
The Differential Metabolic Response of Oral Squamous Cell Carcinoma Cells and Normal Oral Epithelial Cells to Cisplatin Exposure

 and
and 
Abstract
:1. Introduction
2. Results
2.1. Oral Cancer Cells Showed a Dynamic Metabolic Response to Cisplatin
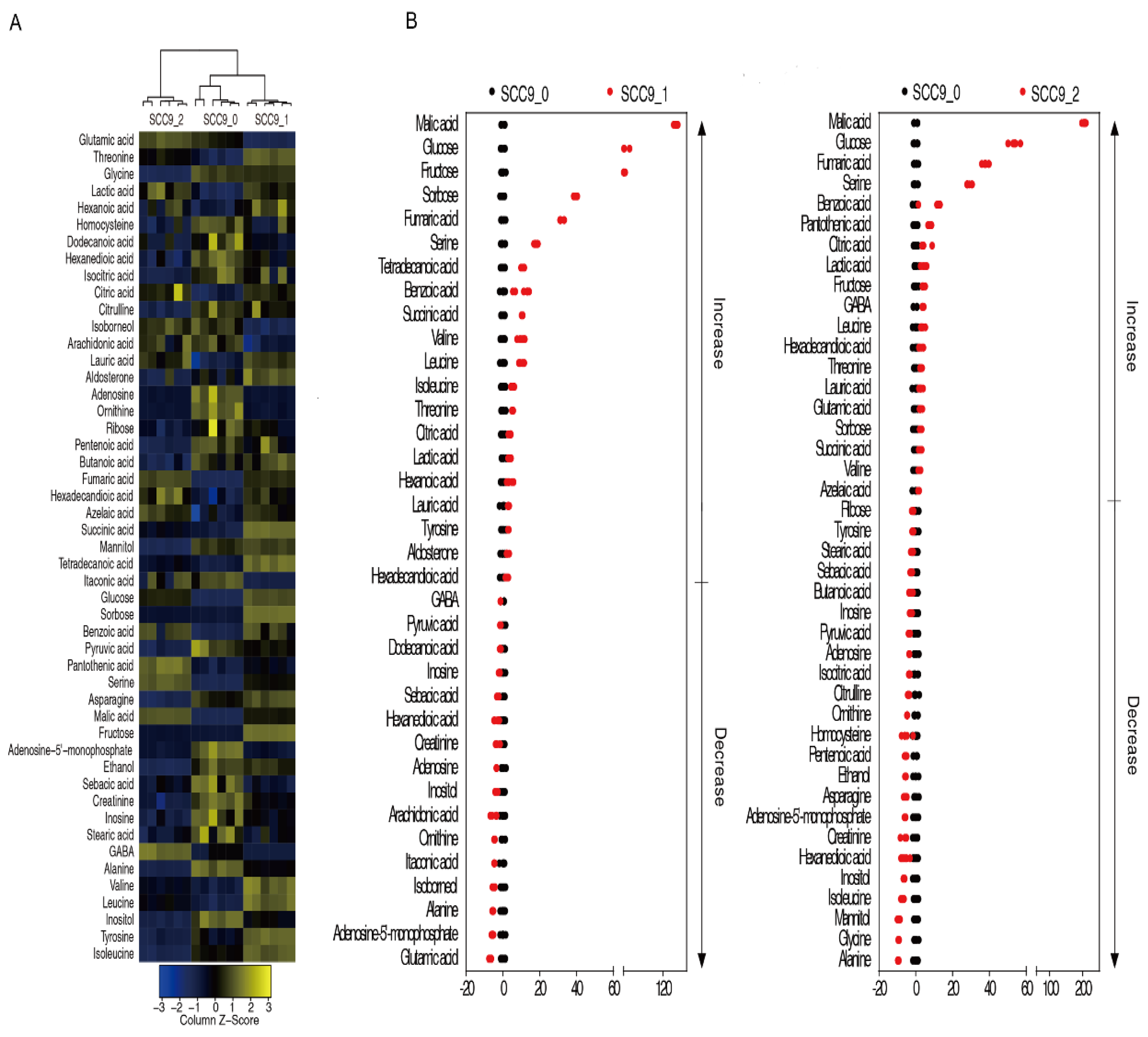
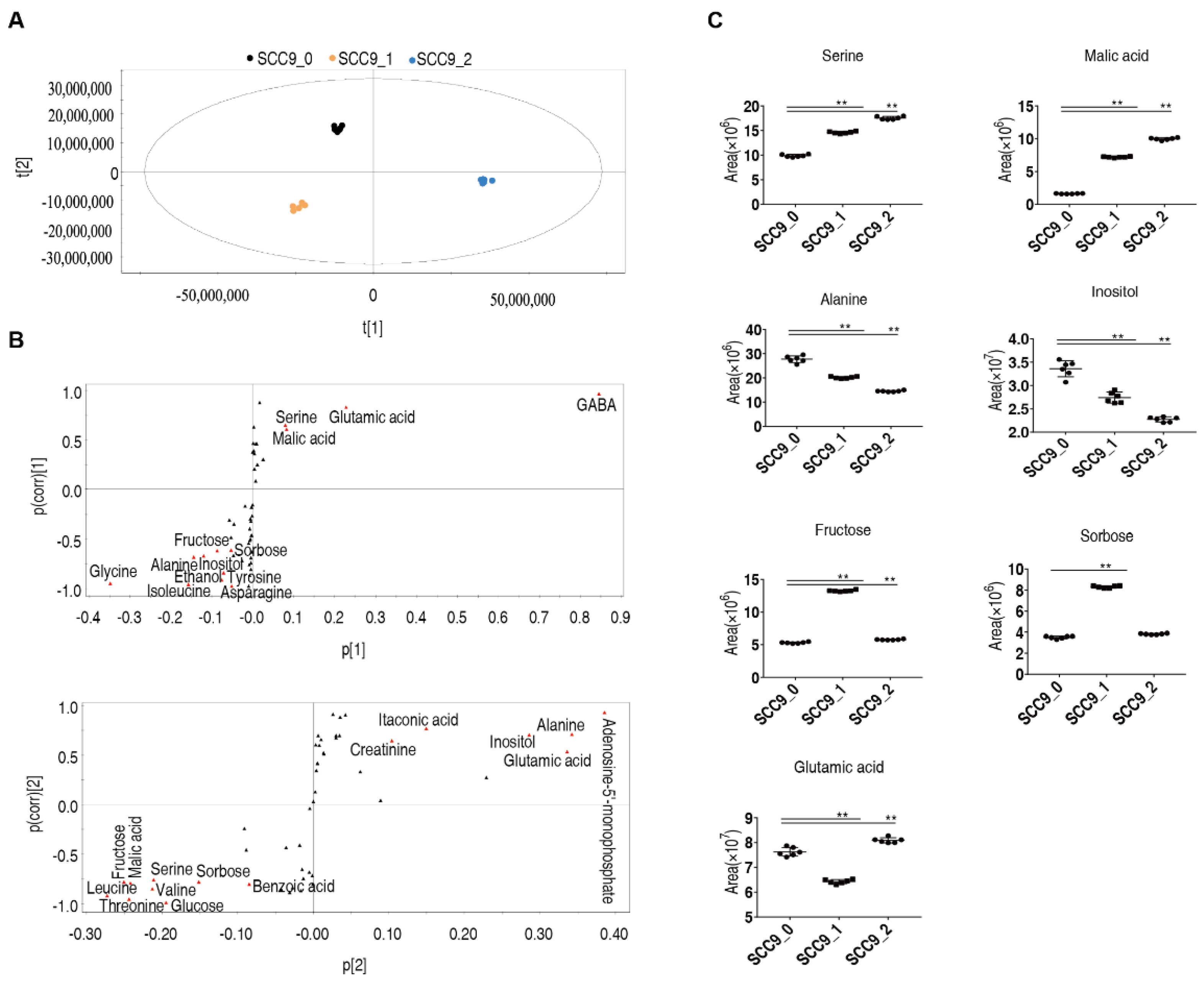
2.2. Effect of Cisplatin on Metabolome of Oral Squamous Cell Carcinoma Cells
2.3. Marker Metabolites of Cisplatin Induced Metabolic Response in SCC-9 Cells
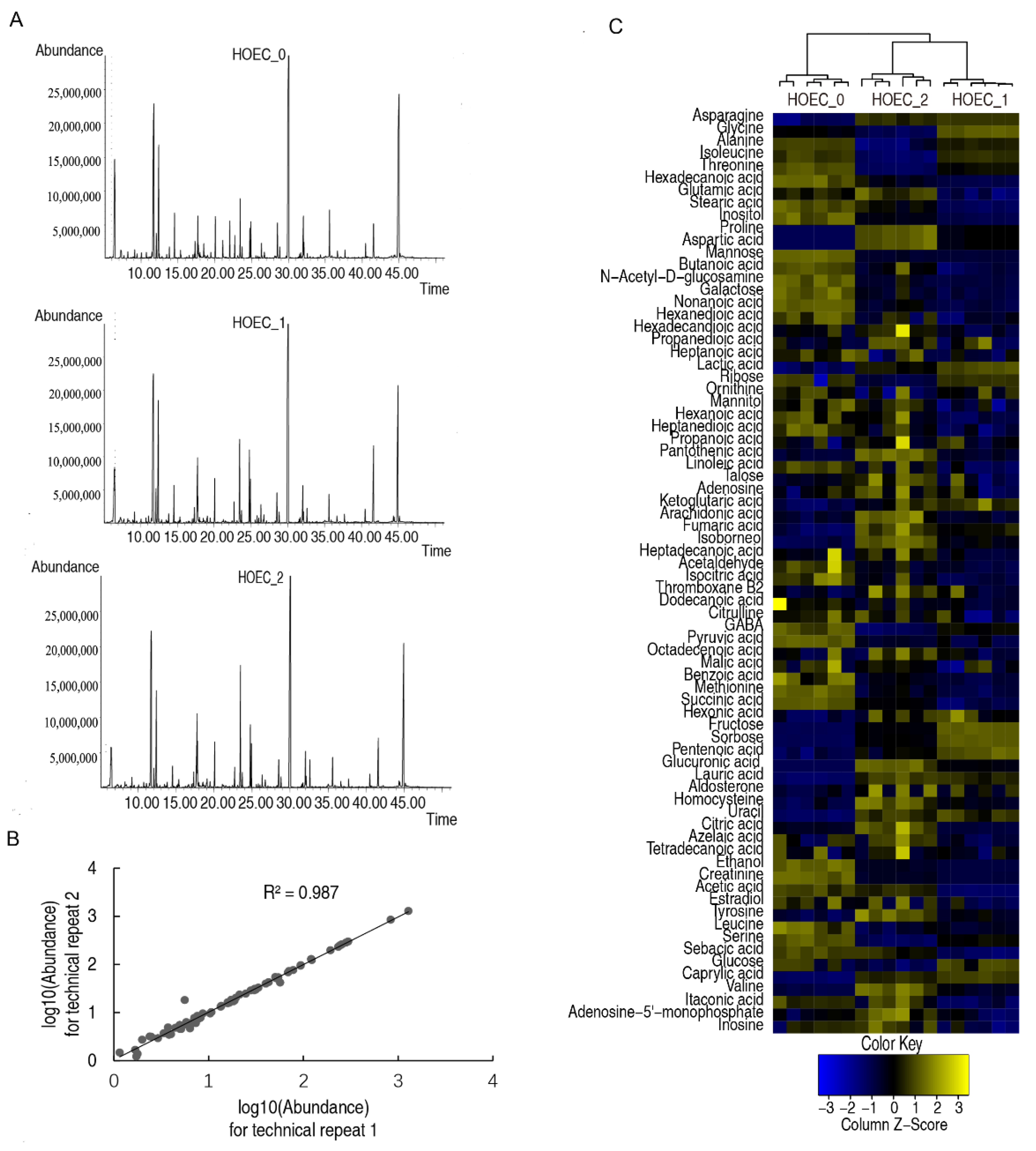
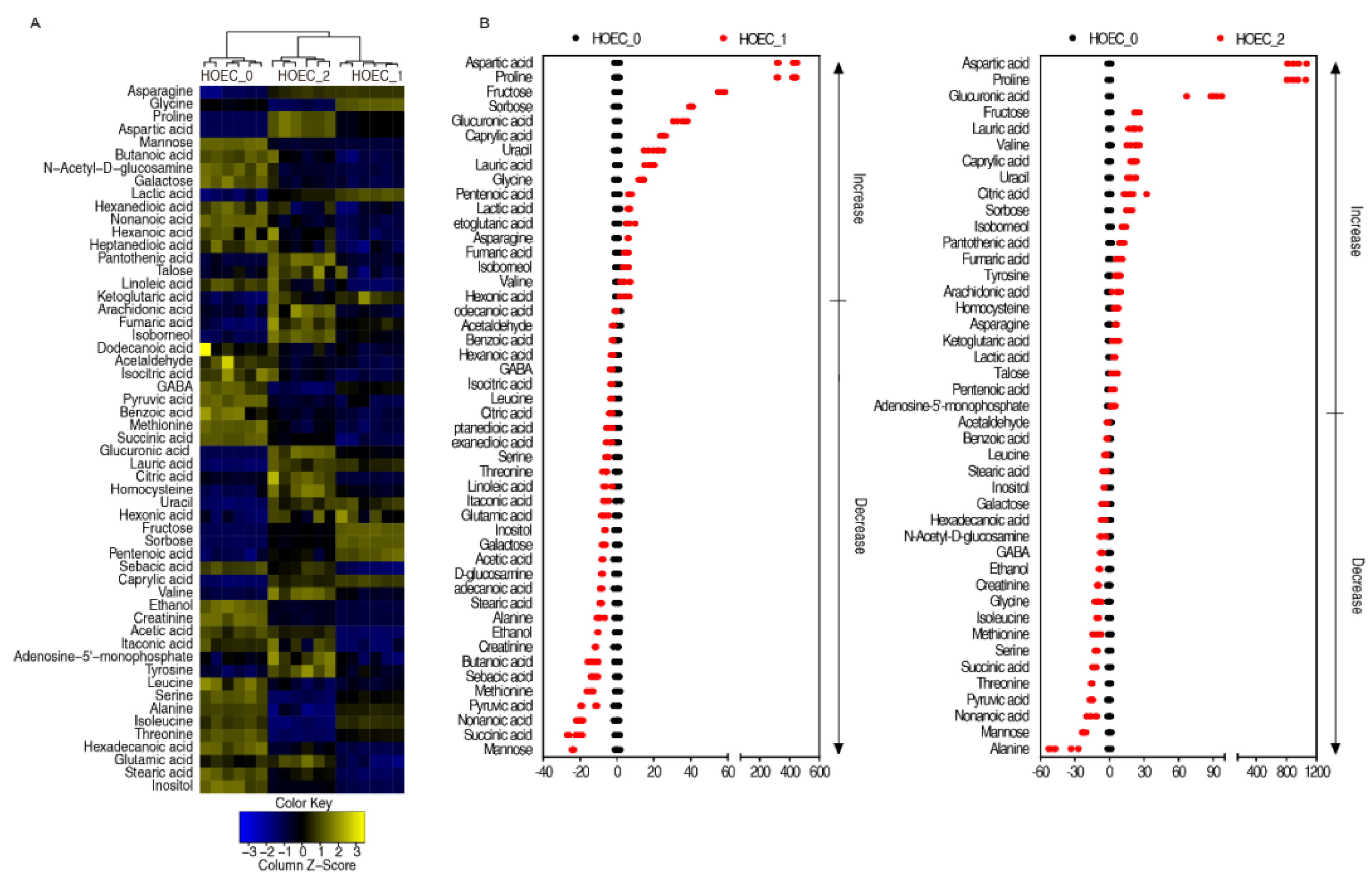
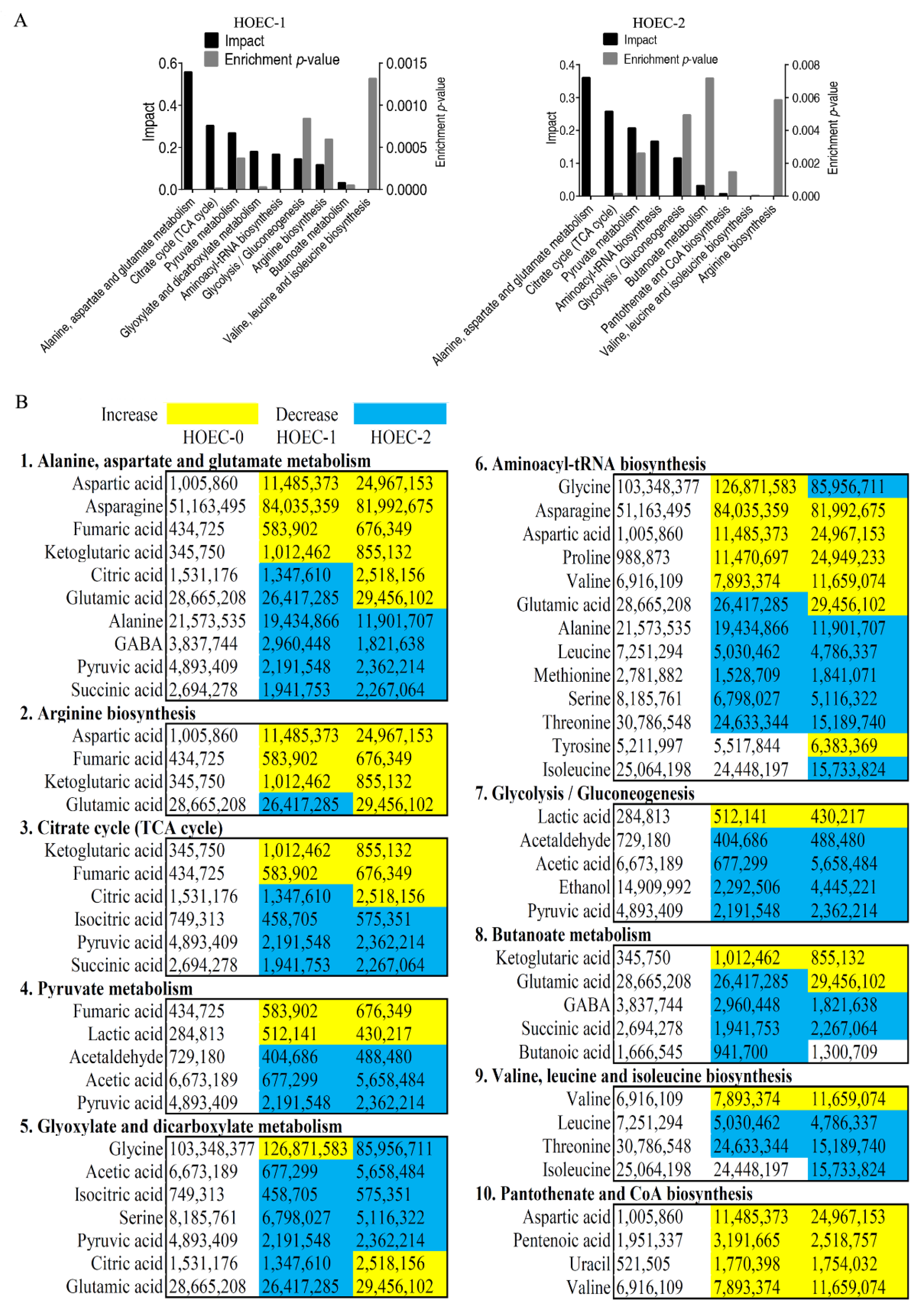
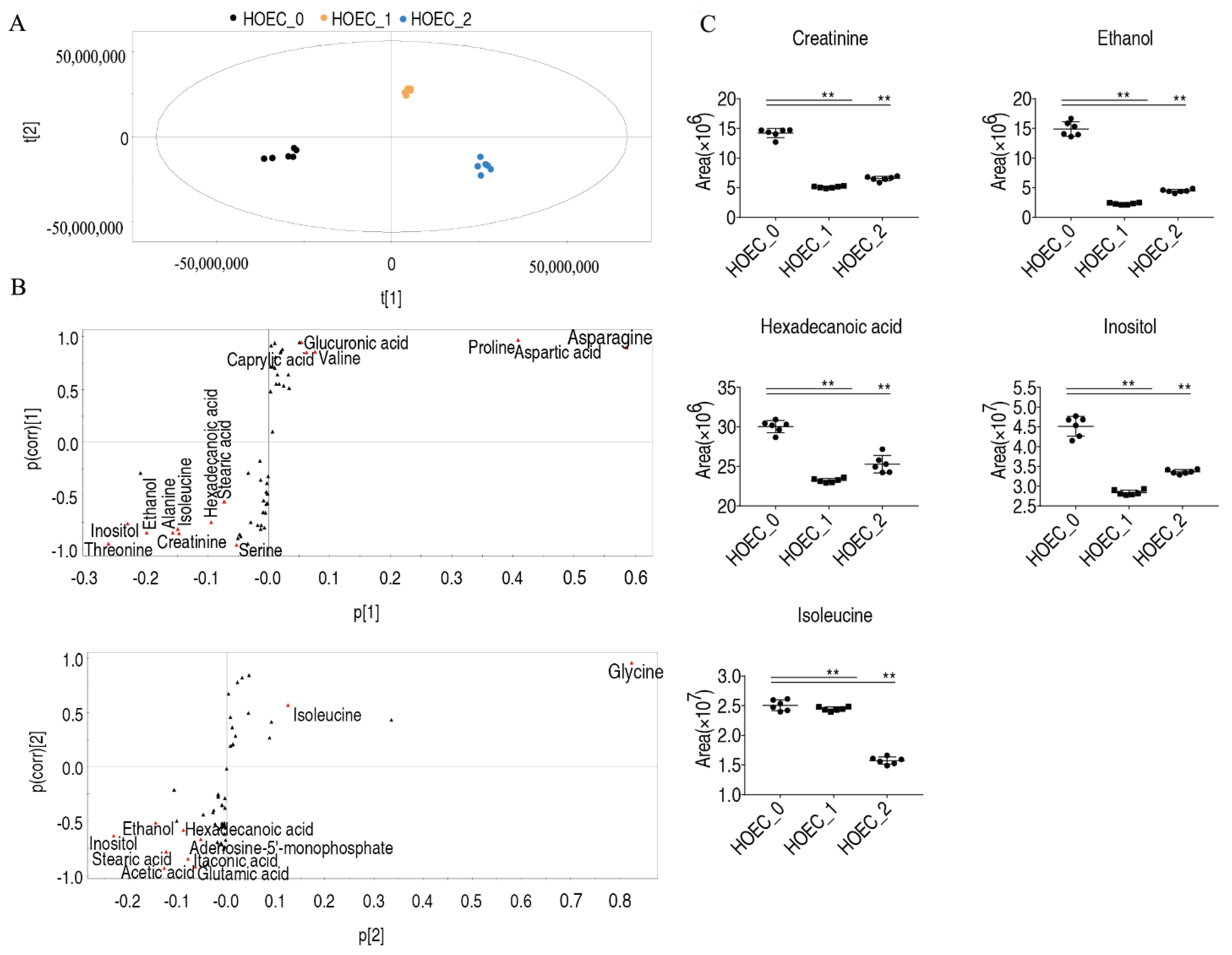
2.4. Metabolic Changes and Marker Metabolites of HOEC Cells Stimulated by Cisplatin
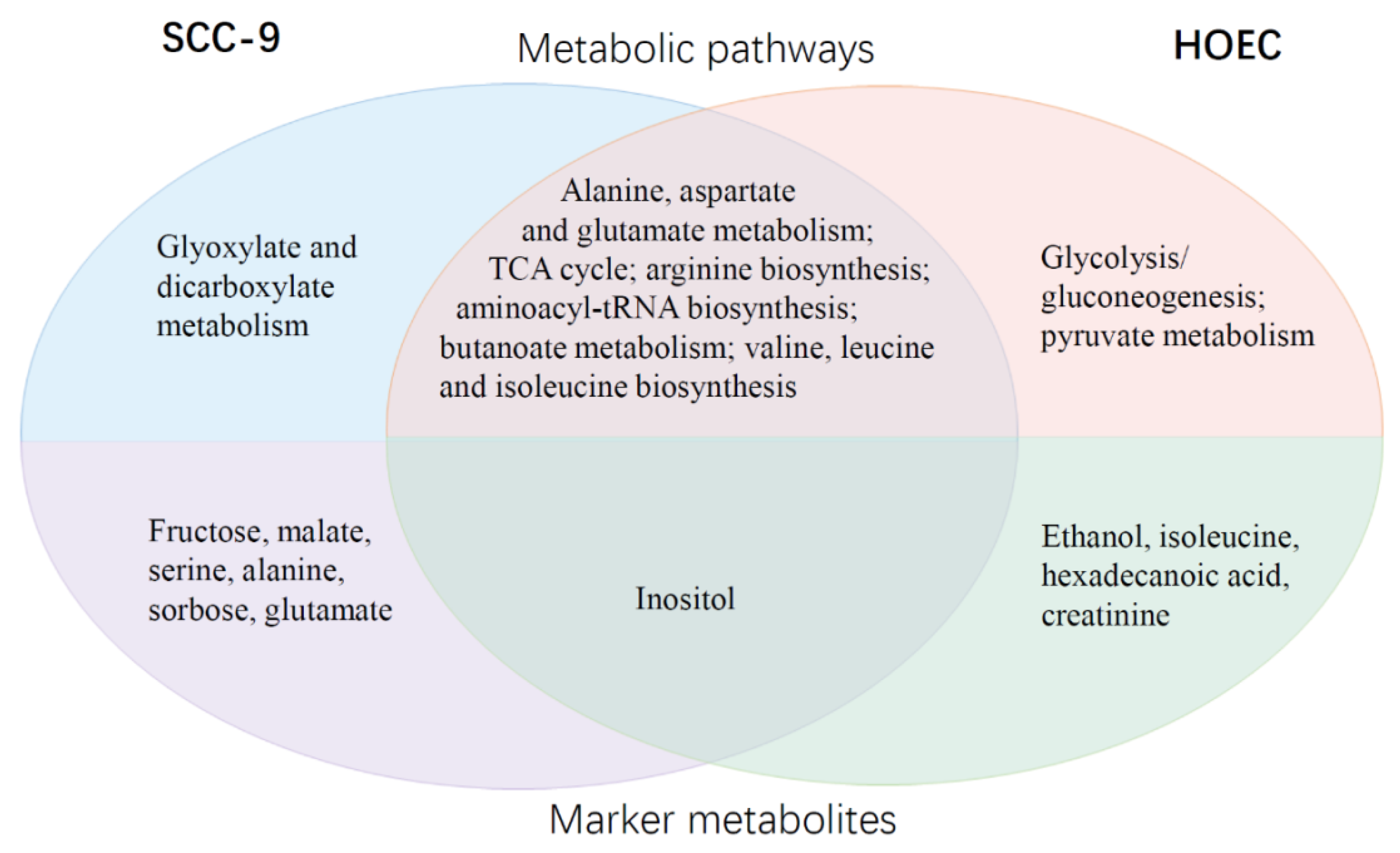
2.5. Differential Metabolic Responses of SCC-9 and HOEC Cells to Cisplatin
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. Cell Viability Assay
4.3. Gas Chromatography-Mass Spectrometry
4.4. Bioinformatics Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Chen, S.W.; Yu, D.S. Metabolic reprogramming of chemoresistant cancer cells and the potential significance of metabolic regulation in the reversal of cancer chemoresistance. Metabolites 2020, 10, 289. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.C.W.; Carella, M.A.; Papa, S.; Bubici, C. High expression of glycolytic genes in cirrhosis correlates with the risk of developing liver cancer. Front. Cell Dev. Biol. 2018, 6, 138. [Google Scholar] [CrossRef] [Green Version]
