meier survival analysis according to EZH2 protein expression in AC patients. (g) Correlation between EZH2 protein expression and KRAS gene mutation in AC samples (p = 0.0039). (h) Kaplan–Meier survival analysis of AC according to EZH2 expression in KRASMUT (p = 0.0419) and (i) KRASWT (p = 0.1226) patients. (j) Correlation between MTHFD2 protein expression grouped by EZH2 protein expression and KRAS mutational status. (k) Kaplan–Meier survival analysis of AC according to both MTHFD2 and EZH2 expression in KRASMUT (p = 0.0128) and (l) KRASWT (p = 0.1152) patients. (d,e) were performed for n = 109; all the other analyses were matched samples, n = 62.
EZH2 was strongly expressed in 38.5% of investigated cases and high expression indicated a significantly shorter OS compared to low EZH2 levels (p = 0.0027) (Figure 1d–f). As for MTHFD2, EZH2 high expression was more common in KRASMUT cases (p = 0.0039, Figure 1g), and patient prognosis was significantly decreased when EZH2 was highly expressed in KRASMUT patients (p = 0.0419, Figure 1h) but not in KRASWT cases (p = 0.1126, Figure 1i).
Next, we have shown that strong expression of MTHFD2 was significantly associated both with KRAS mutations and high levels of EZH2 (p = 0.0005, Figure 1j). Patients with strong expression of both EZH2 and MTHFD2 harboring a KRAS mutation had the worst prognosis (p = 0.0128, Figure 1k), whereas no influence on OS was detected based on protein expression of EZH2 and MTHFD2 in KRASWT and (p = 0.1152, Figure 1l).
In addition, EZH2 expression and KRAS mutation significantly correlated with the occurrence of lymph node metastasis (p = 0.0003 and p = 0.0052, respectively) and poor differentiation grade (p < 0.0001 and p = 0.035) as shown in Table 2.
Table 2.
Correlation of EZH2 expression and KRAS mutation with clinicopathologic parameters.
2.2. Expression of MTHFD2 and EZH2 Depends on the Activity of Mutated KRAS in Human Pulmonary Adenocarcinoma Cell Lines
To further investigate the functional interplay between MTHFD2, EZH2, and mutated KRAS, we used two KRASG12C-mutated (HCC44 and H23) and two KRASWT (H1993 and HCC78) AC cell culture models. Western blot analyses revealed up to 70% increased levels of MTHFD2 and EZH2 in HCC44 and H23 in comparison to H1993 and HCC78 (Figure 2a and Supplementary Figure S1a,b). Treating the cells with 4μM of the KRASG12C-specific inhibitor AMG510 for 48h showed a significant decrease in cellular survival only in the KRAS-mutated cell lines HCC44 and H23 (Figure 2b). Furthermore, EZH2 and MTHFD2 protein levels were decreased only in HCC44 and H23 after AMG510 treatment, whereas there were no changes in H1993 and HCC78 (Figure 2c). Transient overexpression of KRASG12C in H1993 (KRASC12Gvec) also raised the expression of MTHFD2 and EZH2 by about 30% (Figure 2d and Supplementary Figure S1c,d). Treatment with AMG510 (4 μM, 48 h) reduced MTHFD2 and EZH2 expression only in H1993 (KRASC12Gvec) (Figure 2e). Additionally, cellular survival significantly decreased only in KRASG12C-overexpressing H1993 cells and not in cells containing the KRASWT vector (KRASWTvec) (Figure 2f). Altogether, the results show that the aberrant activity of KRASG12C cells increases the expression of MTHFD2 and EZH2 in human pulmonary AC cell lines.
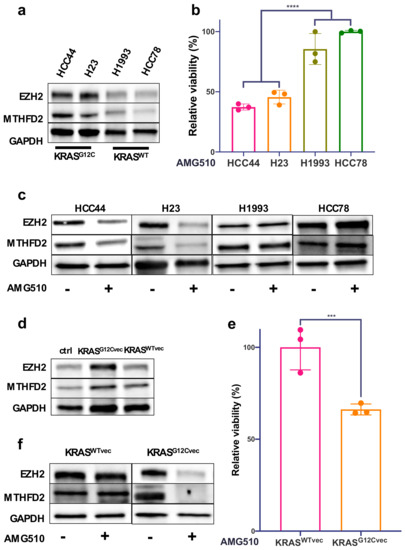
Figure 2.
Expression of MTHFD2 and EZH2 depends on mutated KRAS in human AC cell lines. (a) Western blot analysis of EZH2 and MTHFD2 expression in the AC cell lines HCC44, H23, H1993, and HCC78. (b) Cellular survival of HCC44, H23, H1993, and HCC78 cells after 48 h treatment with the KRASG12C inhibitor AMG510 (4 μM). (c) EZH2 and MTHFD2 expression in AC cells after treatment with AMG510 (4 μM) for 48 h. (d) Western blot analysis of EZH2 and MTHFD2 expression in H1993 cells transfected with KrasG12Cvec or KrasWTvec plasmids. (e) Cellular survival of KrasWTvec- and KrasG12Cvec-transfected H1993 cells after 48 h treatment with AMG510 (4 μM). (f) Western blot analysis of indicated proteins in KrasWTvec- or KrasG12Cvec-transfected H1993 cells that were treated as in (e). (*** p < 0.001, **** p < 0.0001).
