
Endosomal v-ATPase as a Sensor Determining Myocardial Substrate Preference

 ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
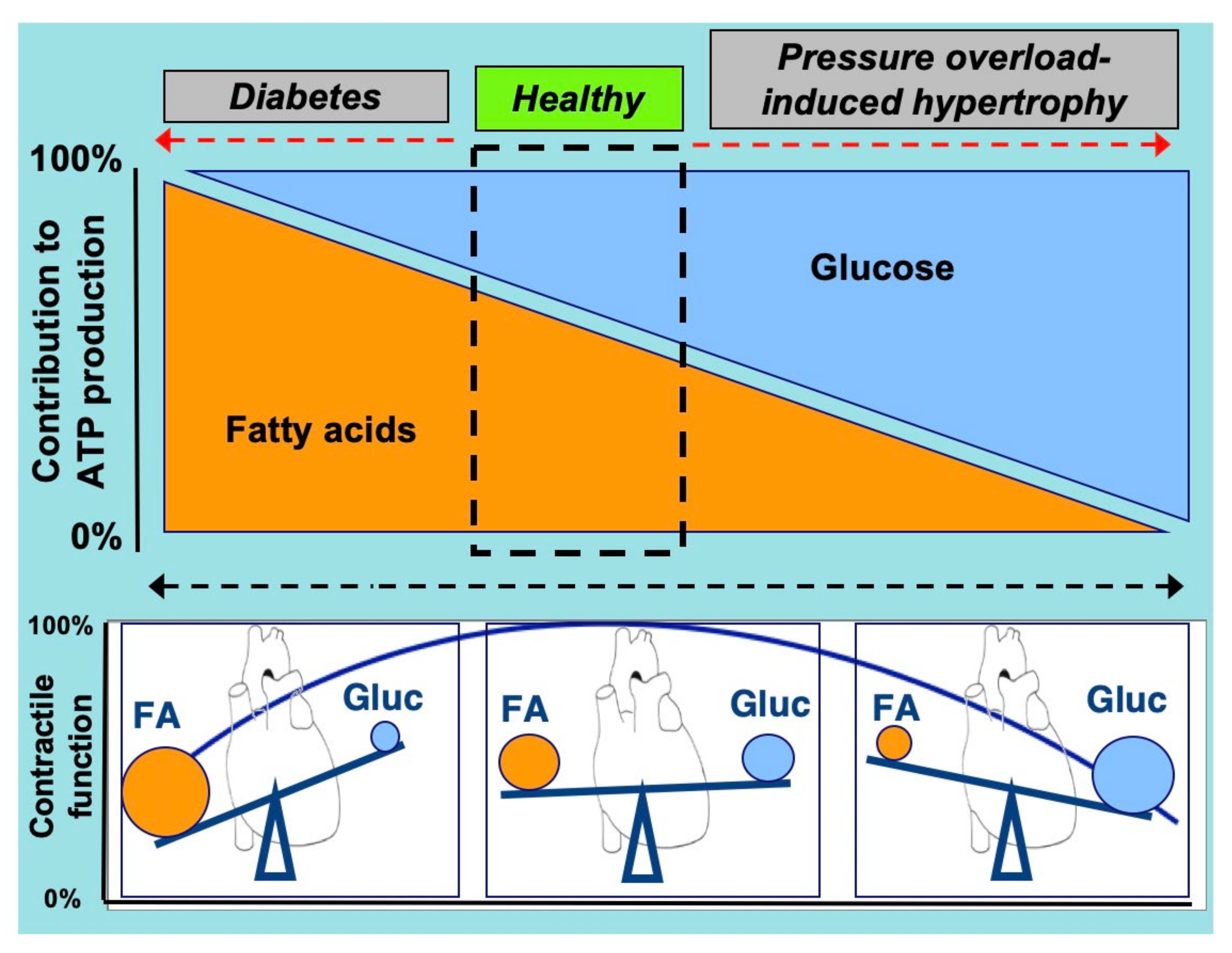
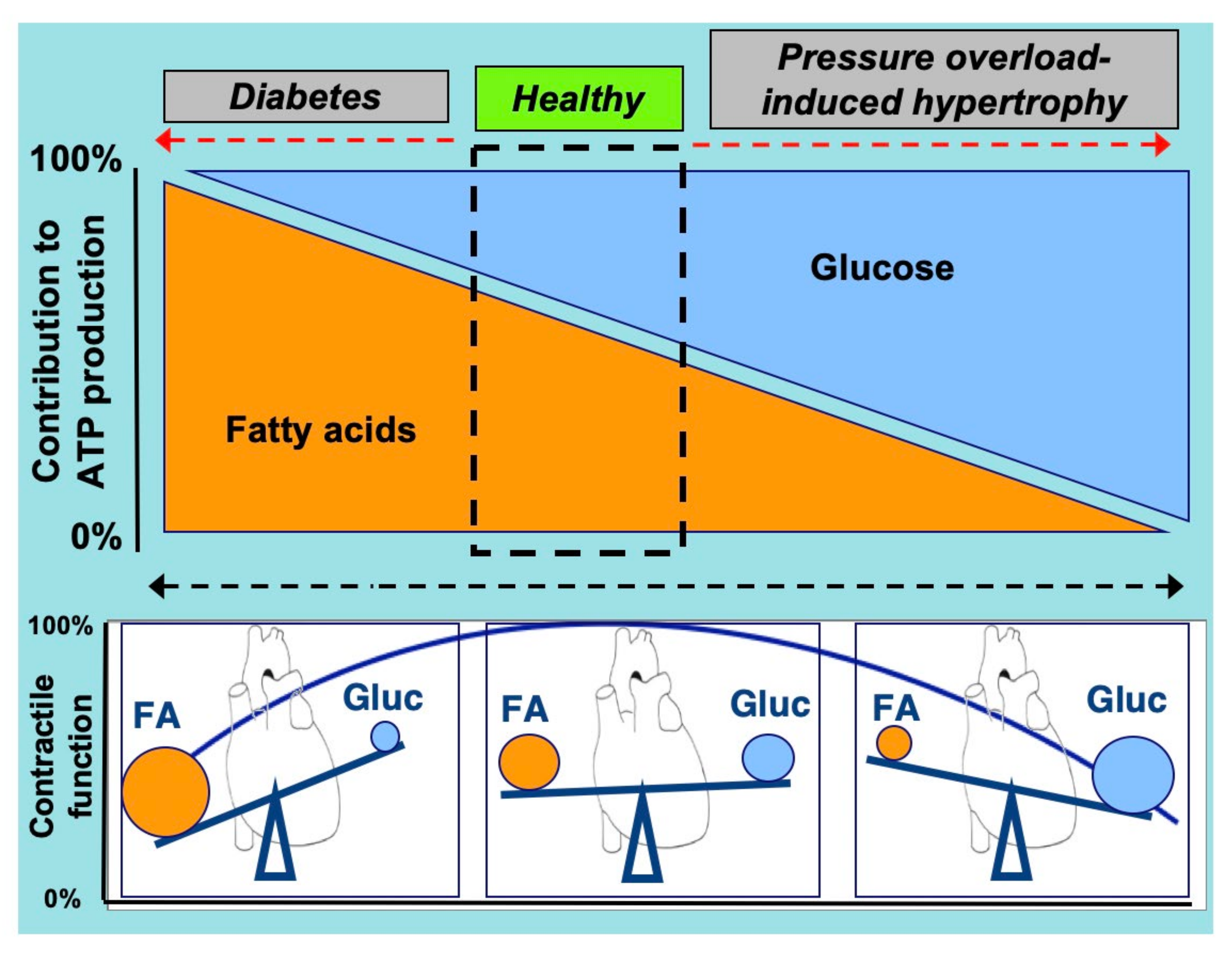
:1. Introduction
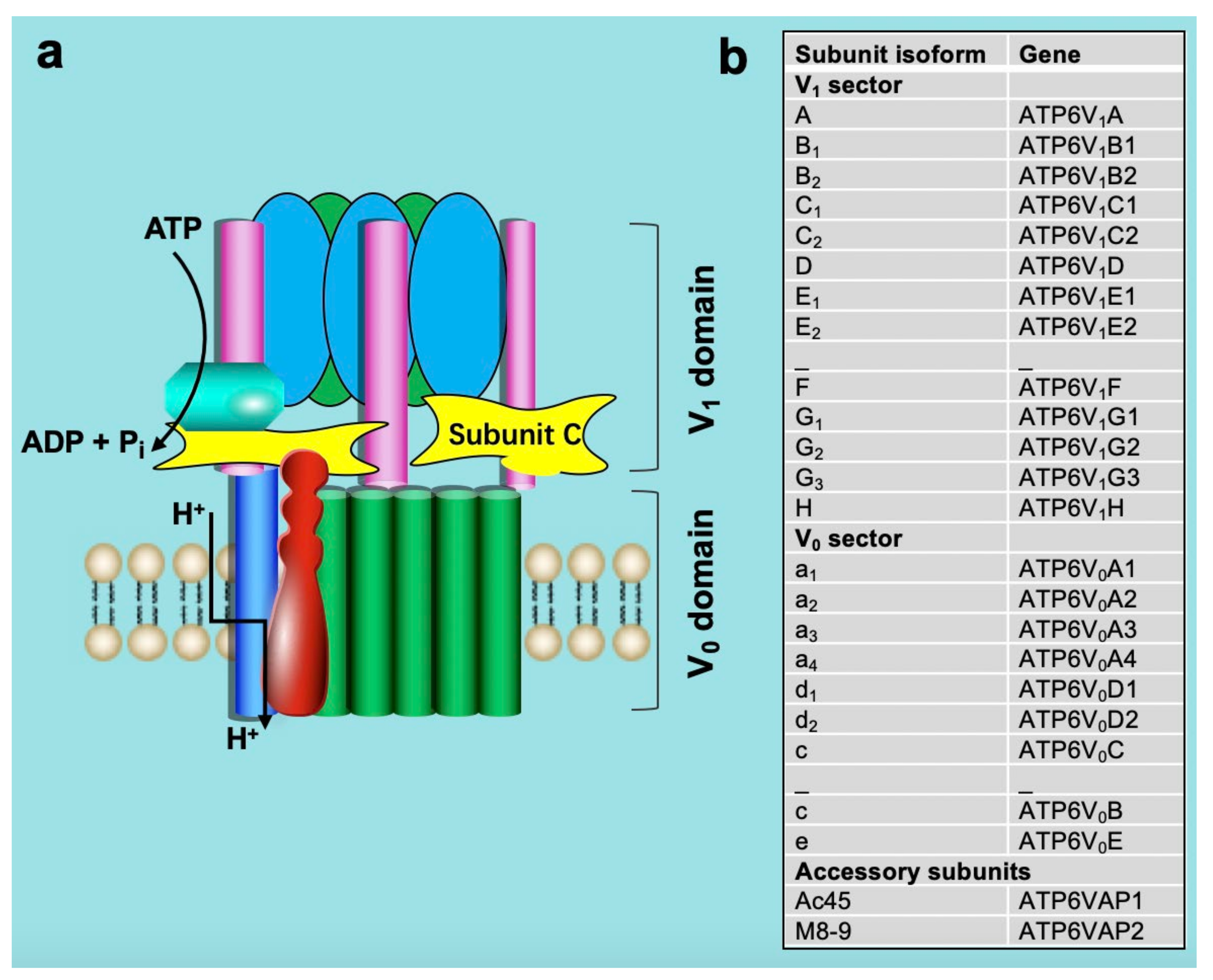
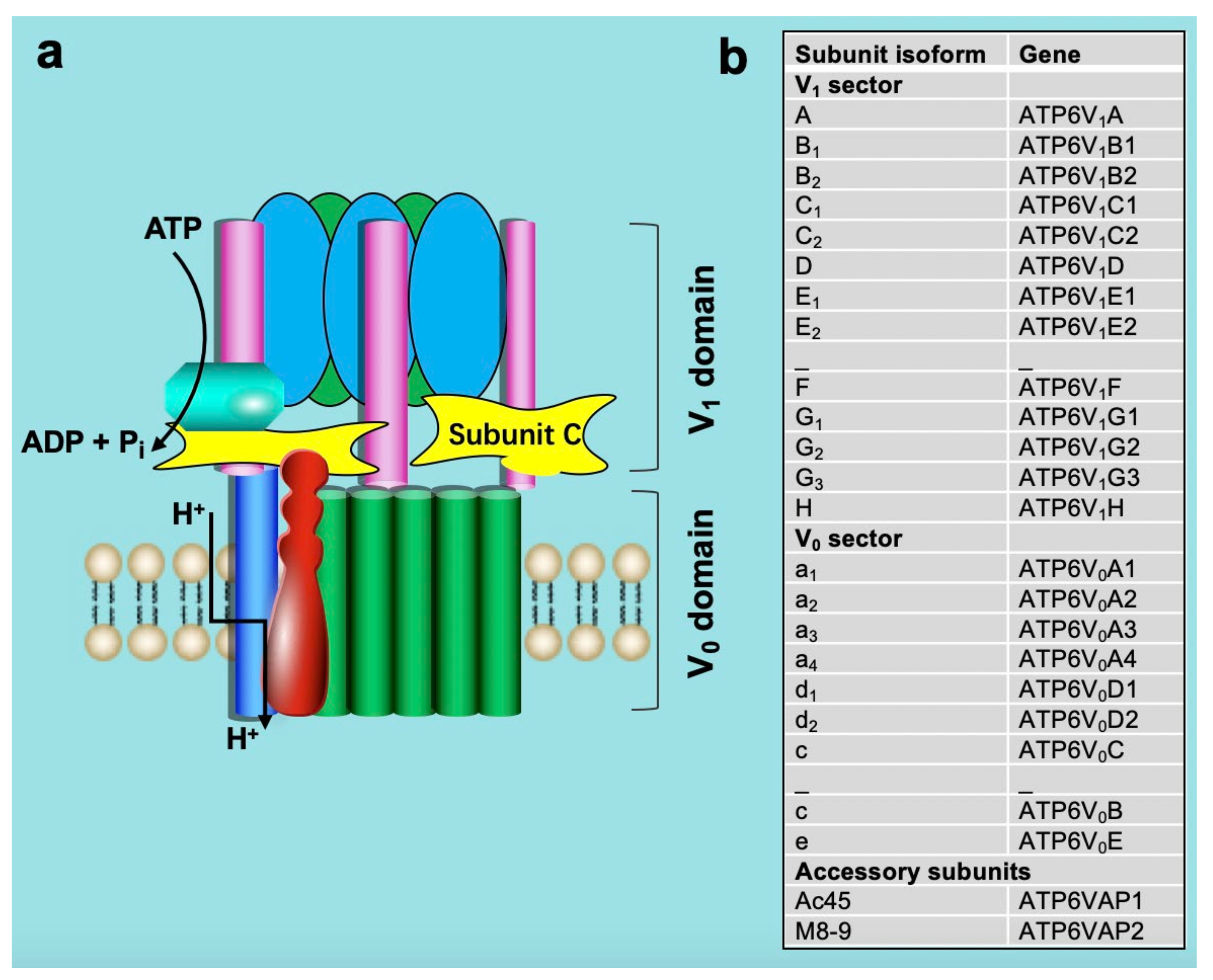
2. Structure and Function of v-ATPase
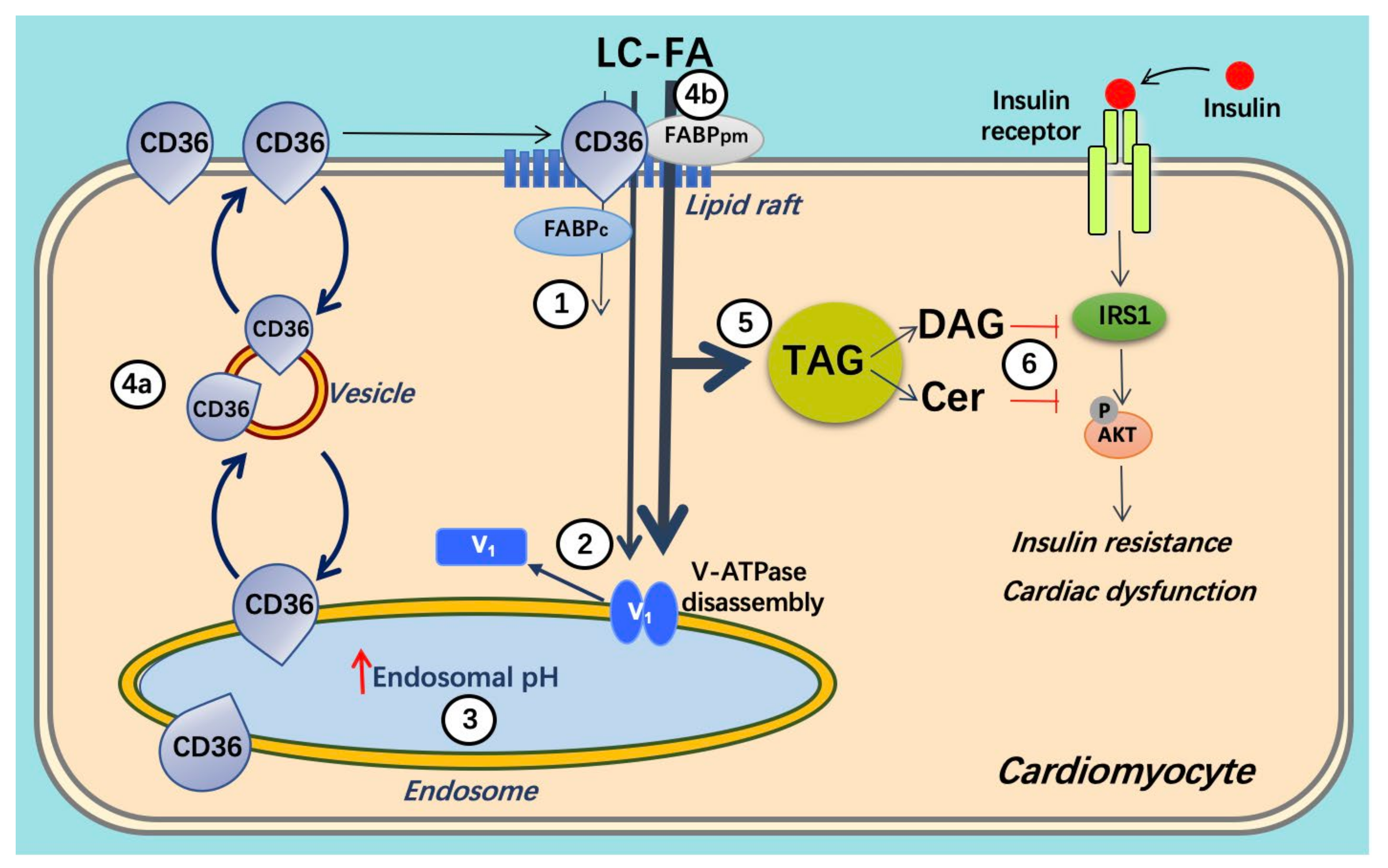
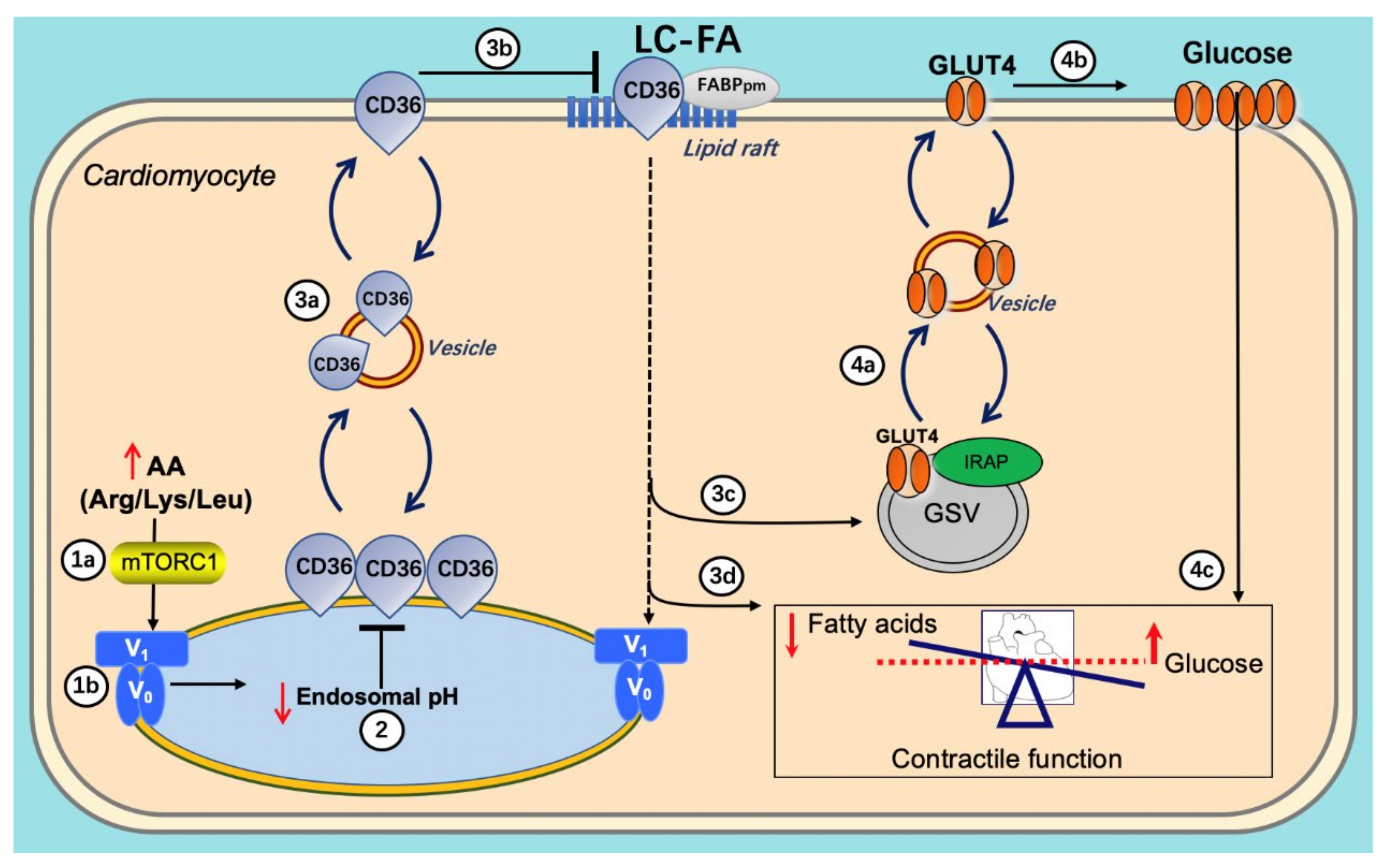
3. Crosstalk between Lipids and v-ATPase
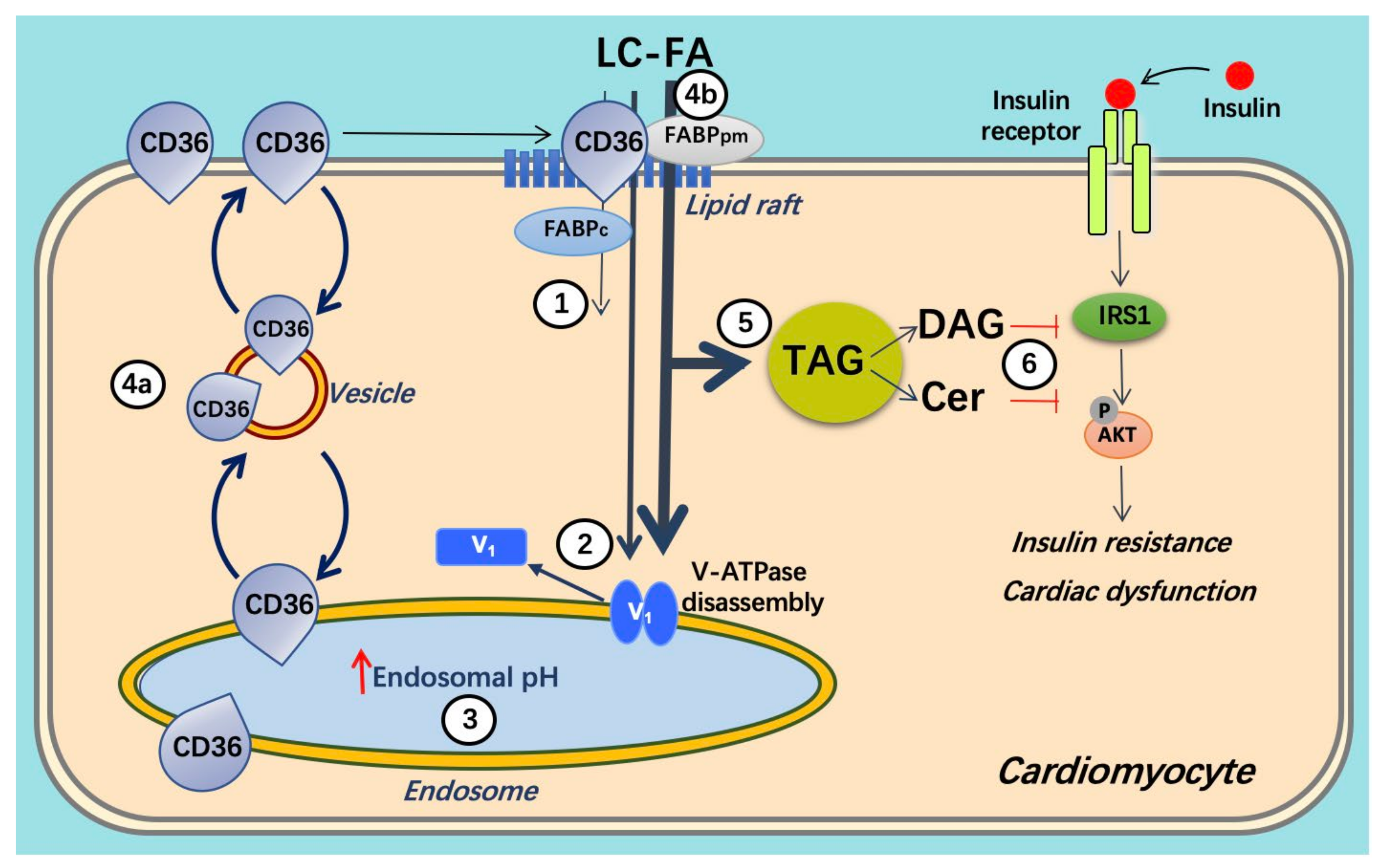
3.1. Lipid and CD36
3.2. v-ATPase and CD36 Traffic
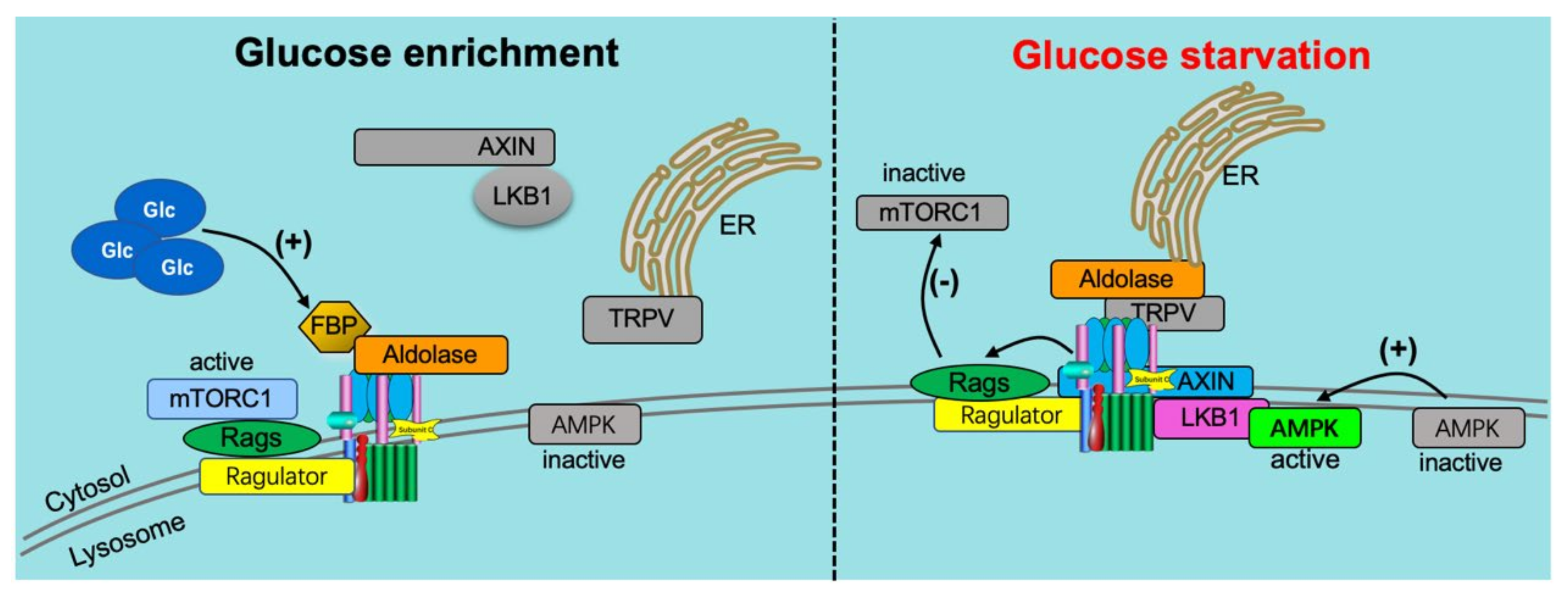
4. Crosstalk between Glucose and v-ATPase
4.1. Glucose Influx Mediates v-ATPase Function
4.2. v-ATPase Influences Glucose Uptake
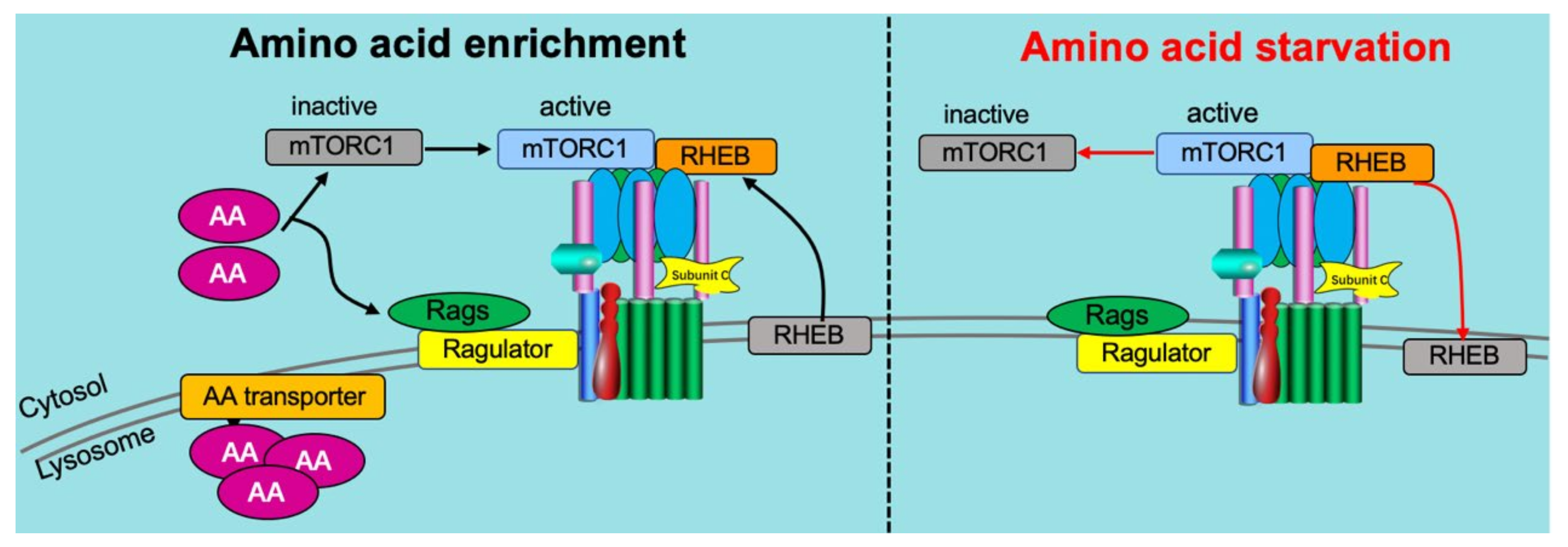
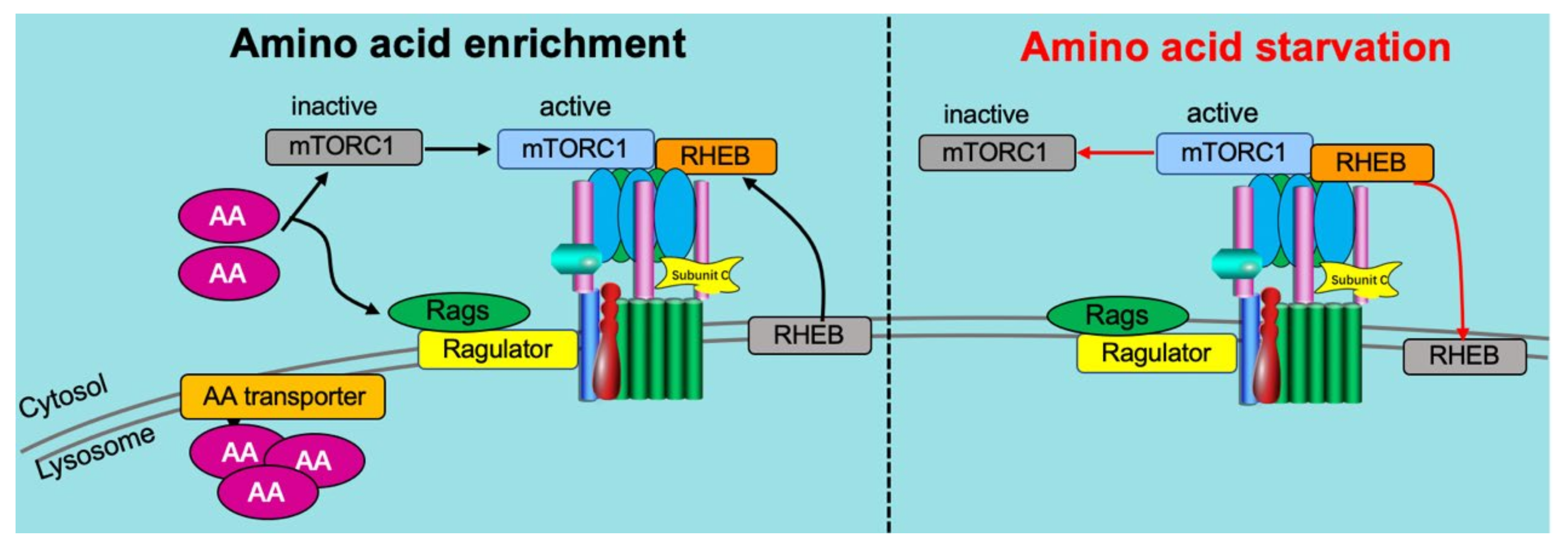
5. Crosstalk between Amino Acids and v-ATPase
5.1. Amino Acid Sensing Needs v-ATPase
5.2. Reciprocal Regulation of v-ATPase on Amino Acid Sensing
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AA | amino acid |
| ACSL | long-chain acyl-CoA synthetase |
| AMPK | AMP-activated protein kinase |
| aRCMs | adult rat cardiomyocytes |
| ATP | adenosine triphosphate |
| FA | long-chain fatty acids |
| FBP | fructose-1,6-bisphosphate |
| GSV | GLUT4 storage vesicles |
| hiPSC-CMs | human-induced pluripotent stem cell-derived cardiomyocytes |
| HP | high palmitate |
| IRAP | insulin-regulated aminopeptidase |
| LP | low palmitate |
| M6P | mannose-6-phosphate |
| mTORC1 | mammalian target of rapamycin complex 1 |
| PI3K | phosphoinositol-3 kinase |
| PKA | protein kinase A |
| Rheb | Ras homolog enriched in brain |
| v-ATPase | vacuolar-type H+-ATPase |
| VAMP | vesicle-associated membrane protein |
References
- Glatz, J.F.; Bonen, A.; Ouwens, D.M.; Luiken, J.J. Regulation of sarcolemmal transport of substrates in the healthy and diseased heart. Cardiovasc. Drugs Ther. 2006, 20, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Glatz, J.F.; Nabben, M.; Young, M.E.; Schulze, P.C.; Taegtmeyer, H.; Luiken, J.J. Re-balancing cellular energy substrate metabolism to mend the failing heart. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165579. [Google Scholar] [CrossRef] [PubMed]
- Taegtmeyer, H. Failing Heart and Starving Brain: Ketone Bodies to the Rescue. Circulation 2016, 134, 265–266. [Google Scholar] [CrossRef] [PubMed]
- Heggermont, W.A.; Papageorgiou, A.P.; Heymans, S.; van Bilsen, M. Metabolic support for the heart: Complementary therapy for heart failure? Eur. J. Heart Fail. 2016, 18, 1420–1429. [Google Scholar] [CrossRef] [Green Version]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Ussher, J.R. Evolving concepts of myocardial energy metabolism: More than just fats and carbohydrates. Circ. Res. 2016, 119, 1173–1176. [Google Scholar] [CrossRef]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Jaswal, J.S.; Keung, W.; Wang, W.; Ussher, J.R.; Lopaschuk, G.D. Targeting fatty acid and carbohydrate oxidation—A novel therapeutic intervention in the ischemic and failing heart. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2011, 1813, 1333–1350. [Google Scholar] [CrossRef] [Green Version]
- Aubert, G.; Martin, O.J.; Horton, J.L.; Lai, L.; Vega, R.B.; Leone, T.C.; Koves, T.; Gardell, S.J.; Kruger, M.; Hoppel, C.L.; et al. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation 2016, 133, 698–705. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac energy metabolism in heart failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Murashige, D.; Jang, C.; Neinast, M.; Edwards, J.J.; Cowan, A.; Hyman, M.C.; Rabinowitz, J.D.; Frankel, D.S.; Arany, Z. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science 2020, 370, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Tomczyk, M.; Olkowicz, M.; Slominska, E.M.; Smolenski, R.T. High Throughput Procedure for Comparative Analysis of In Vivo Cardiac Glucose or Amino Acids Use in Cardiovascular Pathologies and Pharmacological Treatments. Metabolites 2021, 11, 497. [Google Scholar] [CrossRef] [PubMed]