- Wangpaichitr, M.; Wu, C.; Li, Y.Y.; Nguyen, D.J.M.; Kandemir, H.; Shah, S.; Chen, S.; Feun, L.G.; Prince, J.S.; Kuo, M.T.; et al. Exploiting ROS and metabolic differences to kill cisplatin resistant lung cancer. Oncotarget 2017, 8, 49275–49292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wangpaichitr, M.; Kandemir, H.; Li, Y.Y.; Wu, C.; Nguyen, D.; Feun, L.G.; Kuo, M.T.; Savaraj, N. Relationship of metabolic alterations and PD-L1 expression in cisplatin resistant lung cancer. Cell Dev. Biol. 2017, 6, 183. [Google Scholar] [CrossRef] [PubMed]
- Montopoli, M.; Bellanda, M.; Lonardoni, F.; Ragazzi, E.; Dorigo, P.; Froldi, G.; Mammi, S.; Caparrotta, L. “Metabolic reprogramming” in ovarian cancer cells resistant to cisplatin. Curr. Cancer Drug Targets 2011, 11, 226–235. [Google Scholar] [CrossRef]
- Dar, S.; Chhina, J.; Mert, I.; Chitale, D.; Buekers, T.; Kaur, H.; Giri, S.; Munkarah, A.; Rattan, R. Bioenergetic adaptations in chemoresistant ovarian cancer cells. Sci. Rep. 2017, 7, 8760. [Google Scholar] [CrossRef]