2.3. EZH2 Repressive Activity Is Required to Modulate MTHFD2 Expression in KRASG12C Cell Lines
To characterize the relationship between EZH2, MTHFD2, and KRAS in AC cells, we knocked down EZH2 using two specific siRNAs. Interestingly, next to EZH2, MTHFD2, also, was robustly downregulated only in the KRASG12C cell lines HCC44 and H23 (Figure 3a,c and Supplementary Figure S2a,b). Consistently, treatment with the EZH2 methyl transferase-specific inhibitor GSK126 showed a decrease in H3K27me3 in all cells, but a decreased expression of MTHFD2 was only observed in the KRASG12C cell lines (Figure 3b). We show, also, that cellular viability upon GSK126 treatment was significantly decreased in KRASG12C H1993 KRASG12Cvec cell lines compared to KRASWT cell lines (Figure 3d,e).
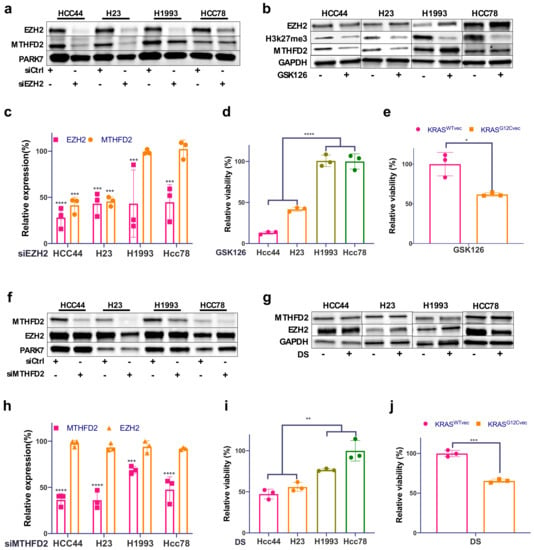
Figure 3.
MTHFD2 expression is dependent on EZH2 in KRASG12C cell lines. (a) Western blot analysis of EZH2 and MTHFD2 in the four AC cell lines HCC44, H23, H1993, and HCC78 after EZH2 siRNA knockdown and (b) after 48 h treatment with the EZH2 inhibitor GSK126 (5 μM). (c) Related quantification of EZH2 and MTHFD2 protein expression after EZH2 knockdown. (d) The cellular survival of the four described cell lines treated as in (b). (e) The cellular survival of KRASWT- and KRASG12C-transfected H1993 cells after 48h treatment with GSK126 (5μM). (f) Western blot analysis of EZH2 and MTHFD2 in the four AC cell lines after MTHFD2 siRNA knockdown and (g) after 48 h treatment with the MTHFD2 inhibitor DS (20 μM). (h) Related quantification of EZH2 and MTHFD2 protein expression after MTHFD2 knockdown. (i) The cellular survival of the four described cell lines treated as in (g). (j) The cellular survival of KRASWTvec and KRASG12Cvec H1993 cells after 48 h treatment with DS (20 μM). (* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001).
On the other hand, there was no relevant impact on EZH2 protein expression upon MTHFD2 knockdown in either KRASG12C or KRASWT cells (Figure 3f,h and Supplementary Figure S2c,d). Similarly, treating cells with the MTHFD2-specific inhibitor DS (DS18561882) did not affect EZH2 expression (Figure 3h), although DS treatment strongly reduced the cellular viability of HCC44, H23, and H1993 KRASG12Cvec cells compared to H1993, Hcc78, and KRASWTvec cells (Figure 3i,j). Altogether, we show that MTHFD2 specifically targets the KRASG12C background and that, in turn, MTHFD2 expression is subjected to KRASG12C and EZH2 activity.
2.4. Combinational Treatment of KRASG12C with EZH2 and MTHFD2 Inhibitors
Based on our results that show a strong dependency of MTHFD2 and EHZ2 on activating KRAS mutations, we decided to test whether a combinational treatment may have a synergistic effect on AC viability. We therefore treated HCC44 with increasing concentrations of AMG510 in combination with (I) GSK126 or (II) DS or (III) GSK126 together with DS. Compared to single treatments, all combinations showed an increased and synergistic reduction in cellular survival (Figure 4a–f). In particular, the combination of GSK126 and DS showed a good response that was comparable to KRASG12C inhibition with relatively low concentrations.
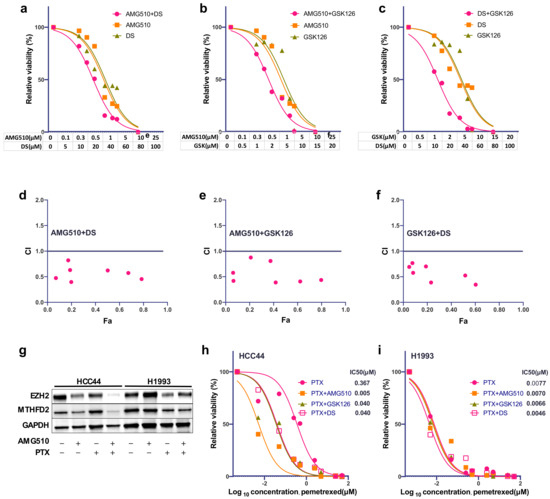
Figure 4.