- Chess, D.J.; Stanley, W.C. Role of diet and fuel overabundance in the development and progression of heart failure. Cardiovasc. Res. 2008, 79, 269–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Adrogue, J.V.; Golfman, L.; Uray, I.; Lemm, J.; Youker, K.; Noon, G.P.; Frazier, O.; Taegtmeyer, H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004, 18, 1692–1700. [Google Scholar] [CrossRef]
- Glatz, J.F.; Nabben, M.; Luiken, J.J. Myocardial Fatty Acid-Glucose Fuel Balance as Target to Treat Cardiac Diseases. Cardiol. Cardiovasc. Med. 2020, 4, 584–590. [Google Scholar] [CrossRef]
- Kane, P.M. Targeting reversible disassembly as a mechanism of controlling V-ATPase activity. Curr. Protein Pept. Sci. 2012, 13, 117–123. [Google Scholar] [CrossRef]
- Collins, M.P.; Stransky, L.A.; Forgac, M. AKT Ser/Thr kinase increases V-ATPase–dependent lysosomal acidification in response to amino acid starvation in mammalian cells. J. Biol. Chem. 2020, 295, 9433–9444. [Google Scholar] [CrossRef]
- Collins, M.P.; Forgac, M. Regulation of V-ATPase Assembly in Nutrient Sensing and Function of V-ATPases in Breast Cancer Metastasis. Front. Physiol. 2018, 9, 902. [Google Scholar] [CrossRef]
- Merkulova, M.; Păunescu, T.G.; Azroyan, A.; Marshansky, V.; Breton, S.; Brown, D. Mapping the H+ (V)-ATPase interactome: Identification of proteins involved in trafficking, folding, assembly and phosphorylation. Sci. Rep. 2015, 5, 14827. [Google Scholar] [CrossRef]
- Banerjee, S.; Kane, P.M. Regulation of V-ATPase activity and organelle pH by phosphatidylinositol phosphate lipids. Front. Cell Dev. Biol. 2020, 8, 510. [Google Scholar] [CrossRef]
- Eaton, A.F.; Merkulova, M.; Brown, D. The H+-ATPase (V-ATPase): From proton pump to signaling complex in health and disease. Am. J. Physiol.-Cell Physiol. 2021, 320, C392–C414. [Google Scholar] [CrossRef]
- Kane, P.M. The where, when, and how of organelle acidification by the yeast vacuolar H+-ATPase. Microbiol. Mol. Biol. Rev. 2006, 70, 177–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, L.A.; Finnigan, G.C.; Kane, P.M. Some assembly required: Contributions of Tom Stevens’ lab to the V-ATPase field. Traffic 2018, 19, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Whitton, B.; Okamoto, H.; Packham, G.; Crabb, S.J. Vacuolar ATPase as a potential therapeutic target and mediator of treatment resistance in cancer. Cancer Med. 2018, 7, 3800–3811. [Google Scholar] [CrossRef] [PubMed]
- Hayek, S.R.; Rane, H.; Parra, K.J. Reciprocal Regulation of V-ATPase and Glycolytic Pathway Elements in Health and Disease. Front. Physiol. 2019, 10, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breton, S.; Brown, D. Regulation of luminal acidification by the V-ATPase. Physiology 2013, 28, 318–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forgac, M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 2007, 8, 917. [Google Scholar] [CrossRef]
- Cotter, K.; Stransky, L.; McGuire, C.; Forgac, M. Recent insights into the structure, regulation, and function of the V-ATPases. Trends Biochem. Sci. 2015, 40, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Stransky, L.; Cotter, K.; Forgac, M. The function of V-ATPases in cancer. Physiol. Rev. 2016, 96, 1071–1091. [Google Scholar] [CrossRef] [Green Version]
