
Integrated Metabolomics and Transcriptomics Analyses Reveal the Metabolic Differences and Molecular Basis of Nutritional Quality in Landraces and Cultivated Rice

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
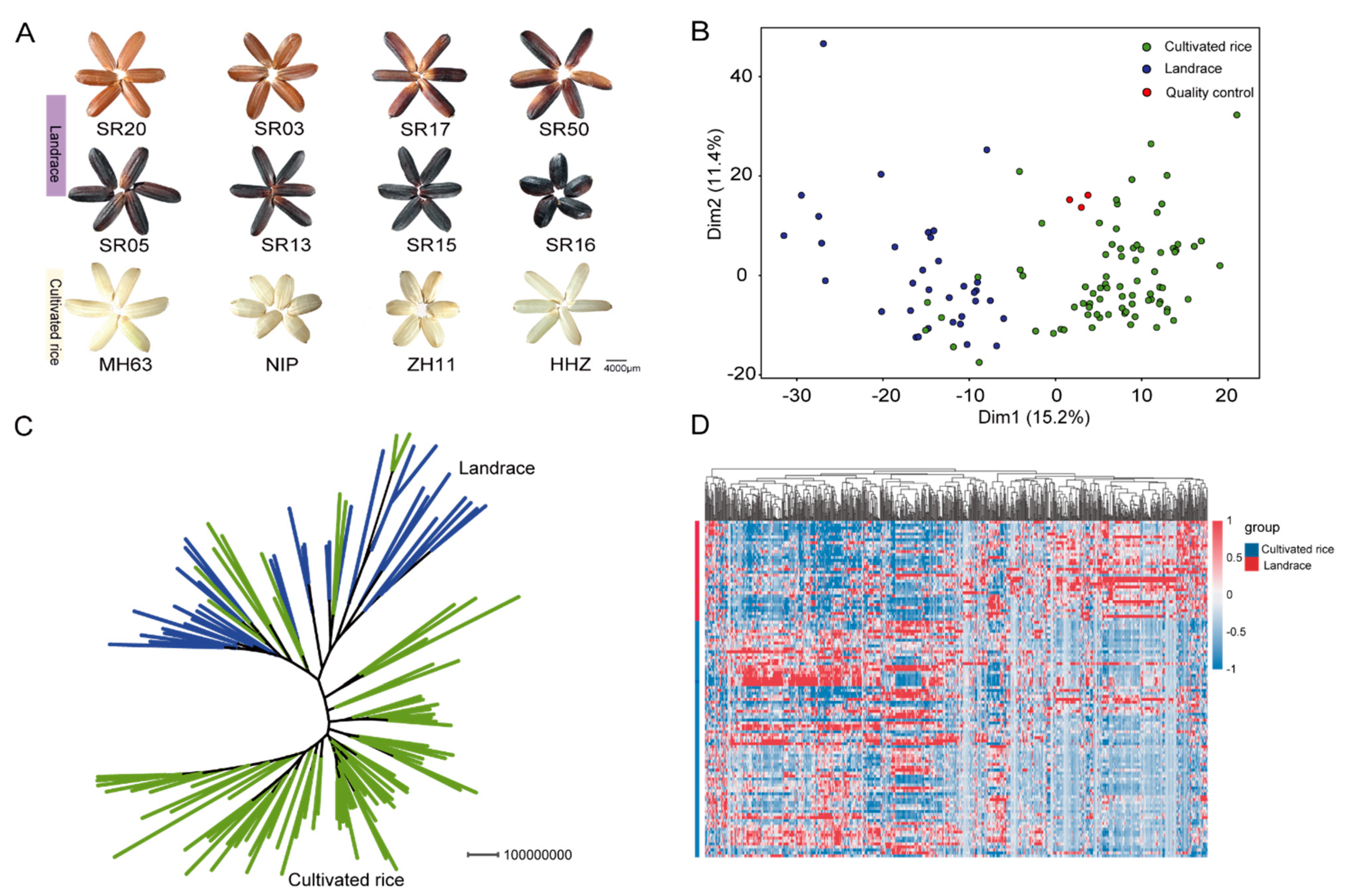
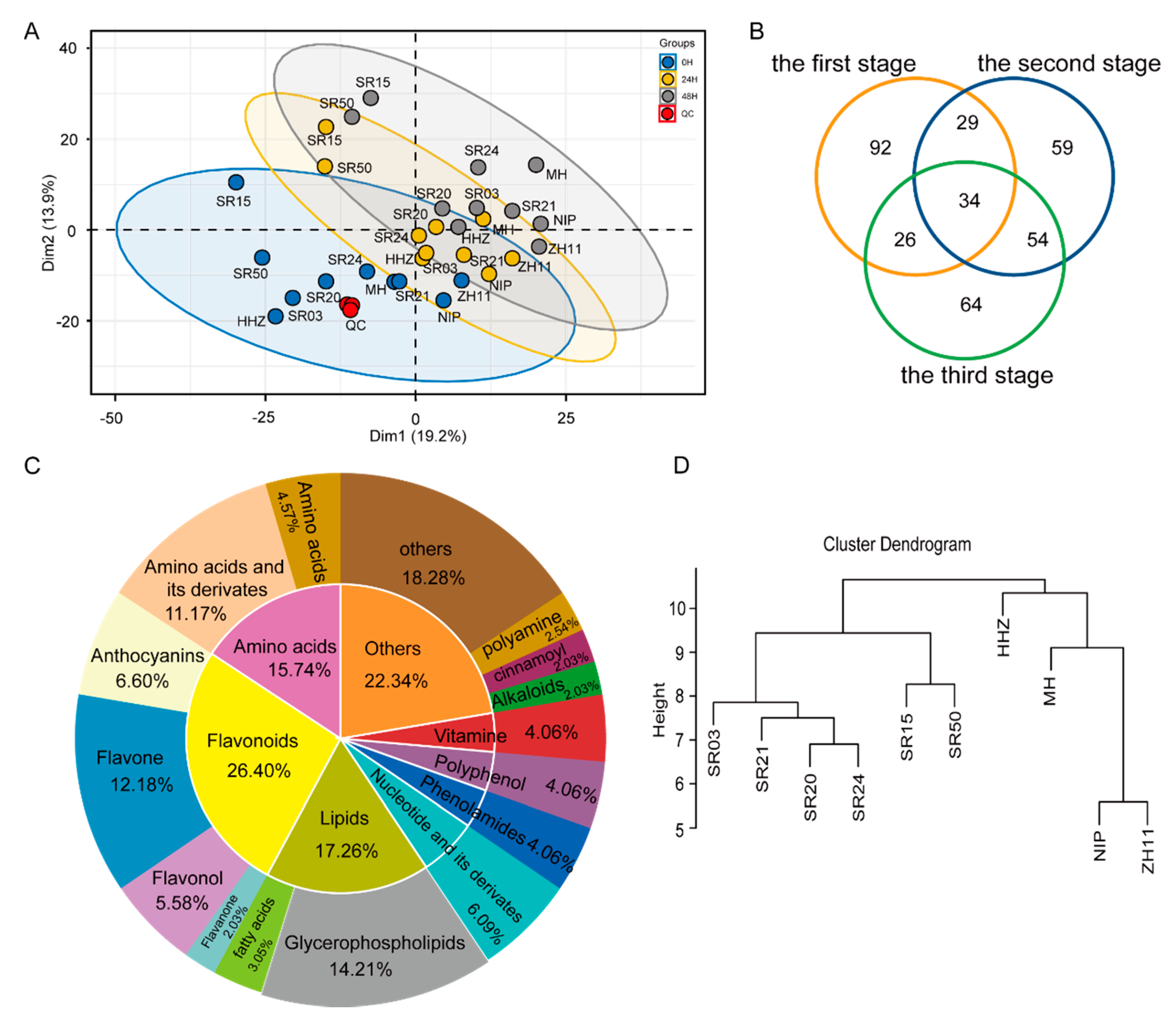
2.1. Comparative Analysis of Metabolic Profiling between Landraces and Cultivated Rice