- Poisson, L.M.; Munkarah, A.; Madi, H.; Datta, I.; Hensley-Alford, S.; Tebbe, C.; Buekers, T.; Giri, S.; Rattan, R. A metabolomic approach to identifying platinum resistance in ovarian cancer. J. Ovarian Res. 2015, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-inducing strategy in anticancer therapy. Oxid. Med. Cell. Longev. 2019, 2019, 5381692. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.J.; Chen, Y.Y.; Putluri, N.; Coarfa, C.; Robertson, M.J.; Putluri, V.; Stossi, F.; Dubrulle, J.; Mancini, M.A.; Pang, J.C.; et al. Acquisition of cisplatin resistance shifts head and neck squamous cell carcinoma metabolism toward neutralization of oxidative stress. Cancers 2020, 12, 1670. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.H.; Wang, Y.Y.; Huang, W.Y.; Wang, Y.; Wang, R.; Yuan, Y.F. Integration of metabolomics and transcriptomics to reveal metabolic characteristics and key targets associated with cisplatin resistance in nonsmall cell lung cancer. J. Proteome Res. 2019, 18, 3259–3267. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Jiang, W.; Lu, W.; Zhang, H.; Yu, T.; Tian, J.; Wen, S.; Garcia-Manero, G.; Huang, P.; Hu, Y. Metabolic reprogramming and redox adaptation in sorafenib-resistant leukemia cells: Detected by untargeted metabolomics and stable isotope tracing analysis. Cancer Commun. 2019, 39, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Stokes, W.; Chater, E.; Roy, R.; de Bruin, E.; Hu, Y.; Liu, Z.; Smit, E.F.; Heynen, G.J.; Downward, J.; et al. Decreased glutathione biosynthesis contributes to EGFR T790M-driven erlotinib resistance in non-small cell lung cancer. Cell Discov. 