
Genome Features and AntiSMASH Analysis of an Endophytic Strain Fusarium sp. R1




Abstract
:1. Introduction
2. Results and Discussion
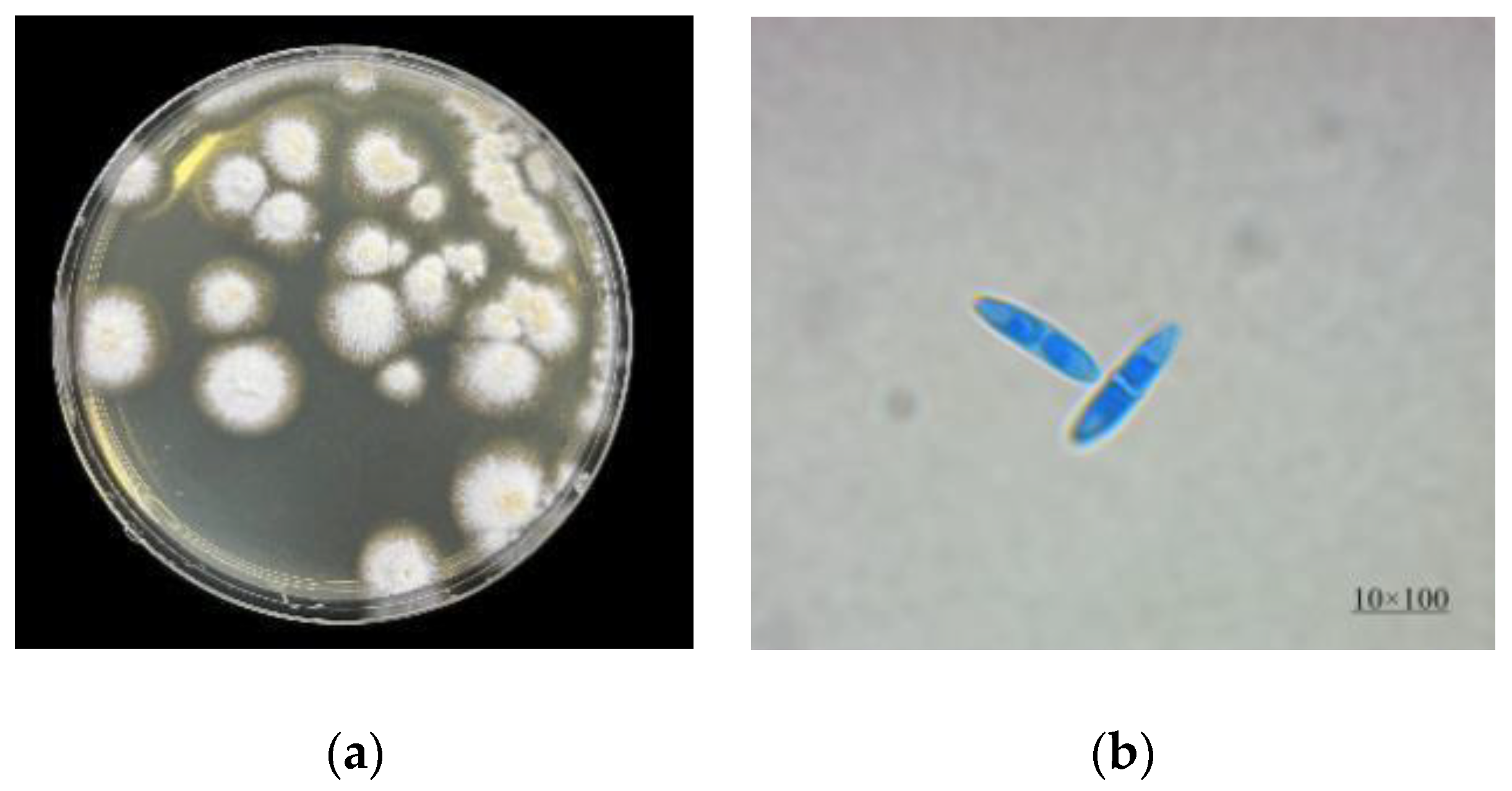
2.1. Identification of Strain R1
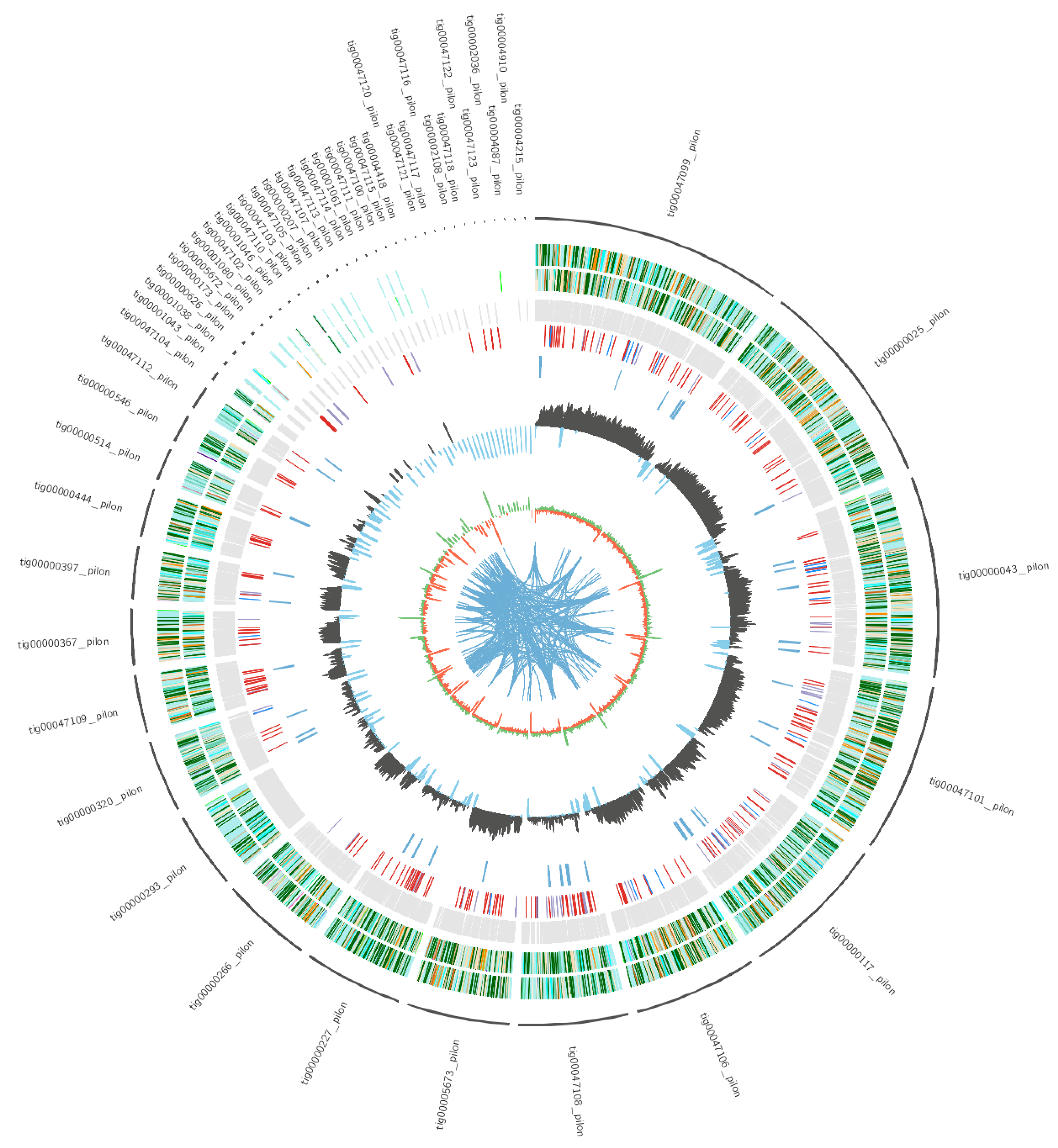
2.2. Genome Sequencing and Assembly
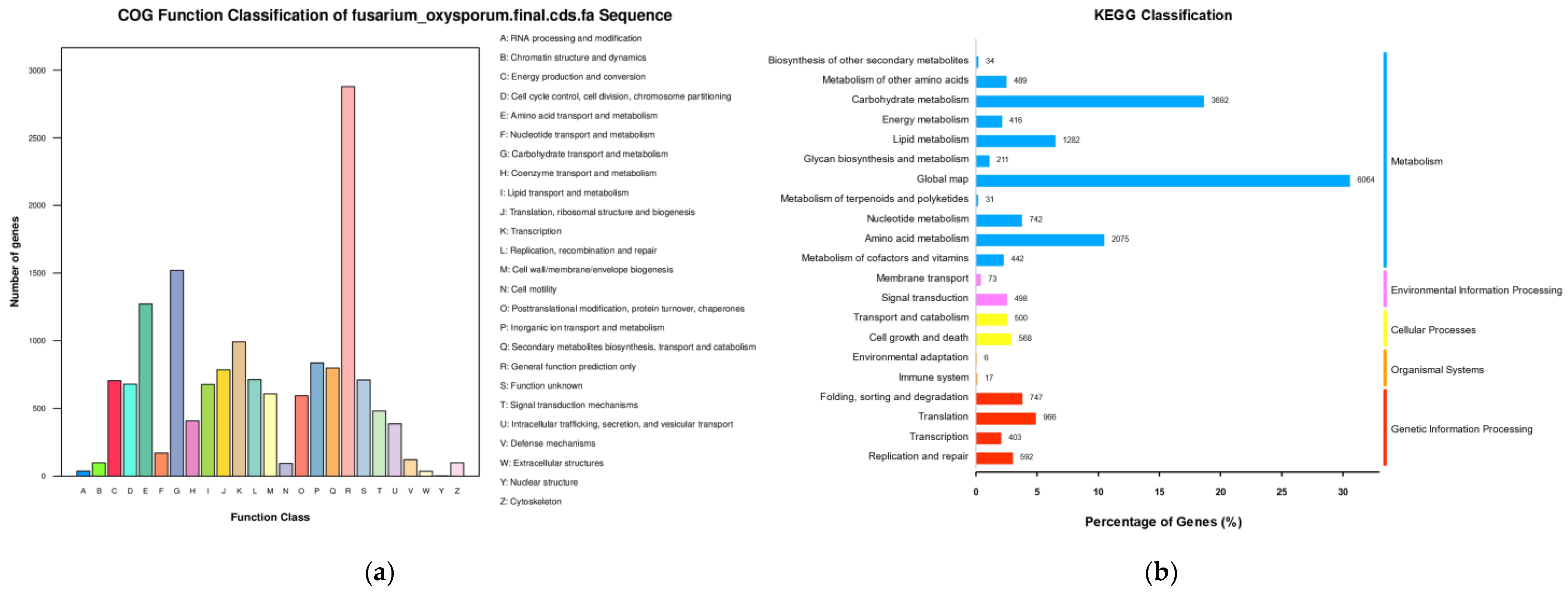
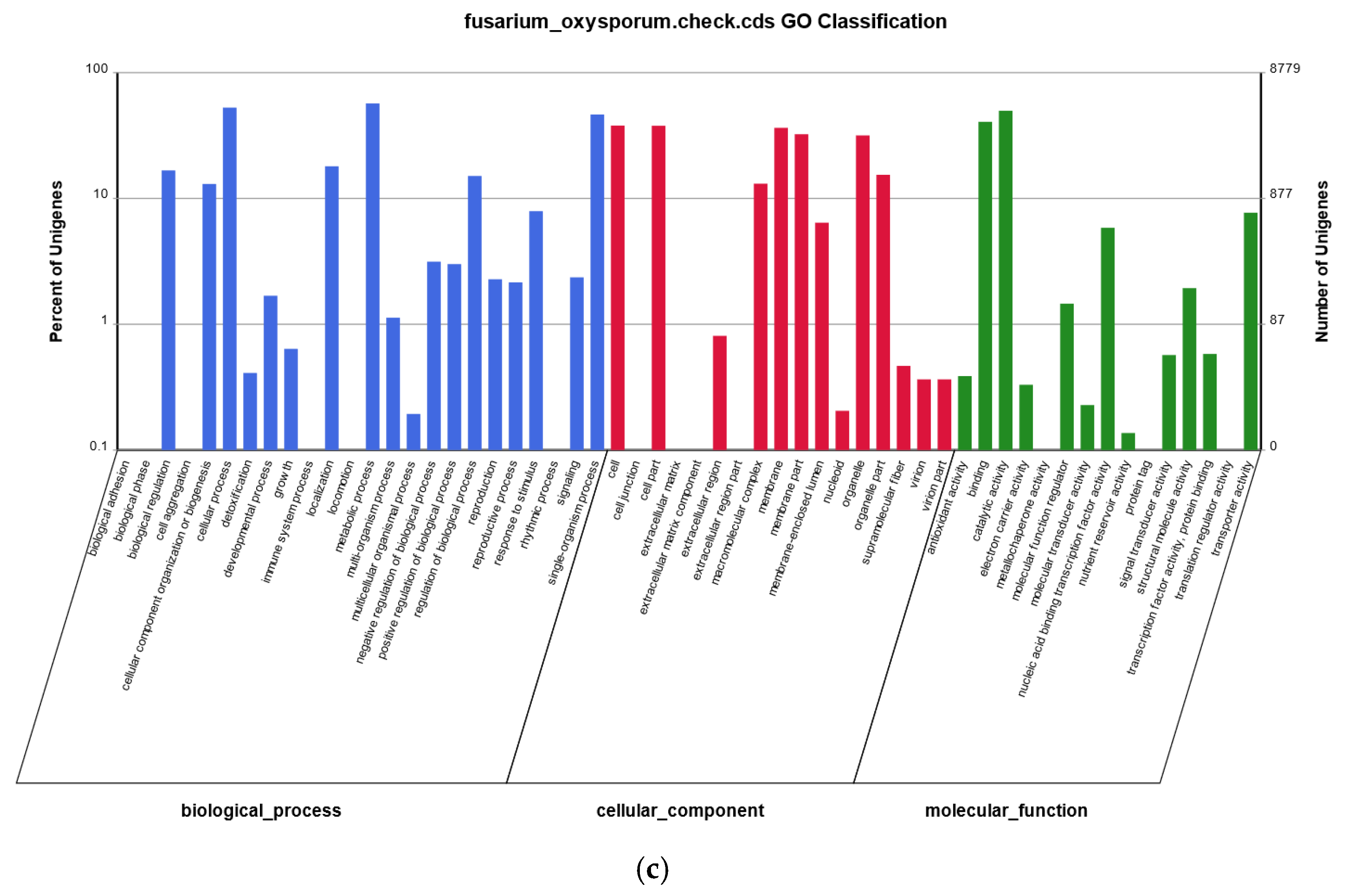
2.3. Genome Sequence Annotation
2.4. Additional Annotation
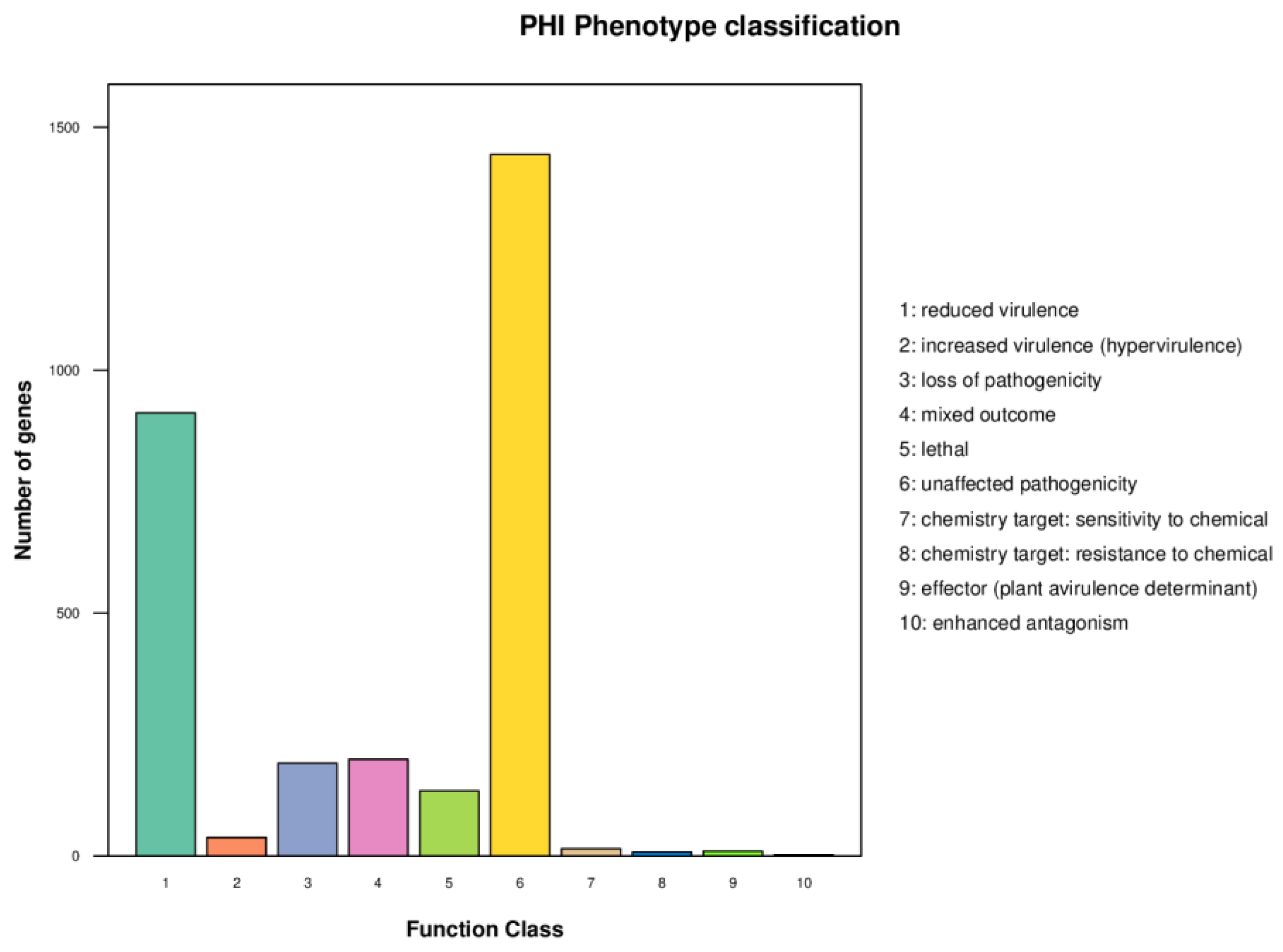
2.4.1. Pathogen Host Interactions (PHI)
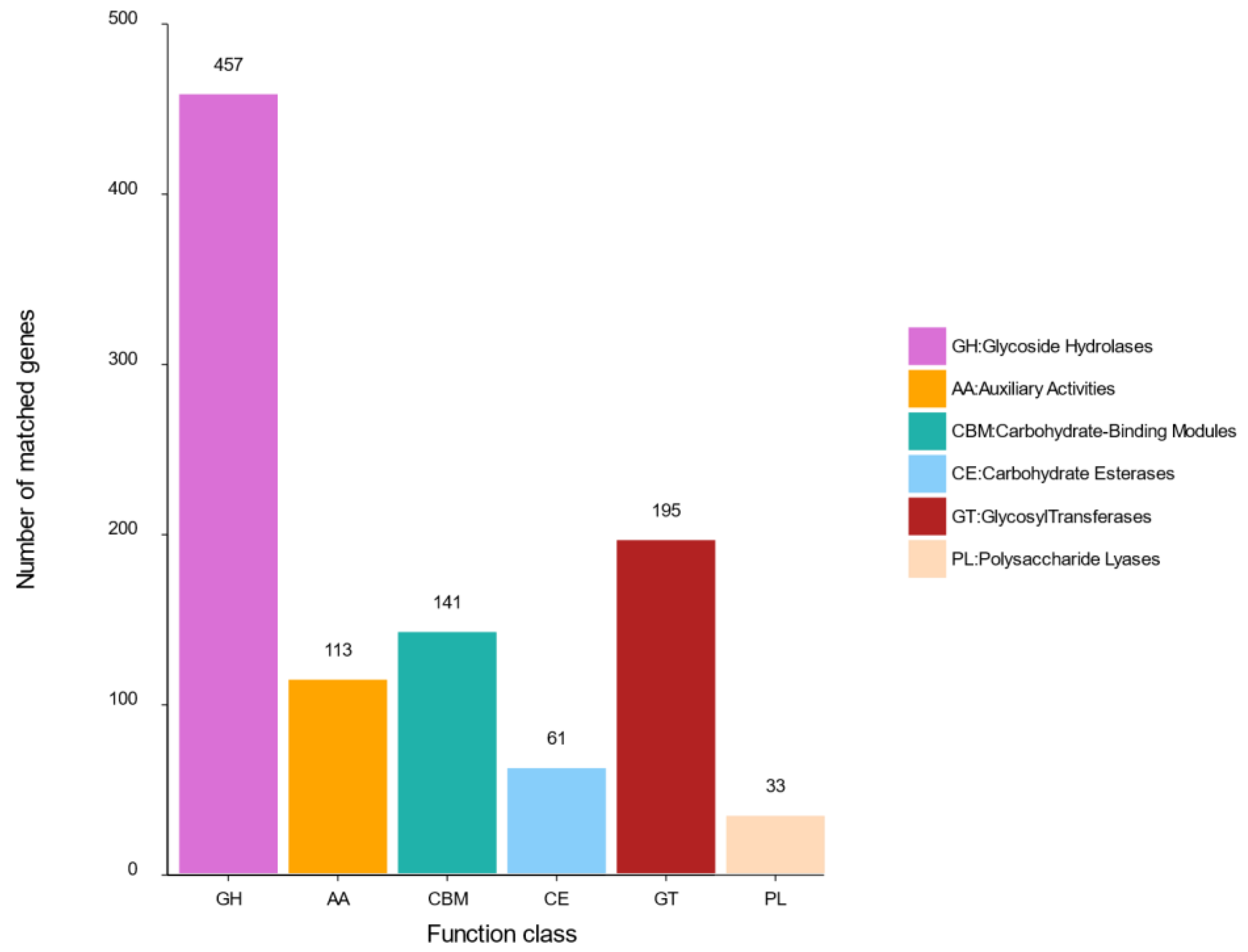
2.4.2. Carbohydrate Genes
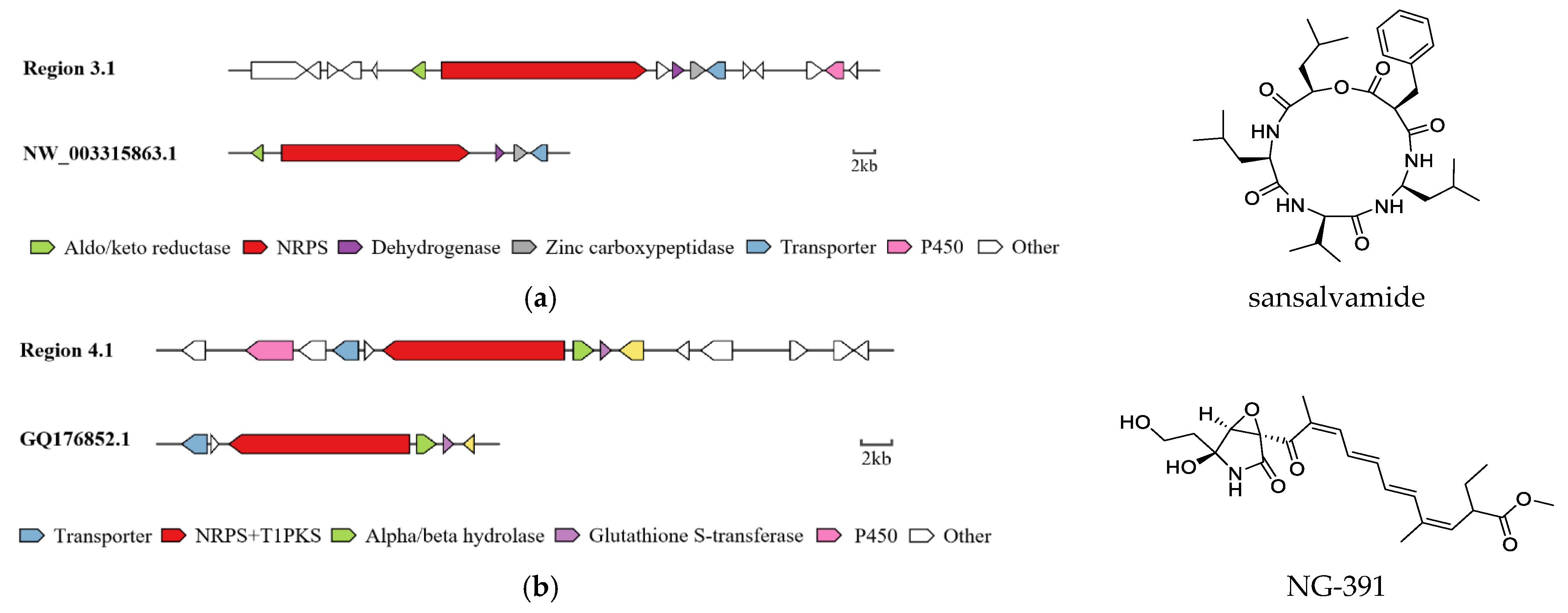
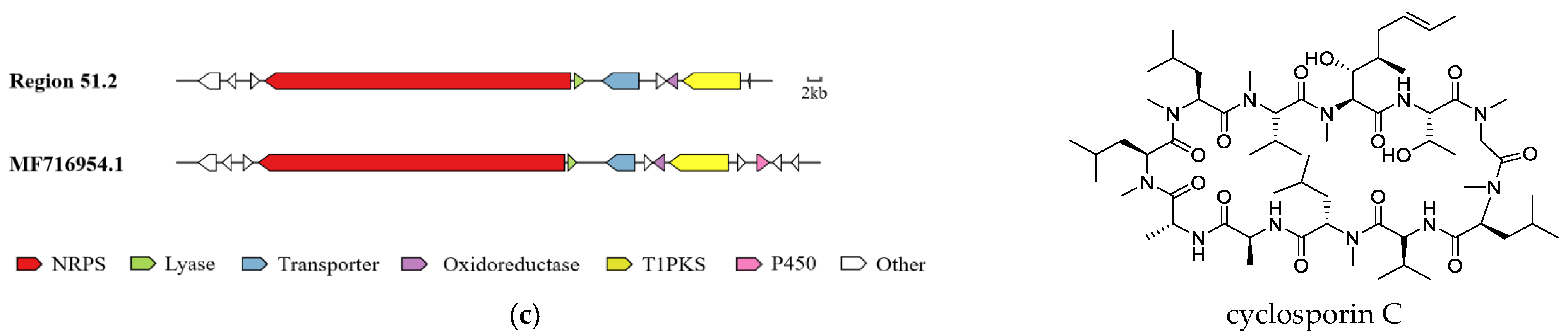
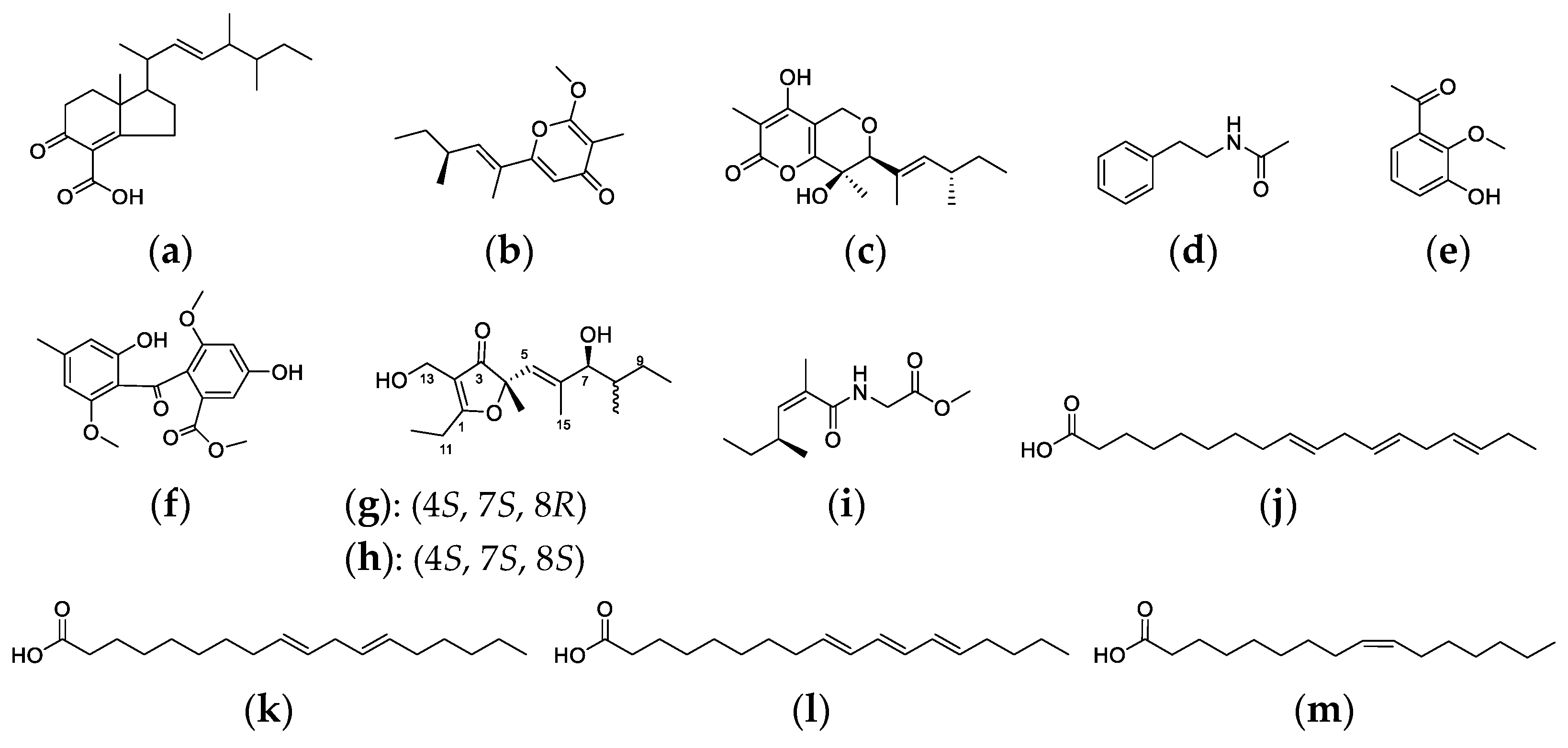
2.5. Analysis of Secondary Metabolite Biosynthetic Gene Clusters
3. Materials and Methods
3.1. Microbes and Cultivation
3.2. Phylogenetic Analysis
3.3. Genome Sequencing and Assembly
3.4. Gene Prediction and Annotation
3.5. Analysis of Secondary Metabolite Biosynthetic Gene Clusters
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2020, 37, 175–223. [Google Scholar] [CrossRef]
- Venugopalan, A.; Srivastava, S. Endophytes as in vitro production platforms of high value plant secondary metabolites. Biotechnol. Adv. 2015, 33, 873–887. [Google Scholar] [CrossRef]
- Zhang, H.; Bai, X.; Zhang, M.; Chen, J.; Wang, H. Bioactive natural products from endophytic microbes. Nat. Prod. J. 2018, 8, 86–108. [Google Scholar] [CrossRef]
- Andrea, S.; Gary, S.; Donald, S. Taxol and taxane production by Taxomyces andreanae, an endophytic fungus of Pacific Yew. Science 1993, 260, 214–216. [Google Scholar]
