
Metabolic Determinants in Cardiomyocyte Function and Heart Regenerative Strategies

 ,
,  , and
, and
Abstract
:1. Introduction
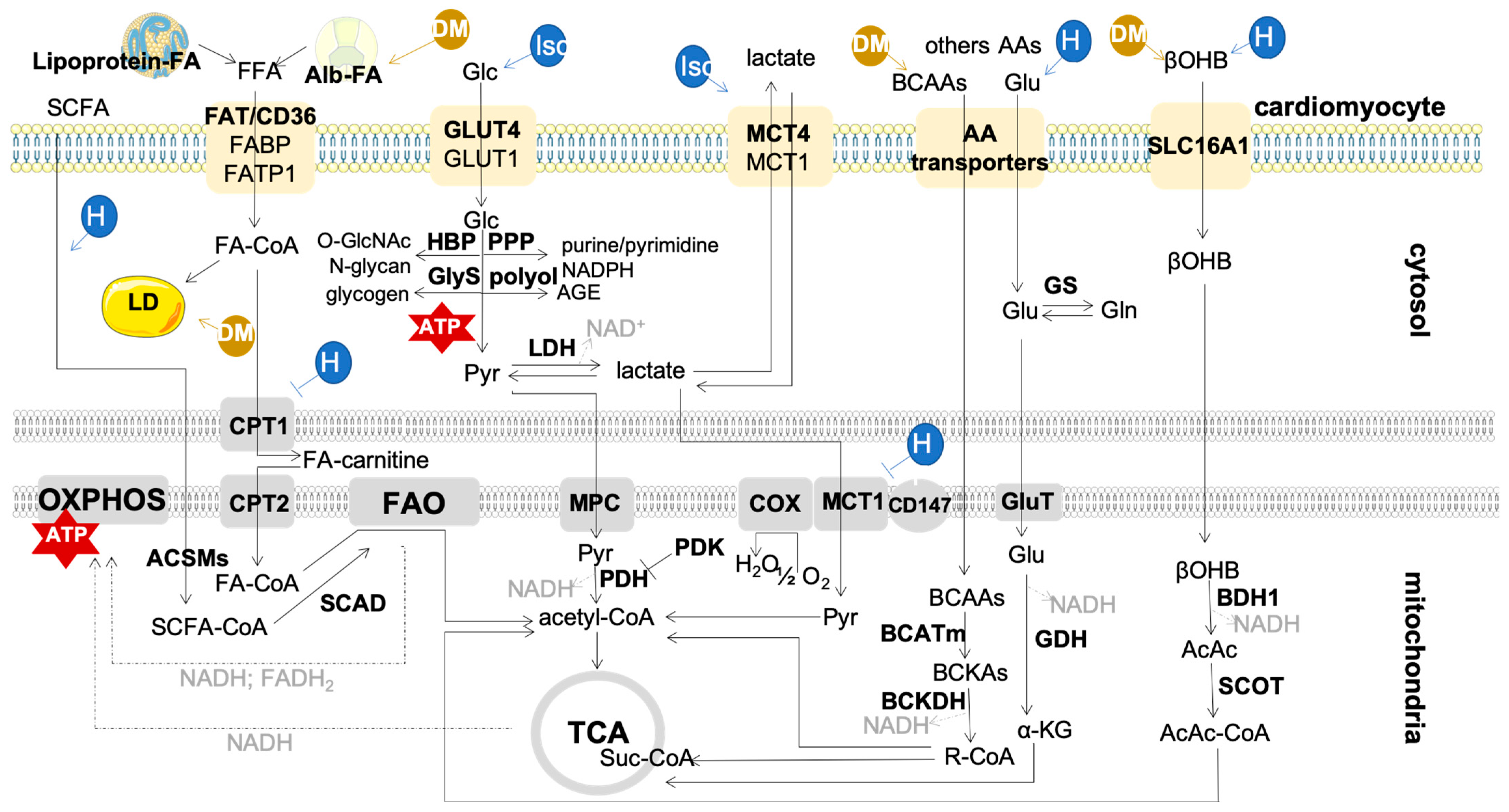
2. Metabolism in the Adult Heart: Mitochondria and Fuels
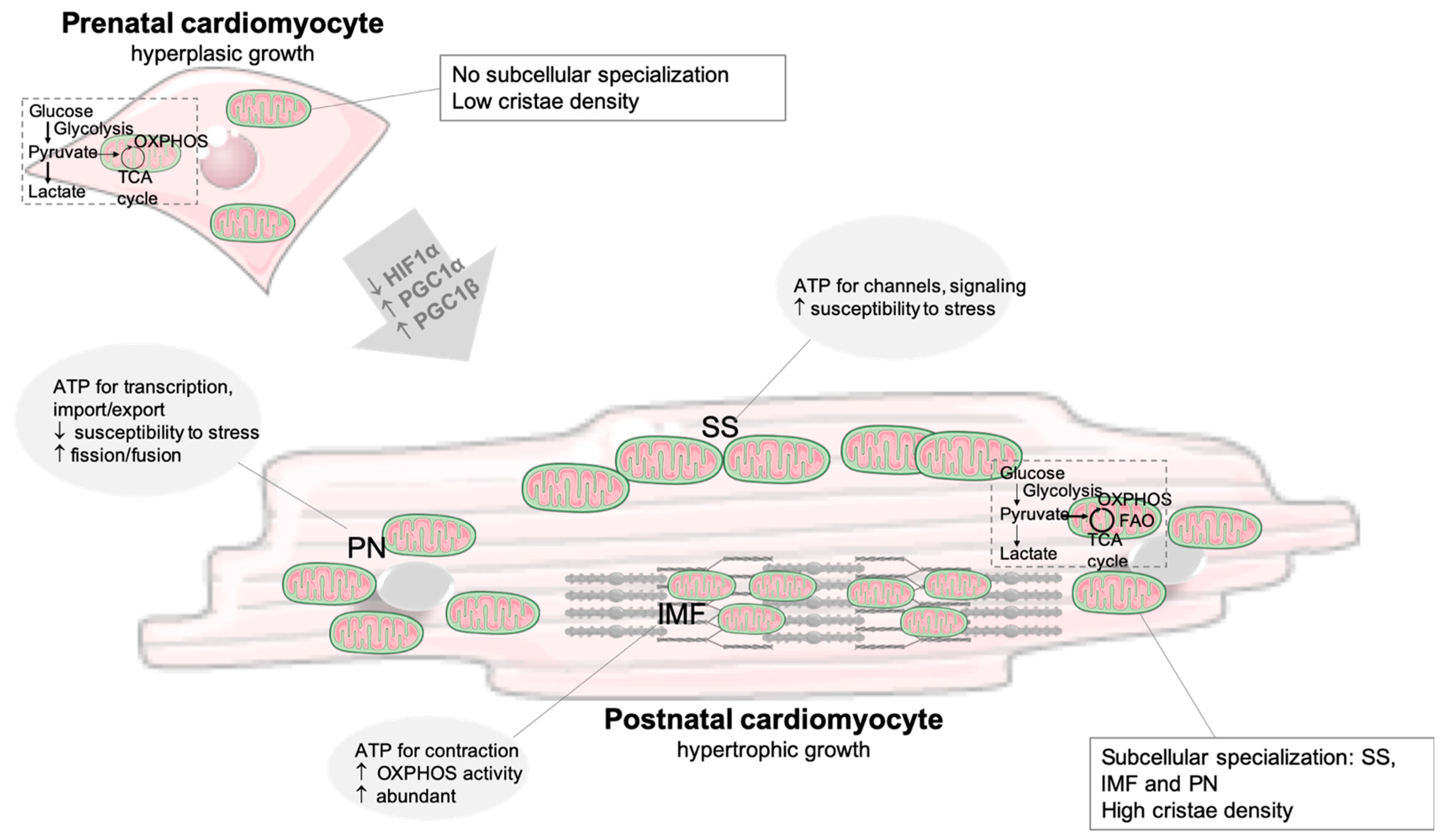
2.1. Developmental Changes and the Fetal Metabolic Switch
2.2. Metabolic Changes in the Injured Heart
3. Metabolic Control of Stem Cells Pluripotency with Implications for Cardiac Regeneration
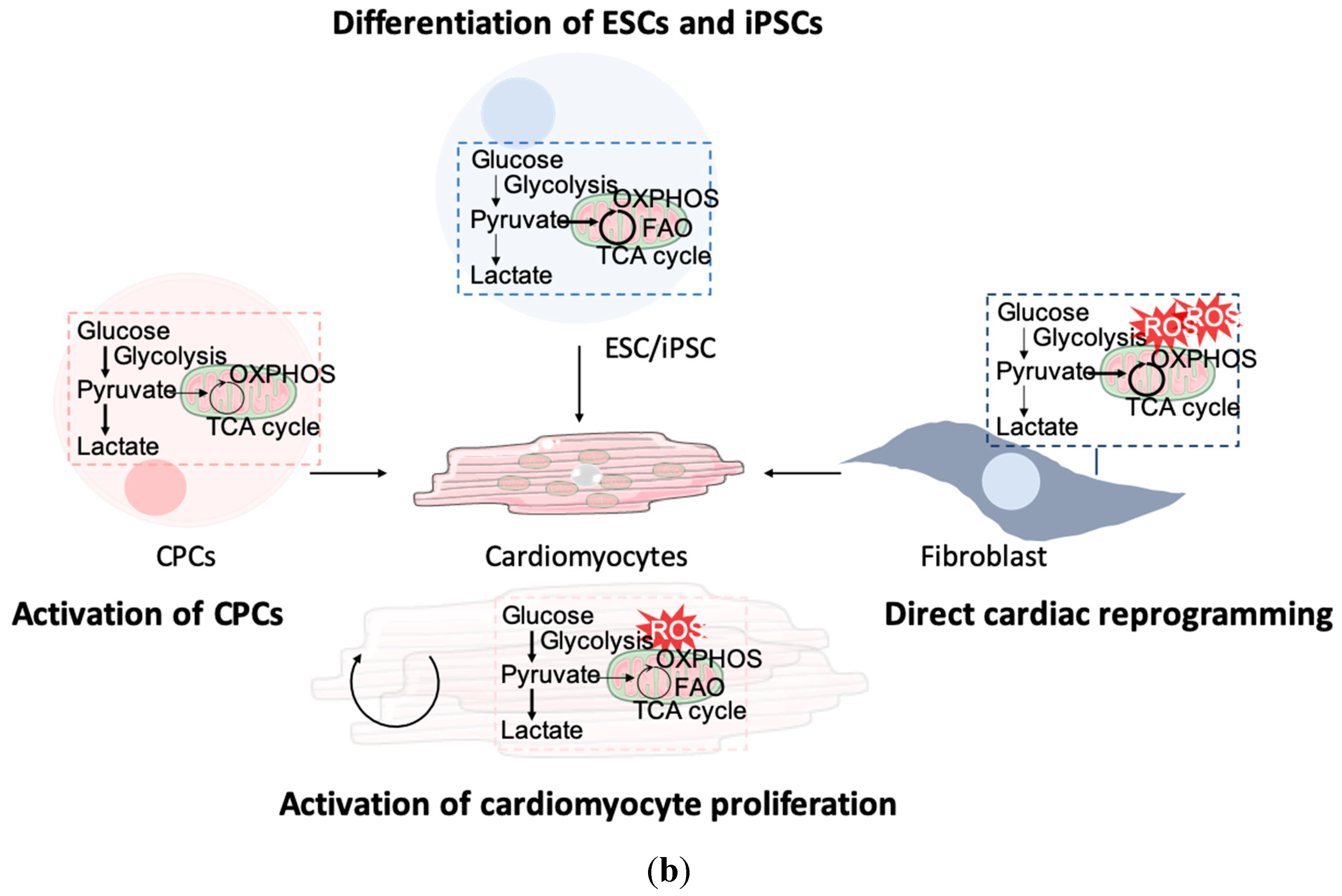
4. Metabolic Reprogramming to Enhance Cardiac Regenerative Strategies
4.1. Activation of Cardiomyocyte Proliferation
4.2. Recruitment of Cardiac Stem or Progenitor Cells
4.3. Delivery of De Novo Cardiomyocytes from Differentiated ESCs/iPSCs
4.4. Direct Reprogramming of Fibroblasts into Induced Cardiomyocytes
4.5. Systemic Metabolic Strategies for Heart Regeneration
4.5.1. Nutrient Signaling
4.5.2. Gene Therapy
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics 2021 Update. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, M.A.; Murry, C.E. Heart regeneration. Nature 2011, 473, 326–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, H.; Olson, E.N.; Bassel-Duby, R. Therapeutic approaches for cardiac regeneration and repair. Nat. Rev. Cardiol. 2018, 15, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Ghiroldi, A.; Piccoli, M.; Cirillo, F.; Monasky, M.M.; Ciconte, G.; Pappone, C.; Anastasia, L. Cell-Based Therapies for Cardiac Regeneration: A Comprehensive Review of Past and Ongoing Strategies. Int. J. Mol. Sci. 2018, 19, 3194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neidig, L.E.; Weinberger, F.; Palpant, N.J.; Mignone, J.; Martinson, A.M.; Sorensen, D.W.; Bender, I.; Nemoto, N.; Reinecke, H.; Pabon, L.; et al. Evidence for minimal cardiogenic potential of stem cell antigen 1-positive cells in the adult mouse heart. Circulation 2018, 138, 2960–2962. [Google Scholar] [CrossRef] [PubMed]
- Soonpaa, M.H.; Kim, K.K.; Pajak, L.; Franklin, M.; Field, L.J. Cardiomyocyte DNA synthesis and binucleation during murine development. Am. J. Physiol. Circ. Physiol. 1996, 271, H2183–H2189. [Google Scholar] [CrossRef]
- Lehman, J.J.; Kelly, D.P. Transcriptional Activation of Energy Metabolic Switches in the Developing and Hypertrophied Heart. Clin. Exp. Pharmacol. Physiol. 2002, 29, 339–345. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Collins-Nakai, R.L.; Itoi, T. Developmental changes in energy substrate use by the heart. Cardiovasc. Res. 1992, 26, 1172–1180. [Google Scholar] [CrossRef]
- Ho, K.L.; Zhang, L.; Wagg, C.; Al Batran, R.; Gopal, K.; Levasseur, J.; Leone, T.; Dyck, J.R.B.; Ussher, J.R.; Muoio, D.M.; et al. Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc. Res. 2019, 115, 1606–1616. [Google Scholar] [CrossRef]
- Ho, K.L.; Karwi, Q.G.; Wagg, C.; Zhang, L.; Vo, K.; Altamimi, T.; Uddin, G.M.; Ussher, J.R.; Lopaschuk, G.D. Ketones can become the major fuel source for the heart but do not increase cardiac efficiency. Cardiovasc. Res. 2020, 117, 1178–1187. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritterhoff, J.; McMillen, T.S.; Villet, O.; Young, S.; Kolwicz, S.C.; Senn, T.; Caudal, A.; Tian, R. Increasing fatty acid oxidation elicits a sex-dependent response in failing mouse hearts. J. Mol. Cell. Cardiol. 2021, 158, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shyh-Chang, N.; Ng, H.-H. The metabolic programming of stem cells. Genes Dev. 2017, 31, 336–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Intlekofer, A.M.; Finley, L.W.S. Metabolic signatures of cancer cells and stem cells. Nat. Metab. 2019, 1, 177–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, J.H.; Chung, J.D.; Lynch, G.S.; Ryall, J.G. The Microenvironment Is a Critical Regulator of Muscle Stem Cell Activation and Proliferation. Front. Cell Dev. Biol. 2019, 7, 254. [Google Scholar] [CrossRef] [PubMed]
- Clémot, M.; Demarco, R.S.; Jones, D.L. Lipid Mediated Regulation of Adult Stem Cell Behavior. Front. Cell Dev. Biol. 2020, 8, 115. [Google Scholar] [CrossRef] [Green Version]
- Karwi, Q.G.; Biswas, D.; Pulinilkunnil, T.; Lopaschuk, G.D. Myocardial Ketones Metabolism in Heart Failure. J. Card. Fail. 2020, 26, 998–1005. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Lopaschuk, G.D. Branched-Chain Amino Acid Metabolism in the Failing Heart. Cardiovasc. Drugs Ther. 2022, 1–8. [Google Scholar] [CrossRef]
- Brown, D.A.; Perry, J.B.; Allen, D.A.B.J.B.P.M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.F.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Jaswal, J.S. Energy Metabolic Phenotype of the Cardiomyocyte during Development, Differentiation, and Postnatal Maturation. J. Cardiovasc. Pharmacol. 2010, 56, 130–140. [Google Scholar] [CrossRef]
- Kolwicz, S.C., Jr.; Purohit, S.; Tian, R. Cardiac Metabolism and its Interactions with Contraction, Growth, and Survival of Cardiomyocytes. Circ. Res. 2013, 113, 603–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haffar, T.; Bérubé-Simard, F.; Bousette, N. Impaired fatty acid oxidation as a cause for lipotoxicity in cardiomyocytes. Biochem. Biophys. Res. Commun. 2015, 468, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Thai, P.N.; Lu, S.; Pu, J.; Bers, D.M. Intrafibrillar and perinuclear mitochondrial heterogeneity in adult cardiac myocytes. J. Mol. Cell. Cardiol. 2019, 136, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-B.; Kalkhoran, S.B.; Hernández-Reséndiz, S.; Samangouei, P.; Hausenloy, D.J. Mitochondrial-Shaping Proteins in Cardiac Health and Disease—The Long and the Short of It! Cardiovasc. Drugs Ther. 2017, 31, 87–107. [Google Scholar] [CrossRef] [Green Version]
- Murashige, D.; Jang, C.; Neinast, M.; Edwards, J.J.; Cowan, A.; Hyman, M.C.; Rabinowitz, J.D.; Frankel, D.S.; Arany, Z. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science 2020, 370, 364–368. [Google Scholar] [CrossRef]
- Heather, L.C.; Pates, K.M.; Atherton, H.J.; Cole, M.A.; Ball, D.R.; Evans, R.D.; Glatz, J.F.; Luiken, J.J.; Griffin, J.L.; Clarke, K. Differential Translocation of the Fatty Acid Transporter, FAT/CD36, and the Glucose Transporter, GLUT4, Coordinates Changes in Cardiac Substrate Metabolism during Ischemia and Reperfusion. Circ. Heart Fail. 2013, 6, 1058–1066. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, D.; Stump, D.; Potter, B.J.; Robinson, R.B.; White, R.; Kiang, C.L.; Berk, P.D. Oleate uptake by cardiac myocytes is carrier mediated and involves a 40-kD plasma membrane fatty acid binding protein similar to that in liver, adipose tissue, and gut. J. Clin. Investig. 1988, 82, 928–935. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, I.K.N.; Tusubira, D.; Ashrafi, H.; Dyrstad, S.E.; Hansen, L.; Liu, X.-Z.; Nilsson, L.I.H.; Løvsletten, N.G.; Berge, K.; Wergedahl, H.; et al. Upregulated PDK4 expression is a sensitive marker of increased fatty acid oxidation. Mitochondrion 2019, 49, 97–110. [Google Scholar] [CrossRef]