2016, 2, 16031. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.Y.; Yeon, A.; Shahid, M.; Cho, E.; Sairam, V.; Figlin, R.; Kim, K.H.; Kim, J. Reprogrammed lipid metabolism in bladder cancer with cisplatin resistance. Oncotarget 2018, 9, 13231–13243. [Google Scholar] [CrossRef] [Green Version]
- Hong, W.; Zhao, Y.; Cao, L.; Cao, D.; Zhao, Z.; Jin, J. Metabolomics study on the differences of endogenous small molecule between A549/DDP and A549 cells based on high solution UPLC-TOF-MS. Zhongguo Fei Ai Za Zhi 2018, 21, 571–577. [Google Scholar] [CrossRef]
- Gunda, V.; Pathania, A.S.; Chava, S.; Prathipati, P.; Chaturvedi, N.K.; Coulter, D.W.; Pandey, M.K.; Durden, D.L.; Challagundla, K.B. Amino acids regulate cisplatin insensitivity in neuroblastoma. Cancers 2020, 12, 2576. [Google Scholar] [CrossRef]
- Sasada, S.; Miyata, Y.; Tsutani, Y.; Tsuyama, N.; Masujima, T.; Hihara, J.; Okada, M. Metabolomic analysis of dynamic response and drug resistance of gastric cancer cells to 5-fluorouracil. Oncol. Rep. 2013, 29, 925–931. [Google Scholar] [CrossRef]
- Chen, X.; Yi, C.; Yang, M.J.; Sun, X.; Liu, X.; Ma, H.; Li, Y.; Li, H.; Wang, C.; He, Y.; et al. Metabolomics study reveals the potential evidence of metabolic reprogramming towards the Warburg effect in precancerous lesions. J. Cancer 2021, 12, 1563–1574. [Google Scholar] [CrossRef]