- Dweba, C.C.; Figlan, S.; Shimelis, H.A.; Motaung, T.E.; Sydenham, S.; Mwadzingeni, L.; Tsilo, T.J. Fusarium head blight of wheat: Pathogenesis and control strategies. Crop Prot. 2017, 91, 114–122. [Google Scholar] [CrossRef]
- Champaco, E.R.; Mrtyn, R.D.; Miller, M.E. Comparison of Fusarium solani and F. oxysporum as causal agents of fruit rot and root rot of muskmelon. Hortscience 1993, 28, 1174–1177. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.X.; Li, S.F.; Zhao, F.; Dai, H.Q.; Bao, L.; Ding, R.; Gao, H.; Zhang, L.X.; Wen, H.A.; Liu, H.W. Chemical constituents from endophytic fungus Fusarium oxysporum. Fitoterapia 2011, 82, 777–781. [Google Scholar] [CrossRef]
- Kyekyeku, J.O.; Kusari, S.; Adosraku, R.K.; Bullach, A.; Golz, C.; Strohmann, C.; Spiteller, M. Antibacterial secondary metabolites from an endophytic fungus, Fusarium solani JK10. Fitoterapia 2017, 119, 108–114. [Google Scholar] [CrossRef]
- Sakai, A.; Suzuki, C.; Masui, Y.; Kuramashi, A.; Takatori, K.; Tanaka, N. The activities of mycotoxins derived from Fusarium and related substances in a short-term transformation assay using v-Ha-ras-transfected BALB/3T3 cells (Bhas 42 cells). Mutat. Res. 2007, 630, 103–111. [Google Scholar] [CrossRef]
- Lee, D.; Shim, S.; Kang, K. 4,6′-anhydrooxysporidinone from Fusarium lateritium SSF2 induces autophagic and apoptosis cell death in MCF-7 breast cancer cells. Biomolecules 2021, 11, 869. [Google Scholar] [CrossRef]
- Dame, Z.T.; Silima, B.; Gryzenhout, M.; van Ree, T. Bioactive compounds from the endophytic fungus Fusarium proliferatum. Nat. Prod. Res. 2016, 30, 1301–1304. [Google Scholar] [CrossRef]
- Alsufiani, H.; Ashour, W. Effectiveness of the natural antioxidant 2,4,4′-Trihydroxychalcone on the oxidation of sunflower oil during storage. Molecules 2021, 26, 1630. [Google Scholar] [CrossRef]