- McGuire, C.; Cotter, K.; Stransky, L.; Forgac, M. Regulation of V-ATPase assembly and function of V-ATPases in tumor cell invasiveness. Biochim. Biophys. Acta 2016, 1857, 1213–1218. [Google Scholar] [CrossRef]
- Geuze, H.J.; Slot, J.W.; Strous, G.J.; Lodish, H.F.; Schwartz, A.L. Intracellular site of asialoglycoprotein receptor-ligand uncoupling: Double-label immunoelectron microscopy during receptor-mediated endocytosis. Cell 1983, 32, 277–287. [Google Scholar] [CrossRef]
- Gu, F.; Gruenberg, J. ARF1 regulates pH-dependent COP functions in the early endocytic pathway. J. Biol. Chem. 2000, 275, 8154–8160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornfeld, S. Structure and function of the mannose 6-phosphate/insulinlike growth factor II receptors. Annu. Rev. Biochem. 1992, 61, 307–330. [Google Scholar] [CrossRef]
- Kozik, P.; Hodson, N.A.; Sahlender, D.A.; Simecek, N.; Soromani, C.; Wu, J.; Collinson, L.M.; Robinson, M.S. A human genome-wide screen for regulators of clathrin-coated vesicle formation reveals an unexpected role for the V-ATPase. Nat. Cell Biol. 2013, 15, 50–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giovanni, J.; Boudkkazi, S.; Mochida, S.; Bialowas, A.; Samari, N.; Lévêque, C.; Youssouf, F.; Brechet, A.; Iborra, C.; Maulet, Y. V-ATPase membrane sector associates with synaptobrevin to modulate neurotransmitter release. Neuron 2010, 67, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Hiesinger, P.R.; Fayyazuddin, A.; Mehta, S.Q.; Rosenmund, T.; Schulze, K.L.; Zhai, R.G.; Verstreken, P.; Cao, Y.; Zhou, Y.; Kunz, J. The v-ATPase V0 subunit a1 is required for a late step in synaptic vesicle exocytosis in Drosophila. Cell 2005, 121, 607–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liégeois, S.; Benedetto, A.; Garnier, J.-M.; Schwab, Y.; Labouesse, M. The V0-ATPase mediates apical secretion of exosomes containing Hedgehog-related proteins in Caenorhabditis elegans. J. Cell Biol. 2006, 173, 949–961. [Google Scholar] [CrossRef] [Green Version]
- Peters, C.; Bayer, M.J.; Bühler, S.; Andersen, J.S.; Mann, M.; Mayer, A. Trans-complex formation by proteolipid channels in the terminal phase of membrane fusion. Nature 2001, 409, 581–588. [Google Scholar] [CrossRef]
- Sun-Wada, G.H.; Toyomura, T.; Murata, Y.; Yamamoto, A.; Futai, M.; Wada, Y. The a3 isoform of V-ATPase regulates insulin secretion from pancreatic beta-cells. J. Cell Sci. 2006, 119, 4531–4540. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.P.; Chen, W.; Liang, Y.; Li, E.; Stashenko, P. Atp6i-deficient mice exhibit severe osteopetrosis due to loss of osteoclast-mediated extracellular acidification. Nat. Genet. 1999, 23, 447–451. [Google Scholar] [CrossRef]
- Adams, D.S.; Masi, A.; Levin, M. H+ pump-dependent changes in membrane voltage are an early mechanism necessary and sufficient to induce Xenopus tail regeneration. Development 2007, 134, 1323–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, D.S.; Robinson, K.R.; Fukumoto, T.; Yuan, S.; Albertson, R.C.; Yelick, P.; Kuo, L.; McSweeney, M.; Levin, M. Early, H+-V-ATPase-dependent proton flux is necessary for consistent left-right patterning of non-mammalian vertebrates. Development 2006, 133, 1657–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Ammar, D.; Ives, H.; Albrecht, F.; Gluck, S.L. Physical interaction between aldolase and vacuolar H+-ATPase is essential for the assembly and activity of the proton pump. J. Biol. Chem. 2007, 282, 24495–24503. [Google Scholar] [CrossRef] [Green Version]