Combinational treatment of KRASG12C cells with EZH2 and MTHFD2 inhibitors. (a) Cellular survival of HCC44 after treatment with AMG510, DS, or the combined treatment at indicated concentrations for 72 h. (b) Cellular survival of HCC44 after treatment with AMG510, GSK126, or the combined treatment at indicated concentrations for 72 h. (c) Cellular survival of HCC44 after treatment with DS, GSK126, or the combined treatment at indicated dosages for 72h. (d–f) Combination index (CI) plot for AMG510, DS, and GSK126 in HCC44. CI values are plotted as a function of the fractional inhibition of cell viability by computer simulation using CompuSyn; the pink dots represent the actual experimental points. CI values: synergism (CI < 0.9), additive effect (CI = 0.9–1.1), and antagonism (CI > 1.1). (g) Western blot analysis of EZH2 and MTHFD2 expression of HCC44 and H1993 after 48 h single treatment with AMG510 (2 μM) or PTX (20 μM) or in combination. (h,i) Dose–response curve of HCC44 and H1993 after 72 H single treatment with PTX (0.005–50 μM) alone or in combination with AMG510 (2 μM), GSK126 (2.5 μM), or DS (10 μM), respectively.
We have previously reported that high MTHFD2 expression induced resistance against Pemetrexed (PTX) [18] and that downregulation of KRASG12C enhances response to PTX [17]. To test whether PTX resistance in HCC44 can be recovered by KRASG12C inhibition, we incubated HCC44 and H1993 cells with either AMG510 (2 μM), PTX (20 μM), or in combination. After a single treatment, EZH2 and MTHFD2 protein levels were only partially reduced. However, the combination almost completely reduced EZH2 and MTHFD2 specifically in HCC44, meanwhile the expression in H1993 was almost unaffected (Figure 4g). Furthermore, the combination of PTX treatment with GSK126 (2.5 μM) and DS (10 μM) was about 10-fold, and the combination of PTX with AMG510 (2 μM) was 80-fold stronger than the single treatment in HCC44 (Figure 4h). On the contrary, in H1993 cells, no additional effect was observed (Figure 4i).
3. Discussion
Metastasis and recurrence after tumor resection and adjuvant therapy occur in almost 70% of pulmonary adenocarcinoma cases and keep the five-year survival rate at approximately 15% [44]. The discovery of the small-molecule inhibitor AMG510 directed against G12C-mutated KRAS significantly improved the prognosis of many affected AC patients; however, besides the mutations other than G12C that cannot be targeted, less than 40% of treated patients responded to therapy [6,7,8]. In this study, we show by immunohistochemical and in vitro approaches that KRASG12C upregulates the epigenetic regulator EZH2 and the 1CM metabolic enzyme MTHFD2, leading to molecular vulnerabilities for combinational treatment approaches.
In our study, we found that expression of both MTHFD2 and EZH2 are increased when KRAS acquires activating mutations in patients with AC. Importantly, only in KRAS-mutated cases, high expression of both MTHFD2 and EZH2 was predictive of inferior patient prognosis. Aberrant KRAS activity has been described previously as rewiring metabolism and epigenetic regulation [35,42,43]. Riquelme et al. showed that in KRASG12C-mutant NSCLC, EZH2 expression was preferentially upregulated through the MEK–ERK signaling pathway [35], and a negative prognostic association of EZH2 overexpression has been proposed in several human malignancies, including NSCLC [29,30,31,32,33,34,35]. Indeed, our data not only show a correlation between KRASG12C and EZH2 expression, but the functional ablation of EZH2 methylase activity also clearly supports its role in AC cancer cell viability. Epigenetic repression of EAF2-HIF1α by EZH2 has been shown to foster metabolic reprogramming in glioblastoma and to promote aerobic glycolysis by upregulating HK2 in prostate cancer [41,45]. In addition, high 1CM activity has been linked to increased tumor aggressiveness and reduced prognosis in several cancer entities, and a dependency of MTHFD2 on KRAS and its prognostic impact was described in AC, colorectal, and pancreatic cancer [18,19,46].
As expected, pharmacological inhibition of KRAS decreased cellular viability and reduced protein levels of MTHFD2 and EZH2 only in KRASG12C cells. By over-expressing KRASG12C in the KRASWT cell line H1993 we could validate a direct influence of KRAS activity on metabolism and the regulation of MTHFD2 and EZH2 expression. Specific knockdown or pharmacological inhibition revealed a KRASG12C-dependent MTHFD2 regulation by EZH2, whereas siRNAs against MTHFD2 had no effect on EZH2 expression. This suggests that KRASG12C activity is responsible for the upregulation of MTHFD2 subjected to the control of EZH2 methyltransferase activity. The fact that our results show MTHFD2 down-regulation in response to EZH2 inhibition indicates that such regulation most likely occurs indirectly. One obvious mechanistic explanation is that EZH2 may target an MTHFD2 transcriptional repressor. Another possibility to consider is that 3D genome organization analyses indicate that Polycomb-bound loci form insulated and self-interacting chromatin domains [47] and that the removal of EZH2 activity may induce rewiring of the MTHFD2 gene locus, leading to the preclusion of interaction with its regulatory sequences. Polycomb marks are highly enriched at CpG islands (CGIs), and H3K27me3 distribution is known to be anti-correlated with DNA methylation [48]; in this regard, EZH2-silenced foci near the MTHFD2 gene may acquire DNA methylation (epigenetic switching) [49], triggering the constitutive silencing of the entire locus in response to PRC repression inhibition.