- Caicedo, N.H.; Davalos, A.F.; Puente, P.A.; Rodriguez, A.Y.; Caicedo, P.A. Antioxidant activity of exo-metabolites produced by Fusarium oxysporum: An endophytic fungus isolated from leaves of Otoba gracilipes. Microbiologyopen 2019, 8, e903. [Google Scholar] [CrossRef] [Green Version]
- Russell, A.H.; Truman, A.W. Genome mining strategies for ribosomally synthesised and post-translationally modified peptides. Comput. Struct. Biotechnol. J. 2020, 18, 1838–1851. [Google Scholar] [CrossRef]
- Blin, K.; Kim, H.U.; Medema, M.H.; Weber, T. Recent development of antiSMASH and other computational approaches to mine secondary metabolite biosynthetic gene clusters. Brief. Bioinform. 2019, 20, 1103–1113. [Google Scholar] [CrossRef]
- Li, M.; Yu, R.; Bai, X.; Wang, H.; Zhang, H. Fusarium: A treasure trove of bioactive secondary metabolites. Nat. Prod. Rep. 2020, 37, 1568–1588. [Google Scholar] [CrossRef]
- Weber, S.D.; Hofmann, A.; Pilhofer, M.; Wanner, G.; Agerer, R.; Ludwig, W.; Schleifer, K.H.; Fried, J. The diversity of fungi in aerobic sewage granules assessed by 18S rRNA gene and ITS sequence analyses. FEMS Microbiol. Ecol. 2009, 68, 246–254. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Fungal Barcoding, C.; Fungal Barcoding Consortium Author, L. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [Green Version]
- Natale, D.A.; Shankavaram, U.T.; Galperin, M.Y.; Wolf, Y.I.; Aravind, L.; Koonin, E.V. Towards understanding the first genome sequence of a crenarchaeon by genome annotation using clusters of orthologous groups of proteins (COGs). Genome Biol. 2000, 1, 5. [Google Scholar] [CrossRef] [Green Version]