- Sautin, Y.Y.; Lu, M.; Gaugler, A.; Zhang, L.; Gluck, S.L. Phosphatidylinositol 3-kinase-mediated effects of glucose on vacuolar H+-ATPase assembly, translocation, and acidification of intracellular compartments in renal epithelial cells. Mol. Cell. Biol. 2005, 25, 575–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, P.M. Disassembly and reassembly of the yeast vacuolar H+-ATPase in vivo. J. Biol. Chem. 1995, 270, 17025–17032. [Google Scholar] [CrossRef]
- Tabke, K.; Albertmelcher, A.; Vitavska, O.; Huss, M.; Schmitz, H.-P.; Wieczorek, H. Reversible disassembly of the yeast V-ATPase revisited under in vivo conditions. Biochem. J. 2014, 462, 185–197. [Google Scholar] [CrossRef]
- Parra, K.J.; Kane, P.M. Reversible association between the V1 and V0 domains of yeast vacuolar H+-ATPase is an unconventional glucose-induced effect. Mol. Cell. Biol. 1998, 18, 7064–7074. [Google Scholar] [CrossRef] [Green Version]
- Cipriano, D.J.; Wang, Y.; Bond, S.; Hinton, A.; Jefferies, K.C.; Qi, J.; Forgac, M. Structure and regulation of the vacuolar ATPases. Biochim. Biophys. Acta (BBA)-Bioenerg. 2008, 1777, 599–604. [Google Scholar] [CrossRef] [Green Version]
- Diab, H.; Ohira, M.; Liu, M.; Cobb, E.; Kane, P.M. Subunit interactions and requirements for inhibition of the yeast V1-ATPase. J. Biol. Chem. 2009, 284, 13316–13325. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Oot, R.A.; Wilkens, S. MgATP hydrolysis destabilizes the interaction between subunit H and yeast V1-ATPase, highlighting H’s role in V-ATPase regulation by reversible disassembly. J. Biol. Chem. 2018, 293, 10718–10730. [Google Scholar] [CrossRef] [Green Version]
- Couoh-Cardel, S.; Milgrom, E.; Wilkens, S. Affinity purification and structural features of the yeast vacuolar ATPase Vo membrane sector. J. Biol. Chem. 2015, 290, 27959–27971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voss, M.; Vitavska, O.; Walz, B.; Wieczorek, H.; Baumann, O. Stimulus-induced phosphorylation of vacuolar H+-ATPase by protein kinase A. J. Biol. Chem. 2007, 282, 33735–33742. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Forgac, M. Microtubules are involved in glucose-dependent dissociation of the yeast vacuolar [H+]-ATPase in vivo. J. Biol. Chem. 2001, 276, 24855–24861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, E.; Forgac, M. Involvement of the nonhomologous region of subunit A of the yeast V-ATPase in coupling and in vivo dissociation. J. Biol. Chem. 2004, 279, 48663–48670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.-S.; Padron, D.; Liao, X.; Wang, J.; Roth, M.G.; De Brabander, J.K. Salicylihalamide A inhibits the V0 sector of the V-ATPase through a mechanism distinct from bafilomycin A1. J. Biol. Chem. 2004, 279, 19755–19763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smardon, A.M.; Nasab, N.D.; Tarsio, M.; Diakov, T.T.; Kane, P.M. Molecular interactions and cellular itinerary of the yeast RAVE (regulator of the H+-ATPase of vacuolar and endosomal membranes) complex. J. Biol. Chem. 2015, 290, 27511–27523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, A.G.; Stock, D. Priming a molecular motor for disassembly. Structure 2012, 20, 1799–1800. [Google Scholar] [CrossRef] [Green Version]
