
Intracellular Toxic Advanced Glycation End-Products May Induce Cell Death and Suppress Cardiac Fibroblasts



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
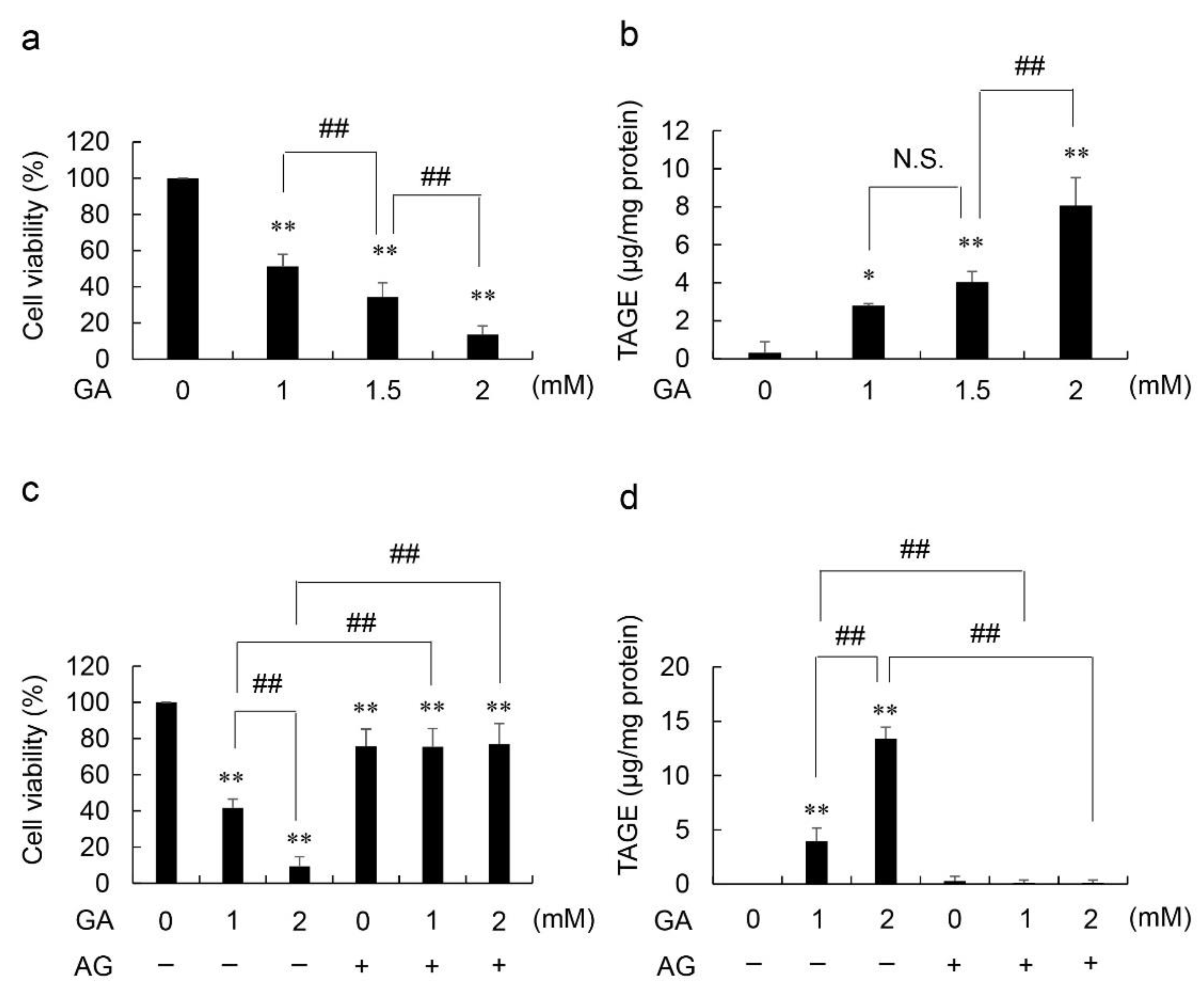
2.1. Viability Based on NADPH Activity and Accumulated Levels of Intracellular TAGE in GA- and Aminoguanidine (AG, an Inhibitor of AGE Production, the Amino Group of Which Reacts with the Carbonyl Group of Carbohydrates)-Treated HCF
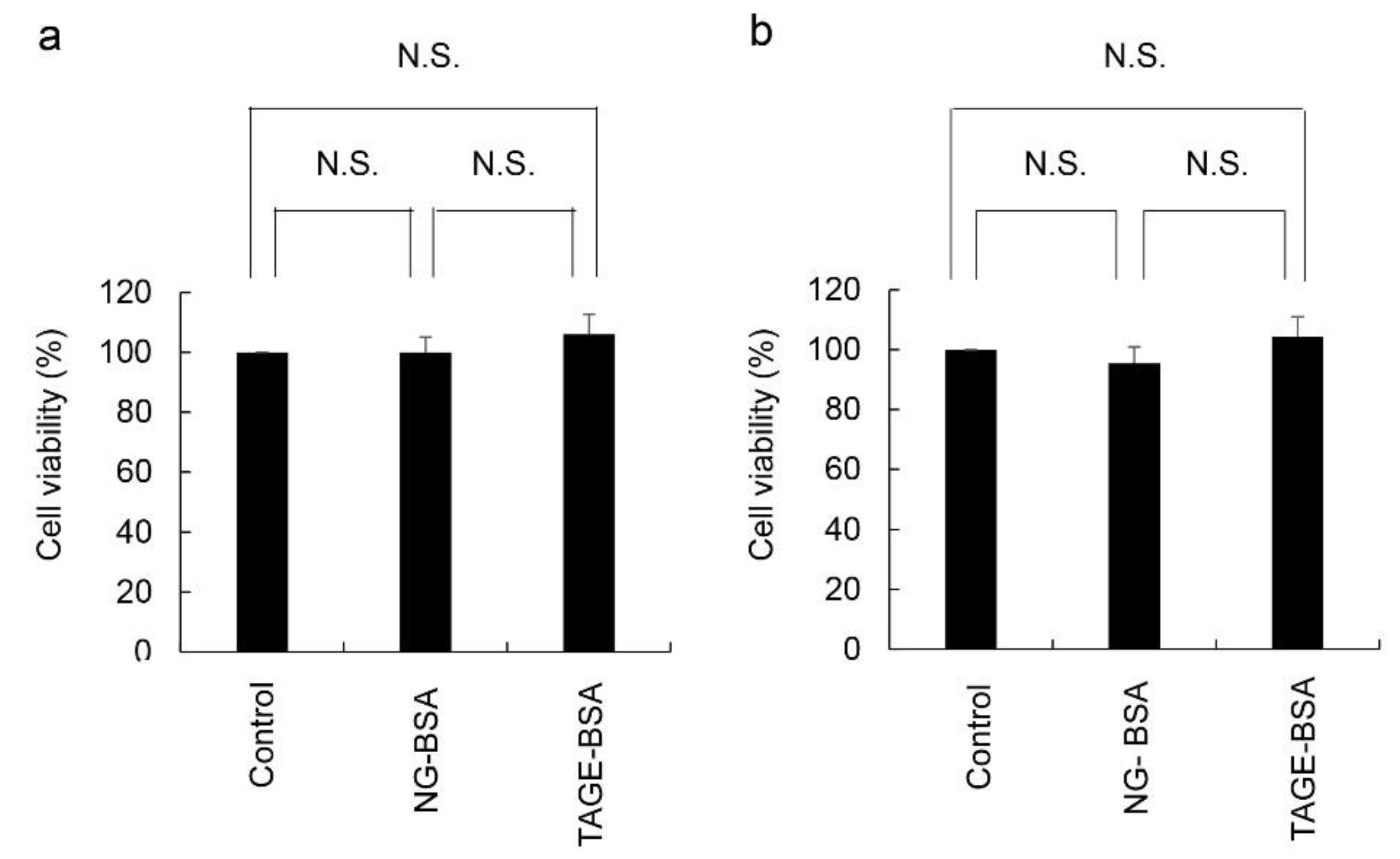
2.2. Cell Viability Based on the NADPH Activity of HCF Treated with Non-Glycated-BSA (NG-BSA) and TAGE-BSA (the Model of Extracellular TAGE)
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Viability of Cells Treated with GA only
4.3. Viability of Cells Treated with GA and AG
4.4. Measurement of Intracellular TAGE Levels in HCF Treated with GA and AG Using a Slot Blot (SB) Analysis
4.5. Treatment of HCF with NG-BSA and TAGE-BSA and Evaluation of Cell Viability
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statements
Data Availability Statement
Conflicts of Interest
Abbreviations
| AG | Aminoguanidine |
| AGEs | Advanced glycation end-products |
| GA | Glyceraldehyde |
| NG-BSA | Non-glycated bovine serum albumin |
| RAGE | Receptor for advanced glycation end-products |
| TAGE | Toxic AGEs |
| TAGE-BSA | TAGE-modified BSA |
References
- Suri, J.S.; Paul, S.; Maindarkar, M.A.; Puvvula, A.; Saxena, S.; Saba, L.; Turk, M.; Laird, J.R.; Khanna, N.N.; Viskovic, K. Cardiovascular/stroke risk stratification in Parkinson’s disease patients using atherosclerosis pathway and artificial intelligence paradigm: A systematic review. Metabolites 2022, 12, 312. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M. Serum levels of toxic AGEs (TAGE) may be a promising novel biomarker for the onset/progression of lifestyle-related diseases. Diagnostics 2016, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Flores-Vergara, R.; Olmedo, I.; Arảnguiz, P.; Riquelme, J.A.; Vivar, R.; Pedrozo, Z. Communication between cardiomyocytes and fibroblasts during cardiac ischemia/reperfusion and remodeling: Roles of TGF-β, CTGF, the renin angiotensin axis, and non-coding RNA molecules. Front. Physiol. 2021, 12, 716721. [Google Scholar] [CrossRef]