- Bertrand, L.; Auquier, J.; Renguet, E.; Angé, M.; Cumps, J.; Horman, S.; Beauloye, C. Glucose transporters in cardiovascular system in health and disease. Pflugers Arch. 2020, 472, 1385–1399. [Google Scholar] [CrossRef]
- Kolwicz, S.C.; Tian, R. Glucose metabolism and cardiac hypertrophy. Cardiovasc. Res. 2011, 90, 194–201. [Google Scholar] [CrossRef] [Green Version]
- Tran, D.H.; Wang, Z.V. Glucose Metabolism in Cardiac Hypertrophy and Heart Failure. J. Am. Heart Assoc. 2019, 8, e012673. [Google Scholar] [CrossRef] [PubMed]
- Ritterhoff, J.; Tian, R. Metabolism in cardiomyopathy: Every substrate matters. Cardiovasc Res. 2017, 113, 411–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walejko, J.M.; Koelmel, J.P.; Garrett, T.J.; Edison, A.S.; Keller-Wood, M. Multiomics approach reveals metabolic changes in the heart at birth. Am. J. Physiol.-Endocrinol. Metab. 2018, 315, E1212–E1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerychova, R.; Pavlinkova, G. HIF-1, Metabolism, and Diabetes in the Embryonic and Adult Heart. Front. Endocrinol. 2018, 9, 460. [Google Scholar] [CrossRef] [Green Version]
- Iruretagoyena, J.I.; Davis, W.; Bird, C.; Olsen, J.; Radue, R.; Broman, A.T.; Kendziorski, C.; Bondurant, S.S.; Golos, T.; Bird, I.; et al. Metabolic gene profile in early human fetal heart development. Mol. Hum. Reprod. 2014, 20, 690–700. [Google Scholar] [CrossRef] [Green Version]
- Goffart, S.; Von Kleist-Retzow, J.-C.; Wiesner, R.J. Regulation of mitochondrial proliferation in the heart: Power-plant failure contributes to cardiac failure in hypertrophy. Cardiovasc. Res. 2004, 64, 198–207. [Google Scholar] [CrossRef] [Green Version]
- Martin, O.J.; Lai, L.; Soundarapandian, M.M.; Leone, T.C.; Zorzano, A.; Keller, M.P.; Attie, A.D.; Muoio, D.M.; Kelly, D.P. A Role for Peroxisome Proliferator-Activated Receptor γ Coactivator-1 in the Control of Mitochondrial Dynamics during Postnatal Cardiac Growth. Circ. Res. 2014, 114, 626–636. [Google Scholar] [CrossRef] [Green Version]
- Menendez-Montes, I.; Escobar, B.; Palacios, B.; Gómez, M.J.; Izquierdo-Garcia, J.L.; Flores, L.; Jiménez-Borreguero, L.J.; Aragones, J.; Ruiz-Cabello, J.; Torres, M.; et al. Myocardial VHL-HIF Signaling Controls an Embryonic Metabolic Switch Essential for Cardiac Maturation. Dev. Cell 2016, 39, 724–739. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.R.; Li, J.; Coven, D.L.; Pypaert, M.; Zechner, C.; Palmeri, M.; Giordano, F.J.; Mu, J.; Birnbaum, M.J.; Young, L.H. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J. Clin. Investig. 2004, 114, 495–503. [Google Scholar] [CrossRef]
- Fillmore, N.; Levasseur, J.L.; Fukushima, A.; Wagg, C.S.; Wang, W.; Dyck, J.R.B.; Lopaschuk, G.D. Uncoupling of glycolysis from glucose oxidation accompanies the development of heart failure with preserved ejection fraction. Mol. Med. 2018, 24, 3. [Google Scholar] [CrossRef] [Green Version]
- Cluntun, A.A.; Badolia, R.; Lettlova, S.; Parnell, K.M.; Shankar, T.S.; Diakos, N.A.; Olson, K.A.; Taleb, I.; Tatum, S.M.; Berg, J.A.; et al. The pyruvate-lactate axis modulates cardiac hypertrophy and heart failure. Cell Metab. 2020, 33, 629–648.e10. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Soto, J.; Anderson, B.; Riehle, C.; Zhang, Y.; Wende, A.; Jones, D.; McClain, D.; Abel, E.D. Regulation of fatty acid metabolism by mTOR in adult murine hearts occurs independently of changes in PGC-1α. Am. J. Physiol. Circ. Physiol.-Heart 2013, 305, H41–H51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, S.; Qian, L.; Cheng, Z.; Chen, C.; Wang, K.; Hu, S.; Zhang, X.; Wu, T. Lactate and Myocadiac Energy Metabolism. Front. Physiol. 2021, 12, 715081. [Google Scholar] [CrossRef] [PubMed]
- Chatham, J.C.; Rosiers, C.D.; Forder, J.R. Evidence of separate pathways for lactate uptake and release by the perfused rat heart. Am. J. Physiol.-Endocrinol. Metab. 2001, 281, E794–E802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfleger, J.; Gross, P.; Johnson, J.; Carter, R.L.; Gao, E.; Tilley, D.G.; Houser, S.R.; Koch, W.J. G protein-coupled receptor kinase 2 contributes to impaired fatty acid metabolism in the failing heart. J. Mol. Cell. Cardiol. 2018, 123, 108–117. [Google Scholar] [CrossRef]
- Carley, A.N.; Maurya, S.K.; Fasano, M.; Wang, Y.; Selzman, C.H.; Drakos, S.G.; Lewandowski, E.D. Short-Chain Fatty Acids Outpace Ketone Oxidation in the Failing Heart. Circulation 2021, 143, 1797–1808. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Liu, J.; Zhu, L.; Tan, F.; Qin, Y.; Huang, H.; Yu, Y. Free fatty acid can induce cardiac dysfunction and alter insulin signaling pathways in the heart. Lipids Health Dis. 2018, 17, 185. [Google Scholar] [CrossRef] [Green Version]
- Karwi, Q.G.; Sun, Q.; Lopaschuk, G.D. The Contribution of Cardiac Fatty Acid Oxidation to Diabetic Cardiomyopathy Severity. Cells 2021, 10, 3259. [Google Scholar] [CrossRef]
- Wang, L.-Y.; Chen, C. Energy metabolism homeostasis in cardiovascular diseases. J. Geriatr. Cardiol. 2021, 18, 1044–1057. [Google Scholar] [CrossRef]
- Lee, S.R.; Heo, J.H.; Jo, S.L.; Kim, G.; Kim, S.J.; Yoo, H.J.; Lee, K.-P.; Kwun, H.-J.; Shin, H.-J.; Baek, I.-J.; et al. Progesterone receptor membrane component 1 reduces cardiac steatosis and lipotoxicity via activation of fatty acid oxidation and mitochondrial respiration. Sci. Rep. 2021, 11, 8781. [Google Scholar] [CrossRef]
- Lynch, C.J.; Adams, S.H. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Rev. Endocrinol. 2014, 10, 723–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fillmore, N.; Wagg, C.S.; Zhang, L.; Fukushima, A.; Lopaschuk, G.D. Cardiac branched-chain amino acid oxidation is reduced during insulin resistance in the heart. Am. J. Physiol.-Endocrinol. Metab. 2018, 315, E1046–E1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, K.J.; Sidorov, V.Y.; Mcguinness, O.P.; Wasserman, D.H.; Wikswo, J.P. Amino acids as metabolic substrates during cardiac ischemia. Exp. Biol. Med. 2012, 237, 1369–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2013, 25, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, S. Pluripotent Stem Cell-Based Cell Therapy—Promise and Challenges. Cell Stem Cell 2020, 27, 523–531. [Google Scholar] [CrossRef]
- Wu, J.; Ocampo, A.; Belmonte, J.C.I. Cellular Metabolism and Induced Pluripotency. Cell 2016, 166, 1371–1385. [Google Scholar] [CrossRef] [Green Version]
- Tsogtbaatar, E.; Landin, C.; Minter-Dykhouse, K.; Folmes, C.D.L. Energy Metabolism Regulates Stem Cell Pluripotency. Front. Cell Dev. Biol. 2020, 8, 87. [Google Scholar] [CrossRef] [Green Version]