- Stransky, L.A.; Forgac, M. Amino acid availability modulates vacuolar H+-ATPase assembly. J. Biol. Chem. 2015, 290, 27360–27369. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Steinbusch, L.K.M.; Nabben, M.; Kapsokalyvas, D.; van Zandvoort, M.; Schonleitner, P.; Antoons, G.; Simons, P.J.; Coumans, W.A.; Geomini, A.; et al. Palmitate-Induced Vacuolar-Type H+-ATPase Inhibition Feeds Forward Into Insulin Resistance and Contractile Dysfunction. Diabetes 2017, 66, 1521–1534. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wong, L.-Y.; Neumann, D.; Liu, Y.; Sun, A.; Antoons, G.; Strzelecka, A.; Glatz, J.F.; Nabben, M.; Luiken, J.J. Augmenting Vacuolar H+-ATPase Function Prevents Cardiomyocytes from Lipid-Overload Induced Dysfunction. Int. J. Mol. Sci. 2020, 21, 1520. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Schianchi, F.; Neumann, D.; Wong, L.-Y.; Sun, A.; van Nieuwenhoven, F.A.; Zeegers, M.P.; Strzelecka, A.; Col, U.; Glatz, J.F. Specific amino acid supplementation rescues the heart from lipid overload-induced insulin resistance and contractile dysfunction by targeting the endosomal mTOR–v-ATPase axis. Mol. Metab. 2021, 53, 101293. [Google Scholar] [CrossRef]
- Glatz, J.F.C.; Luiken, J.J.F.P. From fat to FAT (CD36/SR-B2): Understanding the regulation of cellular fatty acid uptake. Biochimie 2017, 136, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Glatz, J.F.; Luiken, J.J.; Bonen, A. Membrane fatty acid transporters as regulators of lipid metabolism: Implications for metabolic disease. Physiol. Rev. 2010, 90, 367–417. [Google Scholar] [CrossRef] [Green Version]
- Bonen, A.; Luiken, J.J.; Arumugam, Y.; Glatz, J.F.; Tandon, N.N. Acute regulation of fatty acid uptake involves the cellular redistribution of fatty acid translocase. J. Biol. Chem. 2000, 275, 14501–14508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turcotte, L.P.; Srivastava, A.K.; Chiasson, J.-L. Fasting increases plasma membrane fatty acid-binding protein (FABPPM) in red skeletal muscle. Mol. Cell. Biochem. 1997, 166, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Luiken, J.J.; Koonen, D.P.; Willems, J.; Zorzano, A.; Becker, C.; Fischer, Y.; Tandon, N.N.; Van Der Vusse, G.J.; Bonen, A.; Glatz, J.F. Insulin stimulates long-chain fatty acid utilization by rat cardiac myocytes through cellular redistribution of FAT/CD36. Diabetes 2002, 51, 3113–3119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luiken, J.J.; Nabben, M.; Neumann, D.; Glatz, J.F. Understanding the distinct subcellular trafficking of CD36 and GLUT4 during the development of myocardial insulin resistance. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2020, 1866, 165775. [Google Scholar] [CrossRef] [PubMed]
- Glatz, J.F.; Luiken, J.J. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J. Lipid Res. 2018, 59, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Angin, Y.; Steinbusch, L.K.; Simons, P.J.; Greulich, S.; Hoebers, N.T.; Douma, K.; van Zandvoort, M.A.; Coumans, W.A.; Wijnen, W.; Diamant, M.; et al. CD36 inhibition prevents lipid accumulation and contractile dysfunction in rat cardiomyocytes. Biochem. J. 2012, 448, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Sambandam, N.; Han, X.; Gross, R.W.; Courtois, M.; Kovacs, A.; Febbraio, M.; Finck, B.N.; Kelly, D.P. CD36 deficiency rescues lipotoxic cardiomyopathy. Circ. Res. 2007, 100, 1208–1217. [Google Scholar] [CrossRef]
- Steinbusch, L.K.; Luiken, J.J.; Vlasblom, R.; Chabowski, A.; Hoebers, N.T.; Coumans, W.A.; Vroegrijk, I.O.; Voshol, P.J.; Ouwens, D.M.; Glatz, J.F.; et al. Absence of fatty acid transporter CD36 protects against Western-type diet-related cardiac dysfunction following pressure overload in mice. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E618–E627. [Google Scholar] [CrossRef] [Green Version]
- Glatz, J.F.; Luiken, J.J.; Nabben, M. CD36 (SR-B2) as a Target to Treat Lipid Overload-Induced Cardiac Dysfunction. J. Lipid Atheroscler. 2020, 9, 66–78. [Google Scholar] [CrossRef]