- Oguri, G.; Nakajima, T.; Yamamoto, Y.; Takano, N.; Tanaka, T.; Kikuchi, H.; Morita, T.; Nakamura, T.; Yamasoba, T.; Komuro, I. Effects of methylglyoxial on human cardiac fibroblast: Roles of transient receptor potential Ankyrin 1 (TRPA1) channels. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1339–H1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, R.M.; Burgos Villar, K.N.; Small, E.M. Fibroblast contributions to ischemic cardiac remodeling. Cell Signal. 2021, 77, 109824. [Google Scholar] [CrossRef]
- Feng, W.; Ying, Z.; Ke, F.; Mei-Lin, X. Apigenin suppresses TGF-β1-induced cardiac fibroblast differentiation and collagen synthesis trhough the downregulation of HIF-1α expression by miR-122-5p. Phytomedicine 2021, 83, 153481. [Google Scholar] [CrossRef]
- Vang, S.; Corchran, P.; Domingo, J.S.; Krick, S.; Barnes, J.W. The glycobiologyof pulmonary arterial hypertension. Metabolites 2022, 12, 316. [Google Scholar] [CrossRef]
- Takata, T.; Sakasai-Sakai, A.; Takeuchi, M. Intracellular toxic advanced glycation end-products in cardiomyocytes may cause cardiovascular disease. Sci. Rep. 2019, 9, 2121. [Google Scholar] [CrossRef] [Green Version]
- Hwang, Y.C.; Shaw, S.; Kaneko, M.; Redd, H.; Marrero, M.B.; Ramasamy, R. Aldose reductase pathway mediates JAK-STAT signaling: A novel axis in myocardial ischemic injury. FASEB J. 2005, 19, 795–797. [Google Scholar] [CrossRef]
- Bais, R.; James, H.M.; Rofe, A.M.; Conyers, R.A.J. The purification and properties of human liver ketohexokinase. Biochem. J. 1985, 250, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Ramasamy, R.; Schmidt, A.M. Receptor for advanced glycation end products (RAGE) and implications for the pathophysiology of heart failure. Curr. Heart Fail Rep. 2012, 9, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Takata, T.; Sakasai-Sakai, A.; Takeuchi, M. Intracellular toxic advanced glycation end-products in 1.4E7 cell line induce death with reduction of microtubule-associated protein 1 light chain 3 and p62. Nutrients 2022, 14, 332. [Google Scholar] [CrossRef] [PubMed]