- Yamada, Y.; Hirakawa, H.; Hori, K.; Minakuchi, Y.; Toyoda, A.; Shitan, N.; Sato, F. Comparative analysis using the draft genome sequence of California poppy (Eschscholzia californica) for exploring the candidate genes involved in benzylisoquinoline alkaloid biosynthesis. Biosci. Biotechnol. Biochem. 2021, 85, 851–859. [Google Scholar] [CrossRef]
- Huntley, R.P.; Harris, M.A.; Alam-Faruque, Y.; Blake, J.A.; Seth, C.; Dietze, H.; Dimmer, E.C.; Foulger, R.E.; Hill, D.P.; Khodiyar, V.K.; et al. A method for increasing expressivity of Gene Ontology annotations using a compositional approach. BMC Bioinf. 2014, 15, 155. [Google Scholar] [CrossRef] [Green Version]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; Sahu, J.; Iyer, S.V.; Khamari, L.; De Silva, N.; et al. PHI-base in 2022: A multi-species phenotype database for Pathogen–Host Interactions. Nucleic Acids Res. 2022, 50, D837–D847. [Google Scholar] [CrossRef]
- Pernas, L. Cellular metabolism in the defense against microbes. J. Cell Sci. 2021, 134, jcs252023. [Google Scholar] [CrossRef]
- Garron, M.L.; Henrissat, B. The continuing expansion of CAZymes and their families. Curr. Opin. Chem. Biol. 2019, 53, 82–87. [Google Scholar] [CrossRef]
- Drula, E.; Garron, M.L.; Dogan, S.; Lombard, V.; Henrissat, B.; Terrapon, N. The carbohydrate-active enzyme database: Functions and literature. Nucleic Acids Res. 2022, 50, D571–D577. [Google Scholar] [CrossRef]
- Xu, L.; Li, Y.; Biggins, J.B.; Bowman, B.R.; Verdine, G.L.; Gloer, J.B.; Alspaugh, J.A.; Bills, G.F. Identification of cyclosporin C from Amphichorda felina using a Cryptococcus neoformans differential temperature sensitivity assay. Appl. Microbiol. Biotechnol. 2018, 102, 2337–2350. [Google Scholar] [CrossRef] [Green Version]