- Steinbusch, L.K.; Wijnen, W.; Schwenk, R.W.; Coumans, W.A.; Hoebers, N.T.; Ouwens, D.M.; Diamant, M.; Bonen, A.; Glatz, J.F.; Luiken, J.J. Differential regulation of cardiac glucose and fatty acid uptake by endosomal pH and actin filaments. Am. J. Physiol.-Cell Physiol. 2010, 298, C1549–C1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yèagle, P.L. Lipid regulation of cell membrane structure and function. FASEB J. 1989, 3, 1833–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Rodriguez-Calvo, R.; Wang, S.; Zhu, X.; Broers, J.L.; Glatz, J.F.; Luiken, J.J.; Neumann, D. Fluorescent labelling of membrane fatty acid transporter CD36 (SR-B2) in the extracellular loop. PLoS ONE 2019, 14, e0210704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagh, M.; Chandra, G.; Peng, S.; Zhang, Z.; Mukherjee, A. A lysosomal targeting defect of V0a1 suppresses V-ATPase activity elevating lysosomal pH in Ppt1−/− mice: Amelioration by NtBuHA. FASEB J. 2015, 29 (Suppl. 1), 570.6. [Google Scholar] [CrossRef]
- Montigny, C.; Decottignies, P.; Le Maréchal, P.; Capy, P.; Bublitz, M.; Olesen, C.; Møller, J.V.; Nissen, P.; Le Maire, M. S-palmitoylation and s-oleoylation of rabbit and pig sarcolipin. J. Biol. Chem. 2014, 289, 33850–33861. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.J.; Kim, M.; Park, H.-S.; Kim, H.S.; Jeon, M.J.; Oh, K.S.; Koh, E.H.; Won, J.C.; Kim, M.-S.; Oh, G.T. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARα and PGC-1. Biochem. Biophys. Res. Commun. 2006, 340, 291–295. [Google Scholar] [CrossRef]
- Zhang, Y.-L.; Guo, H.; Zhang, C.-S.; Lin, S.-Y.; Yin, Z.; Peng, Y.; Luo, H.; Shi, Y.; Lian, G.; Zhang, C. AMP as a low-energy charge signal autonomously initiates assembly of AXIN-AMPK-LKB1 complex for AMPK activation. Cell Metab. 2013, 18, 546–555. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.-S.; Jiang, B.; Li, M.; Zhu, M.; Peng, Y.; Zhang, Y.-L.; Wu, Y.-Q.; Li, T.Y.; Liang, Y.; Lu, Z. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 2014, 20, 526–540. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Kowalsky, A.H.; Lee, J.H. Sestrins in physiological stress responses. Annu. Rev. Physiol. 2021, 83, 381–403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fan, J.; Li, H.; Chen, C.; Wang, Y. CD36 Signaling in Diabetic Cardiomyopathy. Aging Dis. 2021, 12, 826. [Google Scholar] [CrossRef]
- Shulman, G.I. Cellular mechanisms of insulin resistance. J. Clin. Investig. 2000, 106, 171–176. [Google Scholar] [CrossRef]
- Sumner, J.-P.; Dow, J.A.; Earley, F.G.; Klein, U.; Jäger, D.; Wieczorek, H. Regulation of Plasma Membrane V-ATPase Activity by Dissociation of Peripheral Subunits. J. Biol. Chem. 1995, 270, 5649–5653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuire, C.M.; Forgac, M. Glucose starvation increases V-ATPase assembly and activity in mammalian cells through AMP kinase and phosphatidylinositide 3-kinase/Akt signaling. J. Biol. Chem. 2018, 293, 9113–9123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dechant, R.; Binda, M.; Lee, S.S.; Pelet, S.; Winderickx, J.; Peter, M. Cytosolic pH is a second messenger for glucose and regulates the PKA pathway through V-ATPase. EMBO J. 2010, 29, 2515–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.-Y.; Dominguez, D.; Parra, K.J. Regulation of vacuolar H+-ATPase (V-ATPase) reassembly by glycolysis flow in 6-phosphofructo-1-kinase (PFK-1)-deficient yeast cells. J. Biol. Chem. 2016, 291, 15820–15829. [Google Scholar] [CrossRef] [Green Version]