- Takata, T.; Sakasai-Sakai, A.; Takeuchi, M. Impact of intracellular toxic advanced glycation end-products (TAGE) on murine myoblast cell death. Diabetol. Metab. Syndr. 2020, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Nakashima, N. Sodium 4-phenylbutyrate inhibits protein glycation. Biomed. Rep. 2020, 13, 61. [Google Scholar] [CrossRef] [PubMed]
- Kazashkov, M.; Chen, K.; Babiy, S.; Peter, H.Y. Evidence for in vivo scavenging by aminoguanidine of formaldehyde produced via semicarbazide-sensitive amine oxidase-mediated deamination. J. Pharmacol. Exp. Ther. 2007, 322, 1201–1207. [Google Scholar] [CrossRef]
- Takata, T.; Sakasai-Sakai, A.; Takino, J.; Takeuchi, M. Evidence for toxic advanced glycation end-products generated in the normal rat liver. Nutrients 2019, 11, 1612. [Google Scholar] [CrossRef] [Green Version]
- Sakasai-Sakai, A.; Takata, T.; Takeuchi, M. Intracellular toxic advanced glycation end-products promote the production of reactive oxygen species in HepG2 cells. Nutrients 2020, 21, 4861. [Google Scholar] [CrossRef]
- Takata, T.; Ueda, T.; Sakasai-Sakai, A.; Takeuchi, M. Generation of glyceraldehyde-derived advanced glycation end-products in pancreatic cancer cells and the potential of tumor promotion. World. J. Gastroenterol. 2017, 23, 4910–4919. [Google Scholar] [CrossRef]
- Nasu, R.; Furukawa, A.; Suzuki, K.; Takeuchi, M.; Koriyama, Y. The effect of glyceraldehyde-derived advanced glycation end products on β-tubulin-inhibited neurite outgrowth in SH-SY5Y human neuroblastoma cells. Nutrients 2020, 12, 2958. [Google Scholar] [CrossRef]
- Kuzan, A. Toxicity of advanced glycation end products (Review). Biomed. Rep. 2021, 14, 46. [Google Scholar] [CrossRef]