- Davidson, K.C.; Mason, E.; Pera, M.F. The pluripotent state in mouse and human. Development 2015, 142, 3090–3099. [Google Scholar] [CrossRef] [Green Version]
- Takashima, Y.; Guo, G.; Loos, R.; Nichols, J.; Ficz, G.; Krueger, F.; Oxley, D.; Santos, F.; Clarke, J.; Mansfield, W.; et al. Resetting Transcription Factor Control Circuitry toward Ground-State Pluripotency in Human. Cell 2014, 158, 1254–1269. [Google Scholar] [CrossRef] [Green Version]
- Sperber, H.; Mathieu, J.; Wang, Y.; Ferreccio, A.; Hesson, J.; Xu, Z.; Fischer, K.A.; Devi, A.; Detraux, D.; Gu, H.; et al. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat. Cell Biol. 2015, 17, 1523–1535. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Gaeta, X.; Sahakyan, A.; Chan, A.B.; Hong, C.S.; Kim, R.; Braas, D.; Plath, K.; Lowry, W.E.; Christofk, H.R. Glycolytic Metabolism Plays a Functional Role in Regulating Human Pluripotent Stem Cell State. Cell Stem Cell 2016, 19, 476–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Nóbrega-Pereira, S.; Caiado, F.; Carvalho, T.; Matias, I.; Graça, G.; Gonçalves, L.G.; Silva-Santos, B.; Norell, H.; Dias, S. VEGFR2–Mediated Reprogramming of Mitochondrial Metabolism Regulates the Sensitivity of Acute Myeloid Leukemia to Chemotherapy. Cancer Res. 2018, 78, 731–741. [Google Scholar] [CrossRef] [Green Version]
- Nobrega-Pereira, S.; Dias, S. VEGF signaling in acute leukemia: Mitochondrial connections. Oncoscience 2018, 5, 54–56. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Bahat, A.; Gross, A. Mitochondrial plasticity in cell fate regulation. J. Biol. Chem. 2019, 294, 13852–13863. [Google Scholar] [CrossRef] [Green Version]
- Carey, B.W.; Finley, L.W.S.; Cross, J.; Allis, C.D.; Thompson, C.B. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [Green Version]
- Tohyama, S.; Fujita, J.; Hishiki, T.; Matsuura, T.; Hattori, F.; Ohno, R.; Kanazawa, H.; Seki, T.; Nakajima, K.; Kishino, Y.; et al. Glutamine Oxidation Is Indispensable for Survival of Human Pluripotent Stem Cells. Cell Metab. 2016, 23, 663–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Choi, M.; Margineantu, D.; Margaretha, L.; Hesson, J.; Cavanaugh, C.; Blau, C.A.; Horwitz, M.S.; Hockenbery, D.; Ware, C.; et al. HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 2012, 31, 2103–2116. [Google Scholar] [CrossRef] [PubMed]
- Panopoulos, A.D.; Yanes, O.; Ruiz, S.; Kida, Y.S.; Diep, D.; Tautenhahn, R.; Herrerías, A.; Batchelder, E.M.; Plongthongkum, N.; Lutz, M.; et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012, 22, 168–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto, J.; León, M.; Ponsoda, X.; Sendra, R.; Bort, R.; Ferrer-Lorente, R.; Raya, A.; López-García, C.; Torres, J. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat. Commun. 2016, 7, 11124. [Google Scholar] [CrossRef] [PubMed]
- Van Winkle, L.J.; Ryznar, R. One-Carbon Metabolism Regulates Embryonic Stem Cell Fate Through Epigenetic DNA and Histone Modifications: Implications for Transgenerational Metabolic Disorders in Adults. Front. Cell Dev. Biol. 2019, 7, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azuara, V.; Perry, P.; Sauer, S.; Spivakov, M.; Jørgensen, H.; John, R.; Gouti, M.; Casanova, M.; Warnes, G.; Merkenschlager, M.; et al. Chromatin signatures of pluripotent cell lines. Nat. Cell Biol. 2006, 8, 532–538. [Google Scholar] [CrossRef]
- Gaspar-Maia, A.; Alajem, A.; Meshorer, E.; Ramalho-Santos, M. Open chromatin in pluripotency and reprogramming. Nat. Rev. Mol. Cell Biol. 2010, 12, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhang, T.; Wang, L.; Cai, Y.; Zhong, X.; He, X.; Hu, L.; Tian, S.; Wu, M.; Hui, L.; et al. Fatty acid synthesis is critical for stem cell pluripotency via promoting mitochondrial fission. EMBO J. 2017, 36, 1330–1347. [Google Scholar] [CrossRef] [Green Version]
- Cornacchia, D.; Zhang, C.; Zimmer, B.; Chung, S.Y.; Fan, Y.; Soliman, M.A.; Tchieu, J.; Chambers, S.M.; Shah, H.; Paull, D.; et al. Lipid Deprivation Induces a Stable, Naive-to-Primed Intermediate State of Pluripotency in Human PSCs. Cell Stem Cell 2019, 25, 120–136.e10. [Google Scholar] [CrossRef]
- Jiang, Z.; Guang, L.; Li, L.; Shyh-Chang, N. Putting Stem Cells on a Low-Fat Diet Switches Their Pluripotent State. Cell Stem Cell 2019, 25, 3–5. [Google Scholar] [CrossRef]
- Moussaieff, A.; Rouleau, M.; Kitsberg, D.; Cohen, M.; Levy, G.; Barasch, D.; Nemirovski, A.; Shen-Orr, S.; Laevsky, I.; Amit, M.; et al. Glycolysis-Mediated Changes in Acetyl-CoA and Histone Acetylation Control the Early Differentiation of Embryonic Stem Cells. Cell Metab. 2015, 21, 392–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Khvorostov, I.; Hong, J.S.; Oktay, Y.; Vergnes, L.; Nuebel, E.; Wahjudi, P.N.; Setoguchi, K.; Wang, G.; Do, A.; et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011, 30, 4860–4873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C.-L.; Molkentin, J.D. The Elusive Progenitor Cell in Cardiac Regeneration: Slip Slidin’ Away. Circ. Res. 2017, 120, 400–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yucel, N.; Wang, Y.X.; Mai, T.; Porpiglia, E.; Lund, P.J.; Markov, G.; Garcia, B.A.; Bendall, S.C.; Angelo, M.; Blau, H.M. Glucose Metabolism Drives Histone Acetylation Landscape Transitions that Dictate Muscle Stem Cell Function. Cell Rep. 2019, 27, 3939–3955.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerhart-Hines, Z.; Rodgers, J.T.; Bare, O.; Lerin, C.; Kim, S.-H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 2007, 26, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Ryall, J.G.; Dell’Orso, S.; Derfoul, A.; Juan, A.; Zare, H.; Feng, X.; Clermont, D.; Koulnis, M.; Gutierrez-Cruz, G.; Sartorelli, V.; et al. The NAD+-Dependent SIRT1 Deacetylase Translates a Metabolic Switch into Regulatory Epigenetics in Skeletal Muscle Stem Cells. Cell Stem Cell 2015, 16, 171–183. [Google Scholar] [CrossRef] [Green Version]
- van Gastel, N.; Stegen, S.; Eelen, G.; Schoors, S.; Carlier, A.; Daniëls, V.W.; Baryawno, N.; Przybylski, D.; Depypere, M.; Stiers, P.-J.; et al. Lipid availability determines fate of skeletal progenitor cells via SOX9. Nature 2020, 579, 111–117. [Google Scholar] [CrossRef]
- Cahill, T.J.; Choudhury, R.P.; Riley, P.R. Heart regeneration and repair after myocardial infarction: Translational opportunities for novel therapeutics. Nat. Rev. Drug Discov. 2017, 16, 699–717. [Google Scholar] [CrossRef]
- Testa, G.; Di Benedetto, G.; Passaro, F. Advanced Technologies to Target Cardiac Cell Fate Plasticity for Heart Regeneration. Int. J. Mol. Sci. 2021, 22, 9517. [Google Scholar] [CrossRef]
- Payan, S.M.; Hubert, F.; Rochais, F. Cardiomyocyte proliferation, a target for cardiac regeneration. Biochim. Biophys. Acta-Mol. Cell Res. 2020, 1867, 118461. [Google Scholar] [CrossRef]