- Bond, S.; Forgac, M. The Ras/cAMP/protein kinase A pathway regulates glucose-dependent assembly of the vacuolar (H+)-ATPase in yeast. J. Biol. Chem. 2008, 283, 36513–36521. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, S. Glucose activates H+-ATPase in kidney epithelial cells. Am. J. Physiol.-Cell Physiol. 2004, 287, C97–C105. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Sautin, Y.Y.; Holliday, L.S.; Gluck, S.L. The glycolytic enzyme aldolase mediates assembly, expression, and activity of vacuolar H+-ATPase. J. Biol. Chem. 2004, 279, 8732–8739. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Holliday, L.S.; Zhang, L.; Dunn, W.A.; Gluck, S.L. Interaction between aldolase and vacuolar H+-ATPase evidence for direct coupling of glycolysis to the ATP-hydrolyzing proton pump. J. Biol. Chem. 2001, 276, 30407–30413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Zhang, C.-S.; Feng, J.-W.; Wei, X.; Zhang, C.; Xie, C.; Wu, Y.; Hawley, S.A.; Atrih, A.; Lamont, D.J. Aldolase is a sensor for both low and high glucose, linking to AMPK and mTORC1. Cell Res. 2021, 31, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-S.; Hawley, S.A.; Zong, Y.; Li, M.; Wang, Z.; Gray, A.; Ma, T.; Cui, J.; Feng, J.-W.; Zhu, M. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 2017, 548, 112–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abel, E.D.; Graveleau, C.; Betuing, S.; Pham, M.; Reay, P.A.; Kandror, V.; Kupriyanova, T.; Xu, Z.; Kandror, K.V. Regulation of insulin-responsive aminopeptidase expression and targeting in the insulin-responsive vesicle compartment of glucose transporter isoform 4-deficient cardiomyocytes. Mol. Endocrinol. 2004, 18, 2491–2501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbusch, L.K.; Schwenk, R.W.; Ouwens, D.M.; Diamant, M.; Glatz, J.F.; Luiken, J.J. Subcellular trafficking of the substrate transporters GLUT4 and CD36 in cardiomyocytes. Cell. Mol. Life Sci. 2011, 68, 2525–2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heather, L.C.; Pates, K.M.; Atherton, H.J.; Cole, M.A.; Ball, D.R.; Evans, R.D.; Glatz, J.F.; Luiken, J.J.; Griffin, J.L.; Clarke, K. Differential translocation of the fatty acid transporter, FAT/CD36, and the glucose transporter, GLUT4, coordinates changes in cardiac substrate metabolism during ischemia and reperfusion. Circ. Heart Fail. 2013, 6, 1058–1066. [Google Scholar] [CrossRef] [Green Version]
- Bonen, A.; Jain, S.S.; Snook, L.A.; Han, X.-X.; Yoshida, Y.; Buddo, K.H.; Lally, J.S.; Pask, E.D.; Paglialunga, S.; Beaudoin, M.-S. Extremely rapid increase in fatty acid transport and intramyocellular lipid accumulation but markedly delayed insulin resistance after high fat feeding in rats. Diabetologia 2015, 58, 2381–2391. [Google Scholar] [CrossRef] [Green Version]
- Zeigerer, A.; Lampson, M.A.; Karylowski, O.; Sabatini, D.D.; Adesnik, M.; Ren, M.; McGraw, T.E. GLUT4 retention in adipocytes requires two intracellular insulin-regulated transport steps. Mol. Biol. Cell 2002, 13, 2421–2435. [Google Scholar] [CrossRef] [Green Version]
- Holman, G.D.; Sandoval, I.V. Moving the insulin-regulated glucose transporter GLUT4 into and out of storage. Trends Cell Biol. 2001, 11, 173–179. [Google Scholar] [CrossRef]
- Jordens, I.; Molle, D.; Xiong, W.; Keller, S.R.; McGraw, T.E. Insulin-regulated aminopeptidase is a key regulator of GLUT4 trafficking by controlling the sorting of GLUT4 from endosomes to specialized insulin-regulated vesicles. Mol. Biol. Cell 2010, 21, 2034–2044. [Google Scholar] [CrossRef] [Green Version]