- Wada, R.; Muraoka, N.; Inagawa, K.; Yamakawa, H.; Miyamoto, K.; Sadahiro, T.; Umei, T.; Kaneda, R.; Suzuki, T.; Kamiya, K.; et al. Induction of human cardiomyocyte-like cells from fibroblasts by defined factors. Proc. Natl. Acad. Sci. USA 2013, 110, 12667–12672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajikawa, M.; Nakashima, A.; Fujimura, N.; Maruhashi, T.; Iwamoto, Y.; Iwamoto, A.; Matsumoto, T.; Oda, N.; Hidaka, T.; Kihara, Y. Ratio of serum levels of AGEs to soluble form of RAGE is a predictor of endothelial function. Diabetes Care 2015, 38, 119–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takino, J.; Sato, T.; Nagamine, K.; Sakasai-Sakai, A.; Takeuchi, M.; Hori, T. Suppression of hepatic stellate cell death by toxic advanced glycation end-products. Biol. Pharm. Bull. 2021, 44, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Makita, Z.; Bucala, R.; Suzuki, T.; Koike, T.; Kameda, Y. Immunological evidence that non-carboxymethyllysine adanced glycation end-products are produced from short chain sugars and dicarbonyl compounds in vivo. Mol. Med. 2000, 6, 114–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takata, T.; Sakasai-Sakai, A.; Takeuchi, M. Intracellular Toxic Advanced Glycation End-Products May Induce Cell Death and Suppress Cardiac Fibroblasts. Metabolites 2022, 12, 615. https://doi.org/10.3390/metabo12070615
Takata T, Sakasai-Sakai A, Takeuchi M. Intracellular Toxic Advanced Glycation End-Products May Induce Cell Death and Suppress Cardiac Fibroblasts. Metabolites. 2022; 12(7):615. https://doi.org/10.3390/metabo12070615
Chicago/Turabian StyleTakata, Takanobu, Akiko Sakasai-Sakai, and Masayoshi Takeuchi. 2022. "Intracellular Toxic Advanced Glycation End-Products May Induce Cell Death and Suppress Cardiac Fibroblasts" Metabolites 12, no. 7: 615. https://doi.org/10.3390/metabo12070615
APA StyleTakata, T., Sakasai-Sakai, A., & Takeuchi, M. (2022). Intracellular Toxic Advanced Glycation End-Products May Induce Cell Death and Suppress Cardiac Fibroblasts. Metabolites, 12(7), 615. https://doi.org/10.3390/metabo12070615