However, the fact that MTHFD2 is transcriptionally regulated by epigenetic fluctuations does not represent the only hypothesis. MTHFD2 is relatively lowly expressed in normal tissues [47] and is upregulated in different cancers, including breast, colorectal, and hepatocellular cancers, where it plays a key role in remodeling the folate metabolism of tumor cells [48]. There have been indications that KRAS regulates MTHFD2 at a transcriptional level [49], but post-translational regulation also seems possible. It has been proposed that MTHFD2 activity and proteasomal degradation are regulated by acetylation [50,51]. Zhang et al. [51] described that the acetylation of MTHFD2 by SIRT4 leads to increased proteasomal degradation in breast cancer. They describe SIRT4 as a guardian of cell metabolism and a sensor of folate availability. Since KRAS and EZH2 are known to increase the metabolic state and the availability of folate, it could be suggested that MTHFD2 is regulated by proteasomal degradation dependent on the metabolic state of the cell. In our study, we aimed to target MTHFD and EZH2 as a therapeutic approach for NSCLC cancer patients and further studies will address the mechanism behind their action. Strikingly, the inhibition of EZH2 or MTHFD2 strongly reduces cellular viability only in KRASG12C cell lines. The increased response to inhibition of EZH2 and MTHFD2 of the KRASWT cell line H1993 overexpressing KRASG12C further suggested that KRAS activity is a main driver of the epigenetic regulation and increased 1CM activity in AC. These findings indicate that the inhibition of EZH2 and MTHFD2 will be mainly effective in KRAS-mutated AC. MTHFD2 and EZH2 inhibition have been proposed as therapeutic options in several solid and lymphatic cancers [19,36,37,38,39,40,50,51]. Importantly, here we show that the combined inhibition of mutated KRAS and either EZH2 or MTHFD2 exhibit a synergistic effect in KRASG12C cells, and, surprisingly, the co-inhibition of EZH2 and MTHFD2 had a similar synergistic effect to the combination with AMG510. These results strongly suggest that AC patients with a KRAS mutation other than KRASG12C might also profit from combined inhibition of MTHFD2 and EZH2.
We previously demonstrated that strong expression of MTHFD2, as seen in KRAS-mutated cells, leads to resistance of AC cells against treatment with PTX, which is commonly used as first-line therapy for AC patients [18]. While the combination of PTX with AMG510, GSK126, or DS had no additional effect in the KRASWT cell line H1993, we discovered a synergistic effect when we combined PTX with GSK126 or DS in the KRASG12C cell line HCC44. Since the combination of PTX with AMG510 led to a similar response to PTX in the KRASWT cell line, we also suggest PTX as a treatment option in AMG510-treated KRASG12C AC with a moderate response.
In summary, our findings indicate a causal connection between KRAS mutation status and the expression of the epigenetic regulator EZH2 and the 1CM marker MTHFD2. KRAS-mutated AC cells are vulnerable to inhibition of EZH2 and MTHFD2, and combinational treatment is as efficient as KRASG12C inhibition alone, revealing a potential treatment option for non-KRASG12C-mutated AC. In addition, we suggest PTX as a treatment option in AMG510-treated KRASG12C AC with moderate response. These findings may lead to a better separation of AC patients and an improved response rate by combinational treatment strategies.
4. Materials and Methods
4.1. Human Tissue Samples
The NSCLC tissue samples used in this study were collected from the Department of Thoracic and Cardiovascular Surgery, University Medical Center Göttingen after surgical resections. Tissues were formalin-fixed in 4% buffered formaldehyde and paraffin-embedded for diagnostic purposes. The performed experiments were approved by the ethics committee of the University Medical Center Göttingen (#1-2-08, 24-4-20) and all procedures were performed in accordance with the seventh version of the Declaration of Helsinki [52].
4.2. Immunohistochemical Staining
Tissue samples were assembled in tissue microarrays and EZH2 was immunohistochemically stained as described previously [53,54,55]. Briefly, 2 µm tissue sections were deparaffinized, rehydrated, and subsequently incubated in EnVision Flex Target Retrieval Solution pH high (Dako/Agilent, Santa Clara, CA, USA), followed by incubation with primary antibody against EZH2 (Leica, Wetzlar, Germany, NCL-L-EZH2, 1:50), secondary antibody (EnVision Flex+, Dako), and DAB (Dako) and counterstaining with Hematoxylin. Staining was evaluated under light microscopy according to the signal intensity of the stained cells: zero = no staining; one = weak staining intensity; two = strong staining intensity.
4.3. DNA Isolation and KRAS Exon 2 Profiling
DNA was isolated from 10 μm FFPE tissue sections with the InnuPREP FFPE DNA Extraction Kit (Analytik Jena, Jena, Germany), according to the manufacturer’s instructions. Considering the specificity of Sotarasib towards the G12C mutation in clinical applications, we exclusively profiled exon 2 of KRAS. Primers were designed using Primer3Web (https://primer3.ut.ee, accessed on 10 July 2022) accessed on 11 Dezember 3021. Primers spanning the specific genomic region were chosen (G12C FOR: 5′-GGCCTGCTGAAAATGAC-3′ and G12C REV: 5′-TGTATCAAAGAATGGTCCTGCAC-3′). Two-hundred nanograms of quantified DNA (Nanodrop, Peqlab, Erlangen, Germany) was PCR-amplified using 2 × MyTaqTM HS Mix (PCRBIOSYSTEMS, London, UK) with indicated KRAS primers on a labcycler (Sensoquest, Göttingen, Germany). PCR products were purified and subjected to Sanger sequencing on a 3500 Genetic Analyzer (Applied Biosystems Inc., Waltham, MA, USA) using the Applied BiosystemsTM Sanger Sequencing Kit (Applied Biosystems Inc., Waltham, MA, USA). Sequences were analyzed by comparing the acquired sequences with KRASWT using Geneious 11.1.3 software (http://www.geneious.com, accessed on 10 July 2022).