- Johnson, J.; Mohsin, S.; Houser, S. Cardiomyocyte Proliferation as a Source of New Myocyte Development in the Adult Heart. Int. J. Mol. Sci. 2021, 22, 7764. [Google Scholar] [CrossRef] [PubMed]
- Tane, S.; Kubota, M.; Okayama, H.; Ikenishi, A.; Yoshitome, S.; Iwamoto, N.; Satoh, Y.; Kusakabe, A.; Ogawa, S.; Kanai, A.; et al. Repression of Cyclin D1 Expression Is Necessary for the Maintenance of Cell Cycle Exit in Adult Mammalian Cardiomyocytes. J. Biol. Chem. 2014, 289, 18033–18044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasumarthi, K.B.; Nakajima, H.; Nakajima, H.O.; Soonpaa, M.H.; Field, L.J. Targeted Expression of Cyclin D2 Results in Cardiomyocyte DNA Synthesis and Infarct Regression in Transgenic Mice. Circ. Res. 2005, 96, 110–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhry, H.W.; Dashoush, N.H.; Tang, H.; Zhang, L.; Wang, X.; Wu, E.X.; Wolgemuth, D.J. Cyclin A2 Mediates Cardiomyocyte Mitosis in the Postmitotic Myocardium. J. Biol. Chem. 2004, 279, 35858–35866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, S.D.; Ranjan, A.K.; Kawase, Y.; Cheng, R.K.; Kara, R.J.; Bhattacharya, R.; Guzman-Martinez, G.; Sanz, J.; Garcia, M.J.; Chaudhry, H.W. Cyclin A2 Induces Cardiac Regeneration After Myocardial Infarction Through Cytokinesis of Adult Cardiomyocytes. Sci. Transl. Med. 2014, 6, 224ra27. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, V.; Giacca, M.; Capogrossi, M.C.; Crescenzi, M.; Martelli, F. Knockdown of Cyclin-dependent Kinase Inhibitors Induces Cardiomyocyte Re-entry in the Cell Cycle. J. Biol. Chem. 2011, 286, 8644–8654. [Google Scholar] [CrossRef] [Green Version]
- Bersell, K.; Arab, S.; Haring, B.; Kühn, B. Neuregulin1/ErbB4 Signaling Induces Cardiomyocyte Proliferation and Repair of Heart Injury. Cell 2009, 138, 257–270. [Google Scholar] [CrossRef] [Green Version]
- D’Uva, G.; Aharonov, A.; Lauriola, M.; Kain, D.; Yahalom-Ronen, Y.; Carvalho, S.; Weisinger, K.; Bassat, E.; Rajchman, D.; Yifa, O.; et al. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat. Cell Biol. 2015, 17, 627–638. [Google Scholar] [CrossRef]
- Abbas, N.; Perbellini, F.; Thum, T. Non-coding RNAs: Emerging players in cardiomyocyte proliferation and cardiac regeneration. Basic Res. Cardiol. 2020, 115, 52. [Google Scholar] [CrossRef]
- Santos, F.; Correia, M.; Nóbrega-Pereira, S.; de Jesus, B.B. Age-Related Pathways in Cardiac Regeneration: A Role for lncRNAs? Front. Physiol. 2021, 11, 583191. [Google Scholar] [CrossRef]
- Correia, M.; de Jesus, B.B.; Nobrega-Pereira, S. Novel Insights Linking lncRNAs and Metabolism with Implications for Cardiac Regeneration. Front. Physiol. 2021, 12, 586927. [Google Scholar] [CrossRef] [PubMed]
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A.; et al. The Oxygen-Rich Postnatal Environment Induces Cardiomyocyte Cell-Cycle Arrest through DNA Damage Response. Cell 2014, 157, 565–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menendez-Montes, I.; Abdisalaam, S.; Xiao, F.; Lam, N.T.; Mukherjee, S.; Szweda, L.I.; Asaithamby, A.; Sadek, H.A. Mitochondrial fatty acid utilization increases chromatin oxidative stress in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2021, 118, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Lalowski, M.M.; Björk, S.; Finckenberg, P.; Soliymani, R.; Tarkia, M.; Calza, G.; Blokhina, D.; Tulokas, S.; Kankainen, M.; Lakkisto, P.; et al. Characterizing the Key Metabolic Pathways of the Neonatal Mouse Heart Using a Quantitative Combinatorial Omics Approach. Front. Physiol. 2018, 9, 365. [Google Scholar] [CrossRef]
- Nakada, Y.; Canseco, D.C.; Thet, S.; Abdisalaam, S.; Asaithamby, A.; Santos, C.X.; Shah, A.M.; Zhang, H.; Faber, J.E.; Kinter, M.T.; et al. Hypoxia induces heart regeneration in adult mice. Nature 2017, 541, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Salamon, R.J.; Brandt, E.B.; Paltzer, W.G.; Zhang, Z.; Britt, E.C.; Hacker, T.A.; Fan, J.; Mahmoud, A.I. Malonate Promotes Adult Cardiomyocyte Proliferation and Heart Regeneration. Circulation 2021, 143, 1973–1986. [Google Scholar] [CrossRef] [PubMed]
- Honkoop, H.; de Bakker, D.E.; Aharonov, A.; Kruse, F.; Shakked, A.; Nguyen, P.D.; de Heus, C.; Garric, L.; Muraro, M.J.; Shoffner, A.; et al. Single-cell analysis uncovers that metabolic reprogramming by ErbB2 signaling is essential for cardiomyocyte proliferation in the regenerating heart. eLife 2019, 8, e50163. [Google Scholar] [CrossRef]
- Fajardo, V.M.; Feng, I.; Chen, B.Y.; Perez-Ramirez, C.A.; Shi, B.; Clark, P.; Tian, R.; Lien, C.-L.; Pellegrini, M.; Christofk, H.; et al. GLUT1 overexpression enhances glucose metabolism and promotes neonatal heart regeneration. Sci. Rep. 2021, 11, 8669. [Google Scholar] [CrossRef]
- Cardoso, A.C.; Lam, N.T.; Savla, J.J.; Nakada, Y.; Pereira, A.H.M.; Elnwasany, A.; Menendez-Montes, I.; Ensley, E.L.; Petric, U.B.; Sharma, G.; et al. Mitochondrial substrate utilization regulates cardiomyocyte cell-cycle progression. Nat. Metab. 2020, 2, 167–178. [Google Scholar] [CrossRef]
- Cao, T.; Liccardo, D.; Lacanna, R.; Zhang, X.; Lu, R.; Finck, B.N.; Leigh, T.; Chen, X.; Drosatos, K.; Tian, Y. Fatty Acid Oxidation Promotes Cardiomyocyte Proliferation Rate but Does Not Change Cardiomyocyte Number in Infant Mice. Front. Cell Dev. Biol. 2019, 7, 42. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, L.; Battiprolu, P.K.; Fukushima, A.; Nguyen, K.; Milner, K.; Gupta, A.; Altamimi, T.; Byrne, N.; Mori, J.; et al. Malonyl CoA Decarboxylase Inhibition Improves Cardiac Function Post-Myocardial Infarction. JACC: Basic Transl. Sci. 2019, 4, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Leigh, T.; Gao, E.; Zhang, X.; Tian, Y. Activation or Inhibition of PPARα-Mediated Fatty Acid β-Oxidation Does Not Active Cardiomyocyte Proliferation in Normal or Infarcted Adult Mice. bioRxiv 2019, 667964. [Google Scholar] [CrossRef]
- Lundsgaard, A.-M.; Fritzen, A.M.; Nicolaisen, T.S.; Carl, C.S.; Sjøberg, K.A.; Raun, S.H.; Klein, A.B.; Quant, E.S.S.; Langer, J.; Ørskov, C.; et al. Glucometabolic consequences of acute and prolonged inhibition of fatty acid oxidation. J. Lipid Res. 2020, 61, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Ordoño, J.; Pérez-Amodio, S.; Ball, K.; Aguirre, A.; Engel, E. Lactate Promotes Cardiomyocyte Dedifferentiation through Metabolic Reprogramming. bioRxiv 2020. [Google Scholar] [CrossRef]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for Cardiomyocyte Renewal in Humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malliaras, K.; Zhang, Y.; Seinfeld, J.; Galang, G.; Tseliou, E.; Cheng, K.; Sun, B.; Aminzadeh, M.; Marbán, E. Cardiomyocyte proliferation and progenitor cell recruitment underlie therapeutic regeneration after myocardial infarction in the adult mouse heart. EMBO Mol. Med. 2013, 5, 191–209. [Google Scholar] [CrossRef]