- Obrist, F.; Michels, J.; Durand, S.; Chery, A.; Pol, J.; Levesque, S.; Joseph, A.; Astesana, V.; Pietrocola, F.; Wu, G.S.; et al. Metabolic vulnerability of cisplatin-resistant cancers. EMBO J. 2018, 37, e98597. [Google Scholar] [CrossRef] [PubMed]
- Maria, R.M.; Altei, W.F.; Selistre-de-Araujo, H.S.; Colnago, L.A. Impact of chemotherapy on metabolic reprogramming: Characterization of the metabolic profile of breast cancer MDA-MB-231 cells using H-1 HR-MAS NMR spectroscopy. J. Pharm. Biomed. 2017, 146, 324–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, K.Q.; Cao, Y.; Li, Z.; Zhou, X.X.; Ding, R.; Chen, K.J.; Liu, Y.; Qiu, Y.K.; Wu, Z.; Fang, M.J. Glycine metabolomic changes induced by anticancer agents in A549 cells. Amino Acids 2020, 52, 793–809. [Google Scholar] [CrossRef] [PubMed]
- Geck, R.C.; Foley, J.R.; Stewart, T.M.; Asara, J.M.; Casero, R.A.; Toker, A. Inhibition of the polyamine synthesis enzyme ornithine decarboxylase sensitizes triple-negative breast cancer cells to cytotoxic chemotherapy. J. Biol. Chem. 2020, 295, 6263–6277. [Google Scholar] [CrossRef] [Green Version]