- Donzelli, B.G.G.; Krasnoff, S.B.; Churchill, A.C.L.; Vandenberg, J.D.; Gibson, D.M. Identification of a hybrid PKS–NRPS required for the biosynthesis of NG-391 in Metarhizium robertsii. Curr. Genet. 2010, 56, 151–162. [Google Scholar] [CrossRef]
- Romans-Fuertes, P.; Sondergaard, T.E.; Sandmann, M.I.; Wollenberg, R.D.; Nielsen, K.F.; Hansen, F.T.; Giese, H.; Brodersen, D.E.; Sorensen, J.L. Identification of the non-ribosomal peptide synthetase responsible for biosynthesis of the potential anti-cancer drug sansalvamide in Fusarium solani. Curr. Genet. 2016, 62, 799–807. [Google Scholar] [CrossRef]
- Gilbert, N.B.; Paul, R.J.; William, F. Sansalvamide: A new cytotoxic cyclic depsipeptide produced by a marine fungus of the genus Fusarium. Tetrahedron Lett. 1999, 40, 2913–2916. [Google Scholar]
- Lawrence, A.W.; Leonard, F.B. Fusarin C, a mutagen from Fusarium Moniliforme grown on corn. J. Food Sci. 1981, 46, 1424–1426. [Google Scholar]
- Gelderblom, W.C.; Thiel, P.G.; van der Merwe, K.J. The chemical and enzymatic interaction of glutathione with the fungal metabolite, fusarin C. Mutat. Res. 1988, 199, 207–214. [Google Scholar] [CrossRef]
- Krasnoff, S.B.; Sommers, C.H.; Moon, Y.-S.; Donzelli, B.G.G.; Vandenberg, J.D.; Churchill, A.C.L.; Gibson, D.M. Production of mutagenic metabolites by Metarhizium anisopliae. J. Agric. Food Chem. 2006, 54, 7083–7088. [Google Scholar] [CrossRef]
- Weber, G.; Schorgendorfer, K.; Schneiderscherzer, E.; Leitner, E. The peptide synthetase catalyzing cyclosporine production in Tolypocladium niveum is encoded by a giant 45.8-kilobase open reading fram. Curr. Genet. 1994, 26, 120–125. [Google Scholar] [CrossRef]