- Brade, T.; Pane, L.S.; Moretti, A.; Chien, K.R.; Laugwitz, K.-L. Embryonic Heart Progenitors and Cardiogenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a013847. [Google Scholar] [CrossRef] [Green Version]
- Urbanek, K.; Torella, D.; Sheikh, F.; De Angelis, A.; Nurzynska, D.; Silvestri, F.; Beltrami, C.A.; Bussani, R.; Beltrami, A.P.; Quaini, F.; et al. Myocardial regeneration by activation of multipotent cardiac stem cells in ischemic heart failure. Proc. Natl. Acad. Sci. USA 2005, 102, 8692–8697. [Google Scholar] [CrossRef] [Green Version]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Murakami, M.; Qi, X.; McAnally, J.; Porrello, E.R.; Mahmoud, A.I.; Tan, W.; Shelton, J.M.; et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 13839–13844. [Google Scholar] [CrossRef] [Green Version]
- Smits, A.M.; van Vliet, P.; Metz, C.H.; Korfage, T.; Sluijter, J.P.; Doevendans, P.A.; Goumans, M.-J. Human cardiomyocyte progenitor cells differentiate into functional mature cardiomyocytes: An in vitro model for studying human cardiac physiology and pathophysiology. Nat. Protoc. 2009, 4, 232–243. [Google Scholar] [CrossRef]
- Smits, A.M.; Van Laake, L.W.; den Ouden, K.; Schreurs, C.; Szuhai, K.; van Echteld, C.J.; Mummery, C.L.; Doevendans, P.A.; Goumans, M.-J. Human cardiomyocyte progenitor cell transplantation preserves long-term function of the infarcted mouse myocardium. Cardiovasc. Res. 2009, 83, 527–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torán, J.L.; Aguilar, S.; Lopez, J.A.; Torroja, C.; Quintana, J.A.; Santiago, C.; Abad, J.L.; Gomes-Alves, P.; Gonzalez, A.; Bernal, J.; et al. CXCL6 is an important paracrine factor in the pro-angiogenic human cardiac progenitor-like cell secretome. Sci. Rep. 2017, 7, 12490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastião, M.J.; Serra, M.; Pereira, R.; Palacios, I.; Gomes-Alves, P.; Alves, P.M. Human cardiac progenitor cell activation and regeneration mechanisms: Exploring a novel myocardial ischemia/reperfusion in vitro model. Stem Cell Res. Ther. 2019, 10, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Zhu, W.; Cheng, M.; Chen, L.; Zhang, J.; Sun, T.; Kishore, R.; Phillips, M.I.; Losordo, D.; Qin, G. Hypoxic Preconditioning Enhances the Benefit of Cardiac Progenitor Cell Therapy for Treatment of Myocardial Infarction by Inducing CXCR4 Expression. Circ. Res. 2009, 104, 1209–1216. [Google Scholar] [CrossRef]
- Huang, M.; Nguyen, P.; Jia, F.; Hu, S.; Gong, Y.; de Almeida, P.E.; Wang, L.; Nag, D.; Kay, M.A.; Giaccia, A.J.; et al. Double Knockdown of Prolyl Hydroxylase and Factor-Inhibiting Hypoxia-Inducible Factor with Nonviral Minicircle Gene Therapy Enhances Stem Cell Mobilization and Angiogenesis After Myocardial Infarction. Circulation 2011, 124, S46–S54. [Google Scholar] [CrossRef] [Green Version]
- André, M.E.; De Pauw, A.; Verdoy, M.R.; Brusa, D.; Bouzin, C.; Timmermans, A.; Bertrand, L.; Balligand, J.-L. Changes of Metabolic Phenotype of Cardiac Progenitor Cells during Differentiation: Neutral Effect of Stimulation of AMP-Activated Protein Kinase. Stem Cells Dev. 2019, 28, 1498–1513. [Google Scholar] [CrossRef] [Green Version]
- Salabei, J.K.; Lorkiewicz, P.K.; Holden, C.R.; Li, Q.; Hong, K.U.; Bolli, R.; Bhatnagar, A.; Hill, B.G. Glutamine Regulates Cardiac Progenitor Cell Metabolism and Proliferation. Stem Cells 2015, 33, 2613–2627. [Google Scholar] [CrossRef] [Green Version]
- Talkhabi, M.; Pahlavan, S.; Aghdami, N.; Baharvand, H. Ascorbic acid promotes the direct conversion of mouse fibroblasts into beating cardiomyocytes. Biochem. Biophys. Res. Commun. 2015, 463, 699–705. [Google Scholar] [CrossRef]
- Lewandowski, J.; Kolanowski, T.J.; Kurpisz, M. Techniques for the induction of human pluripotent stem cell differentiation towards cardiomyocytes. J. Tissue Eng. Regen. Med. 2017, 11, 1658–1674. [Google Scholar] [CrossRef]
- Mummery, C.; Zhang, J.; Ng, E.S.; Elliott, D.; Elefanty, A.; Kamp, T. Differentiation of Human Embryonic Stem Cells and Induced Pluripotent Stem Cells to Cardiomyocytes. Circ. Res. 2012, 111, 344–358. [Google Scholar] [CrossRef]
- Kehat, I.; Kenyagin-Karsenti, D.; Snir, M.; Segev, H.; Amit, M.; Gepstein, A.; Livne, E.; Binah, O.; Itskovitz-Eldor, J.; Gepstein, L. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J. Clin. Investig. 2001, 108, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Mummery, C.; Ward, D.; Brink, C.E.V.D.; Bird, S.D.; Doevendans, P.A.; Opthof, T.; De La Riviere, A.B.; Tertoolen, L.; van der Heyden, M.; Pera, M. Cardiomyocyte differentiation of mouse and human embryonic stem cells. J. Anat. 2002, 200, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Police, S.; Rao, N.; Carpenter, M.K. Characterization and Enrichment of Cardiomyocytes Derived from Human Embryonic Stem Cells. Circ. Res. 2002, 91, 501–508. [Google Scholar] [CrossRef] [PubMed]
- He, J.-Q.; Ma, Y.; Lee, Y.; Thomson, J.A.; Kamp, T. Human Embryonic Stem Cells Develop Into Multiple Types of Cardiac Myocytes. Circ. Res. 2003, 93, 32–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laflamme, M.A.; Chen, K.Y.; Naumova, A.V.; Muskheli, V.; Fugate, J.A.; Dupras, S.K.; Reinecke, H.; Xu, C.; Hassanipour, M.; Police, S.; et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat. Biotechnol. 2007, 25, 1015–1024. [Google Scholar] [CrossRef]
- Zhang, J.; Klos, M.; Wilson, G.F.; Herman, A.M.; Lian, X.; Raval, K.K.; Barron, M.R.; Hou, L.; Soerens, A.G.; Yu, J.; et al. Extracellular Matrix Promotes Highly Efficient Cardiac Differentiation of Human Pluripotent Stem Cells: The Matrix Sandwich Method. Circ. Res. 2012, 111, 1125–1136. [Google Scholar] [CrossRef]
- Mummery, C.; Oostwaard, D.W.-V.; Doevendans, P.; Spijker, R.; Brink, S.V.D.; Hassink, R.; van der Heyden, M.; Opthof, T.; Pera, M.; de la Riviere, A.B.; et al. Differentiation of Human Embryonic Stem Cells to Cardiomyocytes: Role of coculture with visceral endoderm-like cells. Circulation 2003, 107, 2733–2740. [Google Scholar] [CrossRef] [Green Version]
- Passier, R.; Oostwaard, D.W.; Snapper, J.; Kloots, J.; Hassink, R.J.; Kuijk, E.; Roelen, B.; de la Riviere, A.B.; Mummery, C. Increased Cardiomyocyte Differentiation from Human Embryonic Stem Cells in Serum—Free Cultures. Stem Cells 2005, 23, 772–780. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Pushp, P.; Nogueira, D.E.S.; Rodrigues, C.A.V.; Ferreira, F.C.; Cabral, J.M.S.; Gupta, M.K. A Concise Review on Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Personalized Regenerative Medicine. Stem Cell Rev. Rep. 2020, 17, 748–776. [Google Scholar] [CrossRef] [PubMed]