- Jang, W.J.; Choi, B.; Song, S.H.; Lee, N.; Kim, D.J.; Lee, S.; Jeong, C.H. Multi-omics analysis reveals that ornithine decarboxylase contributes to erlotinib resistance in pancreatic cancer cells. Oncotarget 2017, 8, 92727–92742. [Google Scholar] [CrossRef] [Green Version]
- Carneiro, T.J.; Araujo, R.; Vojtek, M.; Goncalves-Monteiro, S.; Diniz, C.; de Carvalho, A.L.M.B.; Marques, M.P.M.; Gil, A.M. Multi-organ NMR metabolomics to assess in vivo overall metabolic impact of cisplatin in mice. Metabolites 2019, 9, 279. [Google Scholar] [CrossRef] [Green Version]
- Cao, B.; Li, M.J.; Zha, W.B.; Zhao, Q.J.; Gu, R.R.; Liu, L.S.; Shi, J.; Zhou, J.; Zhou, F.; Wu, X.L.; et al. Metabolomic approach to evaluating adriamycin pharmacodynamics and resistance in breast cancer cells. Metabolomics 2013, 9, 960–973. [Google Scholar] [CrossRef] [Green Version]
- Guerra, A.R.; Paulino, A.F.; Castro, M.M.; Oliveira, H.; Duarte, M.F.; Duarte, I.F. Triple negative breast cancer and breast epithelial cells differentially reprogram glucose and lipid metabolism upon treatment with triterpenic acids. Biomolecules 2020, 10, 1163. [Google Scholar] [CrossRef]
- Sanchez-Castillo, A.; Vooijs, M.; Kampen, K.R. Linking serine/glycine metabolism to radiotherapy resistance. Cancers 2021, 13, 1191. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Fu, J.F.; Tang, W.F.; Yu, L.L.; Xu, W.X. Inhibition of serine metabolism promotes resistance to cisplatin in gastric cancer. Oncotargets Ther. 2020, 13, 4833–4842. [Google Scholar] [CrossRef]