4.4. Cell Culture
The human AC cell lines HCC44, H23, H1993, and HCC78 were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were maintained in RPMI-1640 supplemented with 1% L-Glutamine, 1% Penicillin–Streptomycin, and 10% fetal bovine serum (Gibco / ThermoFisher, Waltham, MA, USA) at 37 ℃ in a 5% CO2 humidified environment. The medium was refreshed three times per week and cells were passaged at approximate confluency of 80%.
4.5. MTS and ATP Assay
Cell survival analysis was performed using the CellTiter-Glo assay and the MTS assay kit (Promega, Madison, WI, USA), according to the manufacturer’s protocol. The CellTiter-Glo assay was used to perform the ATP luminescence assay. Chemiluminescence was measured using a Tecan Reader Infinite 2000 (Tecan, Männedorf, Switzerland).
4.6. Cell Transfection with siRNA and Expression Plasmids
All siRNAs in this study were obtained from Qiagen (Qiagen, Hilden, Germany). For the cell transfection, 3 × 105 cells were transfected with 30 nM siRNA against MTHFD2 (#1 SI02664928, #2 SI02664921) or 80 nM siRNA against EZH2 (#1 SI02633316, #2 SI02665166) using HiPerFect Transfection Reagent (Qiagen), according to the manufacturer’s protocol. Allstars negative siRNA was used as a scrambled control. Plasmid transfection was performed with XtremeGENE HP DNA transfection reagent (Merck, Darmstadt, Germany). In brief, a 100 µL transfection mix containing 4 μL transfection reagent, 2 μg expression vector DNA, and serum-free RPMI-1640 cell culture medium was incubated at room temperature for 15 min and added to 3 × 105 cells seeded on a 6-well plate in 2 mL medium. Either a pCMV6-Entry-KRASG12C vector (Origene Technologies Inc., Rockville, MD, USA) or a pBabe-KRASWT vector (Addgene, Watertown, MA, USA) was transfected into H1993. Cells expressing de novo KRASG12C and KRASWT were selected with G418 and Puromycin at concentrations of 800 µg/mL and 2 µg/mL, respectively, for at least 10 days, and KRAS protein levels were confirmed by Western blotting.
4.7. Western Blotting
Cells were lysed with NP40 buffer at a pH of 7.6 containing 50 mM Tris, 150 mM NaCl, 1% NP40, 0.2% Lauryl Maltoside, 1 mM Sodium orthovanadate (Sigma-Aldrich, St. Louis, MO, USA), and 1 × protease inhibitor cocktail of cOmplete™ (Roche, Basel, Switzerland). Protein concentration was determined using a DC™ Protein Assay kit (Bio-Rad, Hercules, CA, USA), and proteins were immunoblotted as described previously [56]. Then, 15 µg denatured protein was separated on precast Mini Protein TGX gels (Bio-Rad) and transferred to a nitrocellulose membrane using the semi-dry Trans-Blot Turbo™ system. Antibodies and related secondary antibodies (Dako / Agilent, Santa Clara, CA, USA) were used at a dilution of 1:1,000 in TBST for Anti-EZH2 (Cell Signaling Technology, Danvers, MA, USA, #5246), Anti-H3K27me3 (ActiveMotif, Carlsbad, CA, USA, #39155), and Anti-MTHFD2 (Abnova, Heidelberg, Germany, #H00010797-M01). Anti-PARK7 (Abcam, Camebridge, UK, #ab18257) and Anti-GADPH (Cell Signaling Technology, #5174) were used as loading controls.
4.8. Drug Treatment Assays
Fifteen-hundred cells were seeded on a 96-well plate 24 h before drug treatment. The specific drugs used were AMG510 (MedChemExpress, USA, #HY-114277), GSK126 (Selleckchem, Houston, TX, USA, #S7061), and DS (DS18561882) (MedChemExpress, Monmouth Junction, NJ, USA, HY-130251). Cells were treated with indicated concentrations for indicated time periods. For synergistic effect assays, two inhibitors were combined in a series of nine increasing concentrations. Inhibitor concentrations ranged from 0 to 25 μM for AMG510, 0 to 20 μM for GSK126, and 0 to 100 μM for DS. In corresponding control groups, an equivalent amount of DMSO was added. After 72 h treatment, cell viability was analyzed using MTS, as described above. Drug synergy was evaluated using CompuSyn software (ComboSyn Inc, New York, NY, USA) for the combination index (CI) value calculations. CI values: antagonism (CI > 1.1), additive effect (CI < 1.1 and CI > 0.9), and synergism (CI < 0.9) [57].
4.9. Statistical Analysis
Statistical analysis was performed using GraphPad (GraphPad Software LCC, San Diego, CA, USA). Two-group comparisons were performed with Students’ t-tests. Half-maximal inhibitory concentration (IC50) analysis was performed using Pearson’s correlation test. Cell growth and resistance comparisons were analyzed using two-way ANOVA. Survival analyses were performed using the Kaplan–Meier method and the log-rank (Cox–Mantel) test. ImageJ was used for the quantification of WB signal intensities [58]. Three biological replicates were performed. The data depicted are the means ± SEMs. A p-value of <0.05 was considered significant (* p < 0.05, ** p < 0.01, *** p < 0.001).
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/metabo12070652/s1, Figure S1, Quantification of EZH2 and MTHFD2 expression in AC cell lines and H1993 cells transfected with KrasG12Cvec or KrasWTvec; Figure S2, Western blot analysis of the four AC cell lines HCC44, H23, H1993, and HCC78 after EZH2 and MTHFD2 knockdown with siRNA; Table S1, Complete patient characteristics.