- Klip, A.; McGraw, T.E.; James, D.E. Thirty sweet years of GLUT4. J. Biol. Chem. 2019, 294, 11369–11381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogan, J.S.; Rubin, B.R.; Yu, C.; Löffler, M.G.; Orme, C.M.; Belman, J.P.; McNally, L.J.; Hao, M.; Cresswell, J.A. Endoproteolytic cleavage of TUG protein regulates GLUT4 glucose transporter translocation. J. Biol. Chem. 2012, 287, 23932–23947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belman, J.P.; Bian, R.R.; Habtemichael, E.N.; Li, D.T.; Jurczak, M.J.; Alcazar-Roman, A.; McNally, L.J.; Shulman, G.I.; Bogan, J.S. Acetylation of TUG protein promotes the accumulation of GLUT4 glucose transporters in an insulin-responsive intracellular compartment. J. Biol. Chem. 2015, 290, 4447–4463. [Google Scholar] [CrossRef] [Green Version]
- Bogan, J.S. Regulation of glucose transporter translocation in health and diabetes. Annu. Rev. Biochem. 2012, 81, 507–532. [Google Scholar] [CrossRef]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jewell, J.L.; Russell, R.C.; Guan, K.L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Bar-Peled, L.; Schweitzer, L.D.; Zoncu, R.; Sabatini, D.M. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 2012, 150, 1196–1208. [Google Scholar] [CrossRef] [Green Version]
- Wyant, G.A.; Abu-Remaileh, M.; Wolfson, R.L.; Chen, W.W.; Freinkman, E.; Danai, L.V.; Vander Heiden, M.G.; Sabatini, D.M. mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell 2017, 171, 642–654.e612. [Google Scholar] [CrossRef]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Pamarthy, S.; Kulshrestha, A.; Katara, G.K.; Beaman, K.D. The curious case of vacuolar ATPase: Regulation of signaling pathways. Mol. Cancer 2018, 17, 41. [Google Scholar] [CrossRef] [PubMed]
- Abu-Remaileh, M.; Wyant, G.A.; Kim, C.; Laqtom, N.N.; Abbasi, M.; Chan, S.H.; Freinkman, E.; Sabatini, D.M. Lysosomal metabolomics reveals V-ATPase-and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 2017, 358, 807–813. [Google Scholar] [CrossRef] [Green Version]
- Tomczyk, M.; Braczko, A.; Jablonska, P.; Mika, A.; Przyborowski, K.; Jedrzejewska, A.; Krol, O.; Kus, F.; Sledzinski, T.; Chlopicki, S.; et al. Enhanced Muscle Strength in Dyslipidemic Mice and Its Relation to Increased Capacity for Fatty Acid Oxidation. Int. J. Mol. Sci. 2021, 22, 12251. [Google Scholar] [CrossRef]
- Olkowicz, M.; Tomczyk, M.; Debski, J.; Tyrankiewicz, U.; Przyborowski, K.; Borkowski, T.; Zabielska-Kaczorowska, M.; Szupryczynska, N.; Kochan, Z.; Smeda, M.; et al. Enhanced cardiac hypoxic injury in atherogenic dyslipidaemia results from alterations in the energy metabolism pattern. Metabolism 2021, 114, 154400. [Google Scholar] [CrossRef]
- Geraets, I.M.; Coumans, W.A.; Strzelecka, A.; Schönleitner, P.; Antoons, G.; Schianchi, F.; Willemars, M.M.; Kapsokalyvas, D.; Glatz, J.F.; Luiken, J.J. Metabolic Interventions to Prevent Hypertrophy-Induced Alterations in Contractile Properties In Vitro. Int. J. Mol. Sci. 2021, 22, 3620. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Han, Y.; Nabben, M.; Neumann, D.; Luiken, J.J.F.P.; Glatz, J.F.C. Endosomal v-ATPase as a Sensor Determining Myocardial Substrate Preference. Metabolites 2022, 12, 579. https://doi.org/10.3390/metabo12070579
Wang S, Han Y, Nabben M, Neumann D, Luiken JJFP, Glatz JFC. Endosomal v-ATPase as a Sensor Determining Myocardial Substrate Preference. Metabolites. 2022; 12(7):579. https://doi.org/10.3390/metabo12070579
Chicago/Turabian StyleWang, Shujin, Yinying Han, Miranda Nabben, Dietbert Neumann, Joost J. F. P. Luiken, and Jan F. C. Glatz. 2022. "Endosomal v-ATPase as a Sensor Determining Myocardial Substrate Preference" Metabolites 12, no. 7: 579. https://doi.org/10.3390/metabo12070579
APA StyleWang, S., Han, Y., Nabben, M., Neumann, D., Luiken, J. J. F. P., & Glatz, J. F. C. (2022). Endosomal v-ATPase as a Sensor Determining Myocardial Substrate Preference. Metabolites, 12(7), 579. https://doi.org/10.3390/metabo12070579