- Hoffmeister, D.; Keller, N.P. Natural products of filamentous fungi: Enzymes, genes, and their regulation. Nat. Prod. Rep. 2007, 24, 393–416. [Google Scholar] [CrossRef]
- Sallam, L.; El-Refai, A.; Hamdy, A.; El-Minofi, H.; Abdel-Salam, I. Role of some fermentation parameters on cyclosporin A production by a new isolate of Aspergillus terreus. J. Gen. Appl. Microbiol. 2003, 49, 321–328. [Google Scholar]
- Sakamoto, K.; Tsujii, E.; Miyauchi, M.; Nakanishi, T.; Yamashita, M.; Shigematsu, N.; Tada, T.; Izumi, S.; Okuhara, M. FR901459, a novel immunosuppressant isolated from Stachybotrys chartarum No. 19392. Taxonomy of the producing organism, fermentation, isolation, physico-chemical properties and biological activities. J. Antibiot. 1993, 46, 1788–1798. [Google Scholar] [CrossRef]
- Studt, L.; Wiemann, P.; Kleigrewe, K.; Humpf, H.U.; Tudzynski, B. Biosynthesis of fusarubins accounts for pigmentation of Fusarium fujikuroi perithecia. Appl. Environ. Microbiol. 2012, 78, 4468–4480. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, T.B.; Nielsen, M.R.; Kristensen, S.B.; Spedtsberg, E.M.L.; Yasmine, W.; Matthiesen, R.; Kaniki, S.E.K.; Sorensen, T.; Petersen, C.; Muff, J.; et al. Heterologous expression of the core genes in the complex fusarubin gene cluster of Fusarium Solani. Int. J. Mol. Sci. 2020, 21, 7601. [Google Scholar] [CrossRef]
- Fu, Z.; Liu, Y.; Xu, M.; Yao, X.; Wang, H.; Zhang, H. Absolute configuration determination of two diastereomeric neovasifuranones A and B from Fusarium oxysporum R1 by a combination of Mosher’s method and chiroptical approach. J. Fungi 2021, 8, 40. [Google Scholar] [CrossRef]