Author Contributions
Conceptualization, H.B. and S.K.; investigation, Y.L., O.E., and S.Y.; resources, B.C.D., M.H., A.v.H.-E., C.F. and P.S.; writing, Y.L., C.F., S.K., and H.B. All authors have read and agreed to the published version of the manuscript.
Funding
H.B. and O.E. are supported by the Deutsche Krebshilfe Foundation (grant: 70112551). H.B. is supported by the University Medical Center Göttingen and the Else-Kröner-Fresenius-Foundation. Y.L. and S.Y. are supported by the Chinese Scholarship Counsil. C.F. is supported by the DFG (KFO5002).
Institutional Review Board Statement
The study was conducted according to the Declaration of Helsinki and approved by the Institutional Ethics Committee of the University Medical Center Goettingen (#1-2-08, 24-4-20).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available in article and Supplementary Materials.
Acknowledgments
We thank Sandra Rüngeling, Mario Düker, Cornelia Macke, and Jennifer Appelhans for their technical support.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Organization WHO; AfRo Cancer. Global Cancer Observatory (GCO). Available online: https://gco.iarc.fr/ (accessed on 15 May 2021).
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B. The 2015 World Health Organization classification of lung tumors: Impact of genetic, clinical and radiologic advances since the 2004 classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef] [Green Version]
- El Osta, B.; Behera, M.; Kim, S.; Berry, L.D.; Sica, G.; Pillai, R.N.; Owonikoko, T.K.; Kris, M.G.; Johnson, B.E.; Kwiatkowski, D.J. Characteristics and outcomes of patients with metastatic KRAS-mutant lung adenocarcinomas: The lung cancer mutation consortium experience. J. Thorac. Oncol. 2019, 14, 876–889. [Google Scholar] [CrossRef]
- Guibert, N.; Ilie, M.; Long, E.; Hofman, V.; Bouhlel, L.; Brest, P.; Mograbi, B.; Marquette, C.; Didier, A.; Mazieres, J. KRAS mutations in lung adenocarcinoma: Molecular and epidemiological characteristics, methods for detection, and therapeutic strategy perspectives. Curr. Mol. Med. 2015, 15, 418–432. [Google Scholar] [CrossRef]
- US Food and Drug Administration. FDA Grants Accelerated Approval to Sotorasib for KRAS G12C Mutated NSCLC 2021; US Food and Drug Administration: Silver Spring, MD, USA, 2021. [Google Scholar]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S. KRASG12C inhibition with sotorasib in advanced solid tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Zheng, X.; Luo, J.; Liu, W.; Ashby, C.R., Jr.; Chen, Z.-S.; Lin, L. Sotorasib: A treatment for non-small cell lung cancer with the KRAS G12C mutation. Drugs Today 2022, 58, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F. Sotorasib for lung cancers with KRAS p. G12C mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Ren, B.; Yang, G.; Wang, H.; Chen, G.; You, L.; Zhang, T.; Zhao, Y. The enhancement of glycolysis regulates pancreatic cancer metastasis. Cell. Mol. Life Sci. 2020, 77, 305–321. [Google Scholar] [CrossRef]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2021, 21, 141–162. [Google Scholar] [CrossRef]
- Kaelin, W.G.; Thompson, C.B. Clues from cell metabolism. Nature 2010, 465, 562–564. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Tozzi, F.; Chen, J.; Fan, F.; Xia, L.; Wang, J.; Gao, G.; Zhang, A.; Xia, X.; Brasher, H. Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells. Cancer Res. 2012, 72, 304–314. [Google Scholar] [CrossRef] [Green Version]
- Tao, S.; Wang, S.; Moghaddam, S.J.; Ooi, A.; Chapman, E.; Wong, P.K.; Zhang, D.D. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014, 74, 7430–7441. [Google Scholar] [CrossRef] [Green Version]
- Moran, D.M.; Trusk, P.B.; Pry, K.; Paz, K.; Sidransky, D.; Bacus, S.S. KRAS mutation status is associated with enhanced dependency on folate metabolism pathways in non–small cell lung cancer cells. Mol. Cancer Ther. 2014, 13, 1611–1624. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.; Peng, L.; Elakad, O.; Küffer, S.; Hinterthaner, M.; Danner, B.C.; von Hammerstein-Equord, A.; Ströbel, P.; Bohnenberger, H. One carbon metabolism in human lung cancer. Transl. Lung Cancer Res. 2021, 10, 2523. [Google Scholar] [CrossRef]