- Arlt, B.; Zasada, C.; Baum, K.; Wuenschel, J.; Mastrobuoni, G.; Lodrini, M.; Astrahantseff, K.; Winkler, A.; Schulte, J.H.; Finkler, S.; et al. Inhibiting phosphoglycerate dehydrogenase counteracts chemotherapeutic efficacy against MYCN-amplified neuroblastoma. Int. J. Cancer 2021, 148, 1219–1232. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Enokida, H.; Osako, Y.; Nohata, N.; Yonemori, M.; Sugita, S.; Kuroshima, K.; Tsuruda, M.; Tatarano, S.; Nakagawa, M. Characterization of PHGDH expression in bladder cancer: Potential targeting therapy with gemcitabine/cisplatin and the contribution of promoter DNA hypomethylation. Mol. Oncol. 2020, 14, 2190–2202. [Google Scholar] [CrossRef]
- Hummel, J.; Strehmel, N.; Selbig, J.; Walther, D.; Kopka, J. Decision tree supported substructure prediction of metabolites from GC-MS profiles. Metabolomics 2010, 6, 322–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, J.; Xia, J. MetaboAnalystR: An R package for flexible and reproducible analysis of metabolomics data. Bioinformatics 2018, 34, 4313–4314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Up-Regulated | Down-Regulated |
|---|---|