- Yu, R.; Li, M.; Wang, Y.; Bai, X.; Chen, J.; Li, X.; Wang, H.; Zhang, H. Chemical investigation of a co-culture of Aspergillus fumigatus D and Fusarium oxysporum R1. Rec. Nat. Prod. 2020, 15, 130–135. [Google Scholar] [CrossRef]
- Chen, J.; Bai, X.; Hua, Y.; Zhang, H.; Wang, H. Fusariumins C and D, two novel antimicrobial agents from Fusarium oxysporum ZZP-R1 symbiotic on Rumex madaio Makino. Fitoterapia 2019, 134, 1–4. [Google Scholar] [CrossRef]
- Furumoto, T.; Hamasaki, T.; Nakajima, H. Biosynthesis of phytotoxin neovasinin and its related metabolites, neovasipyrones A and B and neovasifuranones A and B, in the phytopathogenic fungus Neocosmospora vasinfecta. J. Chem. Soc. Perkin Trans. 1999, 1, 131–135. [Google Scholar] [CrossRef]
- Bai, X.; Yu, R.; Li, M.; Zhang, H. Antimicrobial assay of endophytic fungi from Rumex madaio and chemical study of strain R1. Bangl. J. Pharmacol. 2019, 14, 129–135. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A.; et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef] [Green Version]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. AntiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Blin, K.; Medema, M.H.; Kottmann, R.; Lee, S.Y.; Weber, T. The antiSMASH database, a comprehensive database of microbial secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 2017, 45, D555–D559. [Google Scholar] [CrossRef] [Green Version]
- Hoogendoorn, K.; Barra, L.; Waalwijk, C.; Dickschat, J.S.; van der Lee, T.A.J.; Medema, M.H. Evolution and diversity of biosynthetic gene clusters in Fusarium. Front. Microbiol. 2018, 9, 1158. [Google Scholar] [CrossRef] [Green Version]