- Kattman, S.J.; Witty, A.D.; Gagliardi, M.; Dubois, N.C.; Niapour, M.; Hotta, A.; Ellis, J.; Keller, G. Stage-Specific Optimization of Activin/Nodal and BMP Signaling Promotes Cardiac Differentiation of Mouse and Human Pluripotent Stem Cell Lines. Cell Stem Cell 2011, 8, 228–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zeng, D.; Ding, L.; Li, X.-L.; Liu, X.-T.; Li, W.; Wei, T.; Yan, S.; Xie, J.-H.; Wei, L.; et al. Three-dimensional poly-(ε-caprolactone) nanofibrous scaffolds directly promote the cardiomyocyte differentiation of murine-induced pluripotent stem cells through Wnt/β-catenin signaling. BMC Cell Biol. 2015, 16, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically Defined and Small Molecule-Based Generation of Human Cardiomyocytes. Nat. Methods. 2014, 11, 855–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.; Dzeja, P.P.; Faustino, R.S.; Perez-Terzic, C.; Behfar, A.; Terzic, A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, S60–S67. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Chen, X.; Kaushal, S.; Reece, E.A.; Yang, P. High glucose suppresses embryonic stem cell differentiation into cardiomyocytes. Stem Cell Res. Ther. 2016, 7, 187. [Google Scholar] [CrossRef] [Green Version]
- Correia, C.; Koshkin, A.; Duarte, P.; Hu, D.; Teixeira, A.; Domian, I.; Serra, M.; Alves, P.M. Distinct carbon sources affect structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Sci. Rep. 2017, 7, 8590. [Google Scholar] [CrossRef]
- Robertson, C.; Tran, D.D.; George, S.C. Concise Review: Maturation Phases of Human Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cells 2013, 31, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Musunuru, K.; Sheikh, F.; Gupta, R.M.; Houser, S.R.; Maher, K.O.; Milan, D.J.; Terzic, A.; Wu, J.C. Induced Pluripotent Stem Cells for Cardiovascular Disease Modeling and Precision Medicine: A Scientific Statement from the American Heart Association. Circ. Genom. Precis. Med. 2018, 11, e000043. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Badur, M.G.; Spiering, S.; Divakaruni, A.; Meurs, N.E.; Yu, M.S.; Colas, A.R.; Murphy, A.N.; Mercola, M.; Metallo, C.M. Lipid Availability Influences the Metabolic Maturation of Human Pluripotent Stem Cell-Derived Cardiomyocytes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Feyen, D.A.; McKeithan, W.L.; Bruyneel, A.A.; Spiering, S.; Hörmann, L.; Ulmer, B.; Zhang, H.; Briganti, F.; Schweizer, M.; Hegyi, B.; et al. Metabolic Maturation Media Improve Physiological Function of Human iPSC-Derived Cardiomyocytes. Cell Rep. 2020, 32, 107925. [Google Scholar] [CrossRef] [PubMed]
- Mills, R.J.; Titmarsh, D.M.; Koenig, X.; Parker, B.L.; Ryall, J.G.; Quaife-Ryan, G.A.; Voges, H.K.; Hodson, M.P.; Ferguson, C.; Drowley, L.; et al. Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc. Natl. Acad. Sci. USA 2017, 114, E8372–E8381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, D.-F.; Danoviz, M.E.; Wiczer, B.; Laflamme, M.A.; Tian, R. Mitochondrial Maturation in Human Pluripotent Stem Cell Derived Cardiomyocytes. Stem Cells Int. 2017, 2017, 5153625. [Google Scholar] [CrossRef] [PubMed]
- Rana, P.; Anson, B.; Engle, S.; Will, Y. Characterization of Human-Induced Pluripotent Stem Cell–Derived Cardiomyocytes: Bioenergetics and Utilization in Safety Screening. Toxicol. Sci. 2012, 130, 117–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Nie, Y.; Chen, S.; Xie, C.; Fan, Q.; Wang, Z.; Long, B.; Yan, G.; Zhong, Q.; Yan, X. Leucine reduces reactive oxygen species levels via an energy metabolism switch by activation of the mTOR-HIF-1α pathway in porcine intestinal epithelial cells. Int. J. Biochem. Cell Biol. 2017, 89, 42–56. [Google Scholar] [CrossRef]
- Garbern, J.C.; Helman, A.; Sereda, R.; Sarikhani, M.; Ahmed, A.; Escalante, G.; Ogurlu, R.; Kim, S.L.; Zimmerman, J.F.; Cho, A.; et al. Inhibition of mTOR Signaling Enhances Maturation of Cardiomyocytes Derived from Human-Induced Pluripotent Stem Cells via p53-Induced Quiescence. Circulation 2020, 141, 285–300. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, X.; Zhou, Q.; Tan, B.; Xu, H.; Yi, Q.; Yan, L.; Xie, M.; Zhang, Y.; Tian, J.; et al. Activation of AMPK Promotes Maturation of Cardiomyocytes Derived from Human Induced Pluripotent Stem Cells. Front. Cell Dev. Biol. 2021, 9, 644667. [Google Scholar] [CrossRef]
- Ieda, M.; Tsuchihashi, T.; Ivey, K.N.; Ross, R.S.; Hong, T.-T.; Shaw, R.M.; Srivastava, D. Cardiac Fibroblasts Regulate Myocardial Proliferation through β1 Integrin Signaling. Dev. Cell 2009, 16, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Ieda, M.; Fu, J.-D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct Reprogramming of Fibroblasts into Functional Cardiomyocytes by Defined Factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef] [Green Version]
- Jopling, C.; Boue, S.; Belmonte, J.C.I. Dedifferentiation, transdifferentiation and reprogramming: Three routes to regeneration. Nat. Rev. Mol. Cell Biol. 2011, 12, 79–89. [Google Scholar] [CrossRef]
- Qian, L.; Huang, Y.; Spencer, C.I.; Foley, A.; Vedantham, V.; Liu, L.; Conway, S.J.; Fu, J.; Srivastava, D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 2012, 485, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Nam, Y.-J.; Luo, X.; Qi, X.; Tan, W.; Huang, G.N.; Acharya, A.; Smith, C.L.; Tallquist, M.D.; Neilson, E.G.; et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012, 485, 599–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, R.; Muraoka, N.; Inagawa, K.; Yamakawa, H.; Miyamoto, K.; Sadahiro, T.; Umei, T.; Kaneda, R.; Suzuki, T.; Kamiya, K.; et al. Induction of human cardiomyocyte-like cells from fibroblasts by defined factors. Proc. Natl. Acad. Sci. USA 2013, 110, 12667–12672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christoforou, N.; Chellappan, M.; Adler, A.F.; Kirkton, R.D.; Wu, T.; Addis, R.C.; Bursac, N.; Leong, K.W. Transcription Factors MYOCD, SRF, Mesp1 and SMARCD3 Enhance the Cardio-Inducing Effect of GATA4, TBX5, and MEF2C during Direct Cellular Reprogramming. PLoS ONE 2013, 8, e63577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Sun, J. A Revolution in Reprogramming: Small Molecules. Curr. Mol. Med. 2019, 19, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Nam, Y.J.; Song, K.; Luo, X.; Daniel, E.; Lambeth, K.; West, K.; Hill, J.A.; di Maio, J.M.; Baker, L.A.; Bassel-Duby, R.; et al. Reprogramming of human fibroblasts toward a cardiac fate. Proc. Natl. Acad. Sci. USA 2013, 110, 5588–5593. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Stone, N.R.; Liu, L.; Spencer, C.I.; Qian, L.; Hayashi, Y.; Olguín, P.D.; Ding, S.; Bruneau, B.G.; Srivastava, D. Direct Reprogramming of Human Fibroblasts toward a Cardiomyocyte-like State. Stem Cell Rep. 2013, 1, 235–247. [Google Scholar] [CrossRef] [Green Version]