- Ju, H.-Q.; Lu, Y.-X.; Chen, D.-L.; Zuo, Z.-X.; Liu, Z.-X.; Wu, Q.-N.; Mo, H.-Y.; Wang, Z.-X.; Wang, D.-S.; Pu, H.-Y. Modulation of redox homeostasis by inhibition of MTHFD2 in colorectal cancer: Mechanisms and therapeutic implications. JNCI J. Natl. Cancer Inst. 2019, 111, 584–596. [Google Scholar] [CrossRef] [Green Version]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, R.; Jain, M.; Madhusudhan, N.; Sheppard, N.G.; Strittmatter, L.; Kampf, C.; Huang, J.; Asplund, A.; Mootha, V.K. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 2014, 5, 3128. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Feng, C.; Lu, Y.; Lin, Y.; Dong, C. PHGDH is an independent prognosis marker and contributes cell proliferation, migration and invasion in human pancreatic cancer. Gene 2018, 642, 43–50. [Google Scholar] [CrossRef]
- Yu, C.; Yang, L.; Cai, M.; Zhou, F.; Xiao, S.; Li, Y.; Wan, T.; Cheng, D.; Wang, L.; Zhao, C. Down-regulation of MTHFD2 inhibits NSCLC progression by suppressing cycle-related genes. J. Cell. Mol. Med. 2020, 24, 1568–1577. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Zhang, J.; Liao, M.; Yang, Y.; Wang, Y.; Yuan, Y.; Ouyang, L. Folate-mediated one-carbon metabolism: A targeting strategy in cancer therapy. Drug Discov. Today 2021, 26, 817–825. [Google Scholar] [CrossRef]
- Shang, M.; Yang, H.; Yang, R.; Chen, T.; Fu, Y.; Li, Y.; Fang, X.; Zhang, K.; Zhang, J.; Li, H. The folate cycle enzyme MTHFD2 induces cancer immune evasion through PD-L1 up-regulation. Nat. Commun. 2021, 12, 1940. [Google Scholar] [CrossRef]
- Sugiura, A.; Andrejeva, G.; Voss, K.; Heintzman, D.R.; Xu, X.; Madden, M.Z.; Ye, X.; Beier, K.L.; Chowdhury, N.U.; Wolf, M.M. MTHFD2 is a metabolic checkpoint controlling effector and regulatory T cell fate and function. Immunity 2022, 55, 65–81. [Google Scholar] [CrossRef]
- Bonagas, N.; Gustafsson, N.; Henriksson, M.; Marttila, P.; Gustafsson, R.; Wiita, E.; Borhade, S.; Green, A.C.; Vallin, K.S.; Sarno, A. Pharmacological targeting of MTHFD2 suppresses acute myeloid leukemia by inducing thymidine depletion and replication stress. Nat. Cancer 2022, 3, 156–172. [Google Scholar] [CrossRef]
- Liu, F.; Liu, Y.; He, C.; Tao, L.; He, X.; Song, H.; Zhang, G. Increased MTHFD2 expression is associated with poor prognosis in breast cancer. Tumor Biol. 2014, 35, 8685–8690. [Google Scholar] [CrossRef]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef]
- Zingg, D.; Debbache, J.; Schaefer, S.M.; Tuncer, E.; Frommel, S.C.; Cheng, P.; Arenas-Ramirez, N.; Haeusel, J.; Zhang, Y.; Bonalli, M. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat. Commun. 2015, 6, 6051. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Hou, P.; Fan, D.; Dong, M.; Ma, M.; Li, H.; Yao, R.; Li, Y.; Wang, G.; Geng, P. The degradation of EZH2 mediated by lncRNA ANCR attenuated the invasion and metastasis of breast cancer. Cell Death Differ. 2017, 24, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Shan, J.; Al-Muftah, M.A.; Al-Kowari, M.K.; Abuaqel, S.W.; Al-Rumaihi, K.; Al-Bozom, I.; Li, P.; Chouchane, L. Targeting Wnt/EZH2/microRNA-708 signaling pathway inhibits neuroendocrine differentiation in prostate cancer. Cell Death Discov. 2019, 5, 1–13. [Google Scholar] [CrossRef]
- Yu, Y.; Qi, J.; Xiong, J.; Jiang, L.; Cui, D.; He, J.; Chen, P.; Li, L.; Wu, C.; Ma, T. Epigenetic co-deregulation of EZH2/TET1 is a senescence-countering, actionable vulnerability in triple-negative breast cancer. Theranostics 2019, 9, 761. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Su, H.; Tan, Y. UNC5B-AS1 promoted ovarian cancer progression by regulating the H3K27me on NDRG2 via EZH2. Cell Biol. Int. 2020, 44, 1028–1036. [Google Scholar] [CrossRef] [PubMed]
- Riquelme, E.; Behrens, C.; Lin, H.Y.; Simon, G.; Papadimitrakopoulou, V.; Izzo, J.; Moran, C.; Kalhor, N.; Lee, J.J.; Minna, J.D. Modulation of EZH2 expression by MEK-ERK or PI3K-AKT signaling in lung cancer is dictated by different KRAS oncogene mutations. Cancer Res. 2016, 76, 675–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Diaz, E.; LaFrance, L.V. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Warholic, N.M.; Wigle, T.J.; Klaus, C.R.; Allain, C.J.; Raimondi, A.; Scott, M.P.; Chesworth, R.; Moyer, M.P.; Copeland, R.A. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 2013, 110, 7922–7927. [Google Scholar] [CrossRef] [Green Version]
- Morschhauser, F.; Tilly, H.; Chaidos, A.; McKay, P.; Phillips, T.; Assouline, S.; Batlevi, C.L.; Campbell, P.; Ribrag, V.; Damaj, G.L. Tazemetostat for patients with relapsed or refractory follicular lymphoma: An open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020, 21, 1433–1442. [Google Scholar] [CrossRef]
- Hoy, S.M. Tazemetostat: First approval. Drugs 2020, 80, 513–521. [Google Scholar] [CrossRef]