- Pan, R.; Bai, X.; Chen, J.; Zhang, H.; Wang, H. Exploring structural diversity of microbe secondary metabolites using OSMAC strategy: A literature review. Front. Microbiol. 2019, 10, 294. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Value | Item | Count | Percentage (%) |
|---|---|---|---|---|
| Total length (bp) | 51,784,516 | All | 17,145 | 88.68 |
| Max length (bp) | 6,563,362 | NR | 16,803 | 86.91 |
| GC content (%) | 50.4 | NT | 15,461 | 79.97 |
| Gene number | 19,333 | Swiss-Prot | 11,026 | 57.03 |
| Gene total length (bp) | 24,687,144 | KEGG | 10,894 | 56.35 |
| Gene average length (bp) | 1276.94 | COG | 8048 | 41.63 |
| GC content in gene region (%) | 55.06 | GO | 8780 | 45.41 |
| Gene/Genome (%) | 47.67 | |||
| Contigs | 542 | |||
| N50 (bp) | 3,209,824 | |||
| N90 (bp) | 1,367,080 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Xu, M.; Tang, Y.; Shao, Y.; Wang, H.; Zhang, H. Genome Features and AntiSMASH Analysis of an Endophytic Strain Fusarium sp. R1. Metabolites 2022, 12, 521. https://doi.org/10.3390/metabo12060521
Liu Y, Xu M, Tang Y, Shao Y, Wang H, Zhang H. Genome Features and AntiSMASH Analysis of an Endophytic Strain Fusarium sp. R1. Metabolites. 2022; 12(6):521. https://doi.org/10.3390/metabo12060521
Chicago/Turabian StyleLiu, Yuanyuan, Meijie Xu, Yuqi Tang, Yilan Shao, Hong Wang, and Huawei Zhang. 2022. "Genome Features and AntiSMASH Analysis of an Endophytic Strain Fusarium sp. R1" Metabolites 12, no. 6: 521. https://doi.org/10.3390/metabo12060521
APA StyleLiu, Y., Xu, M., Tang, Y., Shao, Y., Wang, H., & Zhang, H. (2022). Genome Features and AntiSMASH Analysis of an Endophytic Strain Fusarium sp. R1. Metabolites, 12(6), 521. https://doi.org/10.3390/metabo12060521