- Tani, H.; Sadahiro, T.; Ieda, M. Direct Cardiac Reprogramming: A Novel Approach for Heart Regeneration. Int. J. Mol. Sci. 2018, 19, 2629. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, K.; Akiyama, M.; Tamura, F.; Isomi, M.; Yamakawa, H.; Sadahiro, T.; Muraoka, N.; Kojima, H.; Haginiwa, S.; Kurotsu, S.; et al. Direct In Vivo Reprogramming with Sendai Virus Vectors Improves Cardiac Function after Myocardial Infarction. Cell Stem Cell 2017, 22, 91–103.e5. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, H.; Wang, Z.; Garry, G.A.; Malladi, V.; Botten, G.A.; Ye, W.; Zhou, H.; Osterwalder, M.; Dickel, D.; Visel, A.; et al. Cardiac Reprogramming Factors Synergistically Activate Genome-wide Cardiogenic Stage-Specific Enhancers. Cell Stem Cell 2019, 25, 69–86.e5. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Z.; Yin, C.; Asfour, H.; Chen, O.; Olivia, C.; Bursac, N.; Liu, J.; Qian, L. Stoichiometry of Gata4, Mef2c, and Tbx5 Influences the Efficiency and Quality of Induced Cardiac Myocyte Reprogramming. Circ. Res. 2015, 116, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, J.K.; Bruneau, B.G. Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature 2009, 459, 708–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaseghi, H.; Liu, J.; Qian, L. Molecular barriers to direct cardiac reprogramming. Protein Cell 2017, 8, 724–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Wang, L.; Welch, J.D.; Ma, H.; Zhou, Y.; Vaseghi, H.R.; Yu, S.; Wall, J.B.; Alimohamadi, S.; Zheng, M.; et al. Single-cell transcriptomics reconstructs fate conversion from fibroblast to cardiomyocyte. Nat. 2017, 551, 100–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nóbrega-Pereira, S.; De Jesus, B.B. Cellular Reprogramming and Aging; Springer: Berlin/Heidelberg, Germany, 2020; pp. 73–91. [Google Scholar] [CrossRef]
- Zhou, H.; Morales, M.G.; Hashimoto, H.; Dickson, M.E.; Song, K.; Ye, W.; Kim, M.S.; Niederstrasser, H.; Wang, Z.; Chen, B.; et al. ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression. Genes Dev. 2017, 31, 1770–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, N.R.; Gifford, C.A.; Thomas, R.; Pratt, K.J.; Samse-Knapp, K.; Mohamed, T.M.; Radzinsky, E.M.; Schricker, A.; Ye, L.; Yu, P.; et al. Context-Specific Transcription Factor Functions Regulate Epigenomic and Transcriptional Dynamics during Cardiac Reprogramming. Cell Stem Cell 2019, 25, 87–102.e9. [Google Scholar] [CrossRef]
- Ishida, T.; Ueyama, T.; Baba, A.; Hasegawa, K.; Kawamura, T. The Role of Isocitrate Dehydrogenases in Direct Reprogramming to Cardiomyocytes. Eur. Cardiol. Rev. 2021, 16, e64. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hodgkinson, C.P.; Lu, K.; Payne, A.J.; Pratt, R.E.; Dzau, V.J. Selenium Augments microRNA Directed Reprogramming of Fibroblasts to Cardiomyocytes via Nanog. Sci. Rep. 2016, 6, 23017. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.J.; Shults, N.V. Antioxidant Regulation of Cell Reprogramming. Antioxidants 2019, 8, 323. [Google Scholar] [CrossRef] [Green Version]
- Meng, S.; Chanda, P.; Thandavarayan, R.A.; Cooke, J.P. Transflammation: How Innate Immune Activation and Free Radicals Drive Nuclear Reprogramming. Antioxid. Redox Signal. 2018, 29, 205–218. [Google Scholar] [CrossRef]
- Chen, J.X.; Krane, M.; Deutsch, M.-A.; Wang, L.; Rav-Acha, M.; Gregoire, S.; Engels, M.C.; Rajarajan, K.; Karra, R.; Abel, E.D.; et al. Inefficient Reprogramming of Fibroblasts into Cardiomyocytes Using Gata4, Mef2c, and Tbx5. Circ. Res. 2012, 111, 50–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamakawa, H.; Muraoka, N.; Miyamoto, K.; Sadahiro, T.; Isomi, M.; Haginiwa, S.; Kojima, H.; Umei, T.; Akiyama, M.; Kuishi, Y.; et al. Fibroblast Growth Factors and Vascular Endothelial Growth Factor Promote Cardiac Reprogramming under Defined Conditions. Stem Cell Rep. 2015, 5, 1128–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.-W.; Yilmaz, H. 100 Years of Exploiting Diet and Nutrition for Tissue Regeneration. Cell Stem Cell 2021, 28, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Elmoselhi, A.B.; Hata, T.; Makino, N. Status of myocardial antioxidants in ischemia–reperfusion injury. Cardiovasc. Res. 2000, 47, 446–456. [Google Scholar] [CrossRef]
- Tangney, C.C.; Hankins, J.S.; Murtaugh, M.A.; Piccione, W. Plasma vitamins E and C concentrations of adult patients during cardiopulmonary bypass. J. Am. Coll. Nutr. 1998, 17, 162–170. [Google Scholar] [CrossRef]
- Saisho, Y. Metformin and Inflammation: Its Potential Beyond Glucose-lowering Effect. Endocr. Metab. Immune Disord. Drug Targets 2015, 15, 196–205. [Google Scholar] [CrossRef]
- Ramachandran, R.; Saraswathi, M. Postconditioning with metformin attenuates apoptotic events in cardiomyoblasts associated with ischemic reperfusion injury. Cardiovasc. Ther. 2017, 35, e12279. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, J.; Yan, L.; He, Q.; Xie, H.; Chen, M. Resveratrol Ameliorates Cardiac Remodeling in a Murine Model of Heart Failure with Preserved Ejection Fraction. Front. Pharmacol. 2021, 12, 646240. [Google Scholar] [CrossRef]
- Li, Y.-G.; Zhu, W.; Tao, J.-P.; Xin, P.; Liu, M.-Y.; Li, J.-B.; Wei, M. Resveratrol protects cardiomyocytes from oxidative stress through SIRT1 and mitochondrial biogenesis signaling pathways. Biochem. Biophys. Res. Commun. 2013, 438, 270–276. [Google Scholar] [CrossRef]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Muller, F.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef] [Green Version]
- de Jesus, B.B.; Vera, E.; Schneeberger, K.; Tejera, A.M.; Ayuso, E.; Bosch, F.; Blasco, M.A. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol. Med. 2012, 4, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Bär, C.; de Jesus, B.B.; Serrano, R.M.; Tejera, A.M.; Ayuso, E.; Jimenez, V.; Formentini, I.; Bobadilla, M.; Mizrahi, J.; De Martino, A.; et al. Telomerase expression confers cardioprotection in the adult mouse heart after acute myocardial infarction. Nat. Commun. 2014, 5, 5863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.-P.; Zhai, P.; Yamamoto, T.; Maejima, Y.; Matsushima, S.; Hariharan, N.; Shao, D.; Takagi, H.; Oka, S.; Sadoshima, J. Silent Information Regulator 1 Protects the Heart from Ischemia/Reperfusion. Circulation 2010, 122, 2170–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-Dependent Regulation of FOXO Transcription Factors by the SIRT1 Deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 Regulates Aging and Resistance to Oxidative Stress in the Heart. Circ. Res. 2007, 100, 1512–1521. [Google Scholar] [CrossRef]