| malate, glucose, fructose, sorbose, fumaric acid, serine, benzoic acid, succinic acid, valine, leucine, threonine, citric acid, actate, lauric acid, hexadecanedioic acid | pyruvate, inosine, sebacic acid, hexanedioic acid, creatinine, adenosine, inositol, ornithine, alanine, AMP |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Kuang, S.; He, Y.; Li, H.; Yi, C.; Li, Y.; Wang, C.; Chen, G.; Chen, S.; Yu, D. The Differential Metabolic Response of Oral Squamous Cell Carcinoma Cells and Normal Oral Epithelial Cells to Cisplatin Exposure. Metabolites 2022, 12, 389. https://doi.org/10.3390/metabo12050389
Chen X, Kuang S, He Y, Li H, Yi C, Li Y, Wang C, Chen G, Chen S, Yu D. The Differential Metabolic Response of Oral Squamous Cell Carcinoma Cells and Normal Oral Epithelial Cells to Cisplatin Exposure. Metabolites. 2022; 12(5):389. https://doi.org/10.3390/metabo12050389
Chicago/Turabian StyleChen, Xun, Sufang Kuang, Yi He, Hongyu Li, Chen Yi, Yiming Li, Chao Wang, Guanhui Chen, Shangwu Chen, and Dongsheng Yu. 2022. "The Differential Metabolic Response of Oral Squamous Cell Carcinoma Cells and Normal Oral Epithelial Cells to Cisplatin Exposure" Metabolites 12, no. 5: 389. https://doi.org/10.3390/metabo12050389
APA StyleChen, X., Kuang, S., He, Y., Li, H., Yi, C., Li, Y., Wang, C., Chen, G., Chen, S., & Yu, D. (2022). The Differential Metabolic Response of Oral Squamous Cell Carcinoma Cells and Normal Oral Epithelial Cells to Cisplatin Exposure. Metabolites, 12(5), 389. https://doi.org/10.3390/metabo12050389