- Italiano, A. Targeting epigenetics in sarcomas through EZH2 inhibition. J. Hematol. Oncol. 2020, 13, 33. [Google Scholar] [CrossRef] [Green Version]
- Pang, B.; Zheng, X.-R.; Tian, J.-X.; Gao, T.-H.; Gu, G.-Y.; Zhang, R.; Fu, Y.-B.; Pang, Q.; Li, X.-G.; Liu, Q. EZH2 promotes metabolic reprogramming in glioblastomas through epigenetic repression of EAF2-HIF1α signaling. Oncotarget 2016, 7, 45134. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Zhou, D.; Wang, J.; Jiang, B.; Liu, X. Reflections on drug resistance to KRASG12C inhibitors and gene silencing/editing tools for targeting mutant KRAS in cancer treatment. Biochim. Biophys. Acta BBA-Rev. Cancer 2022, 1877, 188677. [Google Scholar] [CrossRef]
- Kerk, S.A.; Papagiannakopoulos, T.; Shah, Y.M.; Lyssiotis, C.A. Metabolic networks in mutant KRAS-driven tumours: Tissue specificities and the microenvironment. Nat. Rev. Cancer 2021, 21, 510–525. [Google Scholar] [CrossRef]
- Hamada, A.; Soh, J.; Mitsudomi, T. Salvage surgery after definitive chemoradiotherapy for patients with non-small cell lung cancer. Transl. Lung Cancer Res. 2021, 10, 555. [Google Scholar] [CrossRef]
- Tao, T.; Chen, M.; Jiang, R.; Guan, H.; Huang, Y.; Su, H.; Hu, Q.; Han, X.; Xiao, J. Involvement of EZH2 in aerobic glycolysis of prostate cancer through miR-181b/HK2 axis. Oncol. Rep. 2017, 37, 1430–1436. [Google Scholar] [CrossRef] [Green Version]
- Schneider, G.; Wirth, M.; Keller, U.; Saur, D. Rationale for MYC imaging and targeting in pancreatic cancer. EJNMMI Res. 2021, 11, 104. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, H.; Wang, Q.; Zhou, C.; Wei, L.; Liu, X.; Zhang, W.; Zhang, Y.; Du, Z.; Wang, X.; et al. Genome-wide analyses reveal a role of Polycomb in promoting hypomethylation of DNA methylation valleys. Genome Biol. 2018, 19, 18. [Google Scholar] [CrossRef] [Green Version]
- Ferrai, C.; Torlai Triglia, E.; Risner-Janiczek, J.R.; Rito, T.; Rackham, O.J.; de Santiago, I.; Kukalev, A.; Nicodemi, M.; Akalin, A.; Li, M.; et al. RNA polymerase II primes Polycomb-repressed developmental genes throughout terminal neuronal differentiation. Mol. Syst. Biol. 2017, 13, 946. [Google Scholar] [CrossRef]
- Gal-Yam, E.N.; Egger, G.; Iniguez, L.; Holster, H.; Einarsson, S.; Zhang, X.; Lin, J.C.; Liang, G.; Jones, P.A.; Tanay, A. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc. Natl. Acad. Sci. USA 2008, 105, 12979–12984. [Google Scholar] [CrossRef] [Green Version]
- Kawai, J.; Toki, T.; Ota, M.; Inoue, H.; Takata, Y.; Asahi, T.; Suzuki, M.; Shimada, T.; Ono, K.; Suzuki, K. Discovery of a potent, selective, and orally available MTHFD2 inhibitor (DS18561882) with in vivo antitumor activity. J. Med. Chem. 2019, 62, 10204–10220. [Google Scholar] [CrossRef]
- Almeida, L.; Dhillon-LaBrooy, A.; Castro, C.N.; Adossa, N.; Carriche, G.M.; Guderian, M.; Lippens, S.; Dennerlein, S.; Hesse, C.; Lambrecht, B.N. Ribosome-targeting antibiotics impair T cell effector function and ameliorate autoimmunity by blocking mitochondrial protein synthesis. Immunity 2021, 54, 68–83. [Google Scholar] [CrossRef]
- Association, W.M. World Medical Association Declaration of Helsinki: Ethical principles for medical research involving human subjects. JAMA 2013, 310, 2191–2194. [Google Scholar]
- Bohnenberger, H.; Kaderali, L.; Ströbel, P.; Yepes, D.; Plessmann, U.; Dharia, N.V.; Yao, S.; Heydt, C.; Merkelbach-Bruse, S.; Emmert, A. Comparative proteomics reveals a diagnostic signature for pulmonary head-and-neck cancer metastasis. EMBO Mol. Med. 2018, 10, e8428. [Google Scholar] [CrossRef]
- Buentzel, J.; Yao, S.; Elakad, O.; Lois, A.-M.; Brünies, J.; König, J.; Hinterthaner, M.; Danner, B.C.; Ströbel, P.; Emmert, A. Expression and prognostic impact of alpha thalassemia/mental retardation X-linked and death domain-associated protein in human lung cancer. Medicine 2019, 98, e16712. [Google Scholar] [CrossRef]
- Bremer, S.C.; Conradi, L.-C.; Mechie, N.-C.; Amanzada, A.; Mavropoulou, E.; Kitz, J.; Ghadimi, M.; Ellenrieder, V.; Ströbel, P.; Hessmann, E. Enhancer of zeste homolog 2 in colorectal cancer development and progression. Digestion 2021, 102, 227–235. [Google Scholar] [CrossRef]
- Elakad, O.; Lois, A.M.; Schmitz, K.; Yao, S.; Hugo, S.; Lukat, L.; Hinterthaner, M.; Danner, B.C.; von Hammerstein-Equord, A.; Reuter-Jessen, K. Fibroblast growth factor receptor 1 gene amplification and protein expression in human lung cancer. Cancer Med. 2020, 9, 3574–3583. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.-C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).