- Planavila, A.; Iglesias, R.; Giralt, M.; Villarroya, F. Sirt1 acts in association with PPARα to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc. Res. 2010, 90, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Feng, Z.; Wang, X.; Zeng, M.; Liu, J.; Han, S.; Xu, J.; Chen, L.; Cao, K.; Long, J.; et al. SIRT3/SOD2 maintains osteoblast differentiation and bone formation by regulating mitochondrial stress. Cell Death Differ. 2017, 25, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Li, D.; Dong, X.; Zhang, X.; Liu, J.; Peng, L.; Meng, B.; Hua, Q.; Pei, X.; Zhao, L.; et al. LncDACH1 promotes mitochondrial oxidative stress of cardiomyocytes by interacting with sirtuin3 and aggravates diabetic cardiomyopathy. Sci. China Life Sci. 2021. [Google Scholar] [CrossRef]
- Gorski, P.A.; Jang, S.P.; Jeong, D.; Lee, A.; Lee, P.; Oh, J.G.; Chepurko, V.; Yang, D.K.; Kwak, T.H.; Eom, S.H.; et al. Role of SIRT1 in Modulating Acetylation of the Sarco-Endoplasmic Reticulum Ca 2+ -ATPase in Heart Failure. Circ. Res. 2019, 124, e63–e80. [Google Scholar] [CrossRef]
- Chen, Y.; Li, S.; Zhang, Y.; Wang, M.; Li, X.; Liu, S.; Xu, D.; Bao, Y.; Jia, P.; Wu, N.; et al. The lncRNA Malat1 regulates microvascular function after myocardial infarction in mice via miR-26b-5p/Mfn1 axis-mediated mitochondrial dynamics. Redox Biol. 2021, 41, 101910. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, F.; Yan, P.; Zhang, S.; Lou, Y.; Geng, Z.; Li, Z.; Zhang, Y.; Xu, Y.; Lu, Y.; et al. LARP7 Protects against Heart Failure by Enhancing Mitochondrial Biogenesis. Circulation 2021, 143, 2007–2022. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Atmanli, A.; Morales, M.G.; Tan, W.; Chen, K.; Xiao, X.; Xu, L.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Nrf1 promotes heart regeneration and repair by regulating proteostasis and redox balance. Nat. Commun. 2021, 12, 5270. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cardiac Regenerative Approach | Metabolic Target | Strategy | Species | Cell Type | Impact | References |
|---|---|---|---|---|---|---|
| Activation of cardiomyocyte proliferation | Glycolysis | Glut 1 overexpression in vivo | Mouse | Neonatal cardiomyocytes | Increases proliferation and decreases fibrosis post-injury | [90] |
| Glycolysis | PDK4 knockout in vivo | Mouse | Adult cardiomyocytes | Increases proliferation and improves heart function | [91] | |
| FAO | FAs -deficient milk in vivo | Mouse | Neonatal cardiomyocytes | Extends the post-natal regenerative window | [91] | |
| CPT1 | Etomoxir supplementation in vivo | Mouse Rat | Neonatal and adult cardiomyocytes | Enhances cardiac efficiency post-injury | [92,93] | |
| Lactate | Supplementation in vitro | Mouse | Cardiac fibroblasts | Pro-regenerative environment for cardiomyocytes post-injury | [94] | |
| ROS | NAC, malonate supplementation in vivo | Mouse | Neonatal and adult cardiomyocytes | Extends the post-natal regenerative window | [95,96] | |
| Recruitment of cardiac stem or progenitor cells | HIF-1α | Hypoxia in vivo and in vitro | Mouse | CPCs | Promotes migration and recruitment | [97,98] |
| Glucose, glutamine | Supplementation in vitro | Mouse | CPCs | Increases proliferation and prevents cell death induced by oxidative stress | [99,100] | |
| ROS | Ascorbic acid supplementation in vitro | Mouse | CPCs | Increases proliferation | [101] | |
| Delivery of De Novo cardiomyocytes from differentiated ESCs/iPSCs | Glucose | Low | Human | ESCs-CMs iPSCs-CMs | Physiological support for cardiac development | [102] |
| Glucose | High | Mouse | ESCs-CMs | Suppresses mesoderm and cardiac transcription genes | [103] | |
| mTOR AMPK | Torin1 AICAR supplementation in vitro | Human | iPSCs-CMs | Cardiomyocyte maturation | [104,105] | |
| Galactose, FAs | Supplementation in vitro | Human | iPSCs-CMs ESCS-CMs | Improves contractile capacity and maturation | [106,107] | |
| FAO | MM in vitro | Human Mouse | iPSCs-CMs | Metabolic maturation | [107] | |
| Direct reprogramming of fibroblasts into iCMs | Glycolysis | HIF-1α knockdown in vitro | Mouse | Neonatal cardiac fibroblasts | Enhances reprogramming efficiency | [108] |
| OXPHOS TCA cycle | Rotenone, IDH3A knockdown in vitro | Mouse | Embryonic fibroblasts | Decreases reprogramming efficiency | [109] | |
| ROS | Selenium, ascorbic acid supplementation in vitro | Mouse | Embryonic, neonatal cardiac and tail tip fibroblasts | Enhances reprogramming efficiency | [101,110] | |
| ROS | Vitamin E nicotinate supplementation in vivo | Rat | Cardiac fibroblasts | Improves heart damage repair through reprogramming | [111] | |
| Nutrient signaling | AMPK | Metformin supplementation in vitro | Rat | H9C2 cardiomyoblasts | Nitrate-dependent decrease in oxidative damage | [112] |
| SIRT1 | Resveratrol supplementation in vivo | Mouse | Neonatal cardiomyocytes | Ejection fraction preservation, decreases cardiac stiffness and oxidative stress | [113] | |
| Gene therapy | Mitochondria | AAV9- Tert overexpression in vivo | Mouse | Adult heart | Improves mitochondrial fitness and activity, protects against heart failure after MI | [114,115,116] |
| Mitochondria | Ad5-CMV-Sirt1 overexpression in vitro | Rat | Neonatal cardiomyocytes | Protects against oxidative stress, FAO inhibition and cell size enlargement | [117] | |
| Mitochondria | Malat1 knockdown in vitro knockout in vivo | Mouse | CMECs | Mitochondrial dysfunction, apoptosis and microvascular injuries | [118] | |
| Mitochondria | AAV9- LARP7 overexpression in vivo | Mouse | Adult heart | Protects against heart failure, improves pump function | [119] | |
| ROS | lncDACH1 knockdown in vitro knockout in vivo | Mouse | NMCVs Adult heart | SIRT3-mediated attenuation of mitochondrial oxidative stress | [120] | |
| ROS | AAV9-Nrf1 overexpression in vivo | Mouse | Adult heart | Protects against I/R injury by activating ROS scavengers | [121] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Correia, M.; Santos, F.; da Silva Ferreira, R.; Ferreira, R.; Bernardes de Jesus, B.; Nóbrega-Pereira, S. Metabolic Determinants in Cardiomyocyte Function and Heart Regenerative Strategies. Metabolites 2022, 12, 500. https://doi.org/10.3390/metabo12060500
Correia M, Santos F, da Silva Ferreira R, Ferreira R, Bernardes de Jesus B, Nóbrega-Pereira S. Metabolic Determinants in Cardiomyocyte Function and Heart Regenerative Strategies. Metabolites. 2022; 12(6):500. https://doi.org/10.3390/metabo12060500
Chicago/Turabian StyleCorreia, Magda, Francisco Santos, Rita da Silva Ferreira, Rita Ferreira, Bruno Bernardes de Jesus, and Sandrina Nóbrega-Pereira. 2022. "Metabolic Determinants in Cardiomyocyte Function and Heart Regenerative Strategies" Metabolites 12, no. 6: 500. https://doi.org/10.3390/metabo12060500
APA StyleCorreia, M., Santos, F., da Silva Ferreira, R., Ferreira, R., Bernardes de Jesus, B., & Nóbrega-Pereira, S. (2022). Metabolic Determinants in Cardiomyocyte Function and Heart Regenerative Strategies. Metabolites, 12(6), 500. https://doi.org/10.3390/metabo12060500