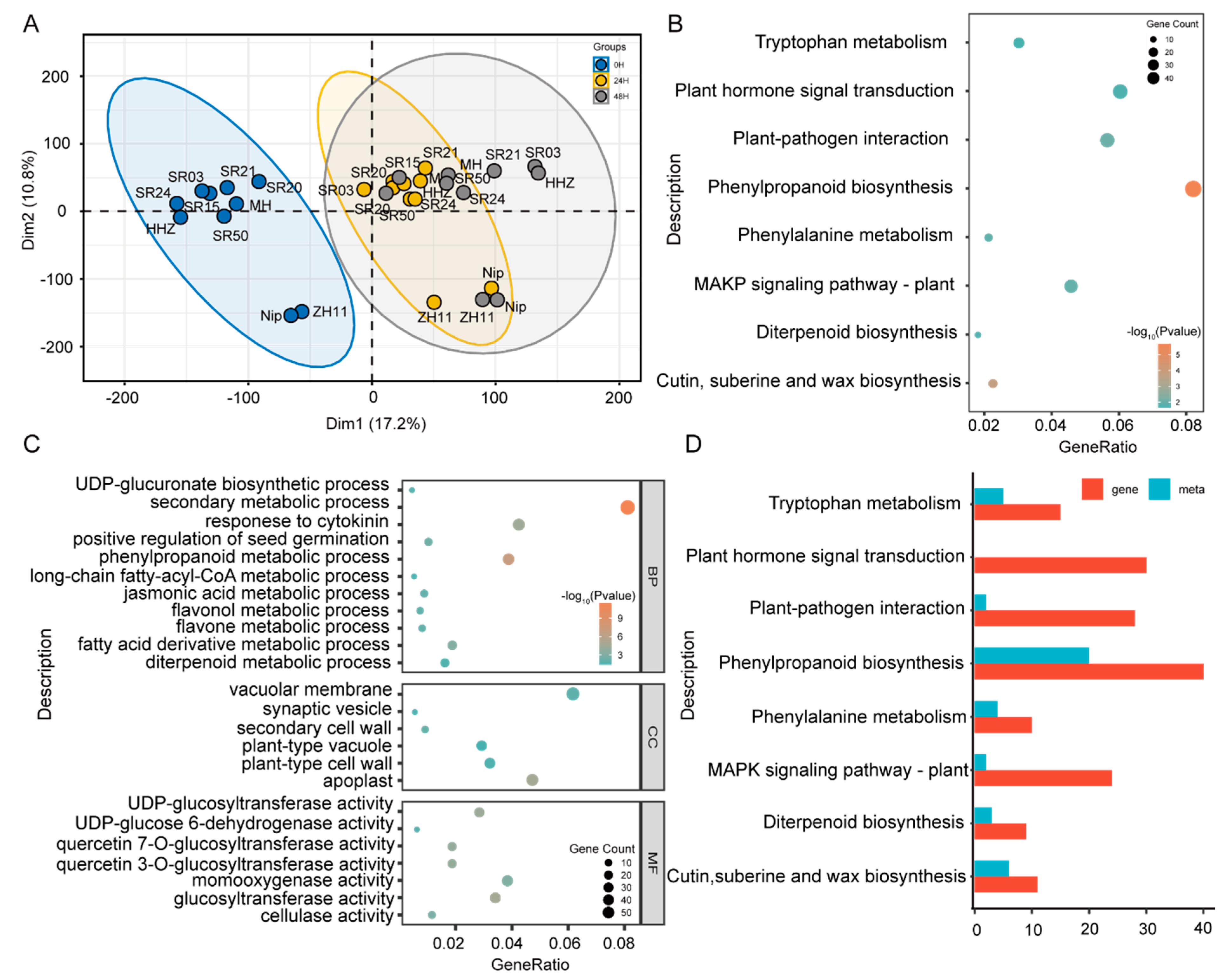
2.2. Analysis of DAM and DEG in Seeds of between Landrace and Cultivated Rice during Germination
2.3. Correlation Network Based on Genes and Metabolites during Seed Germination
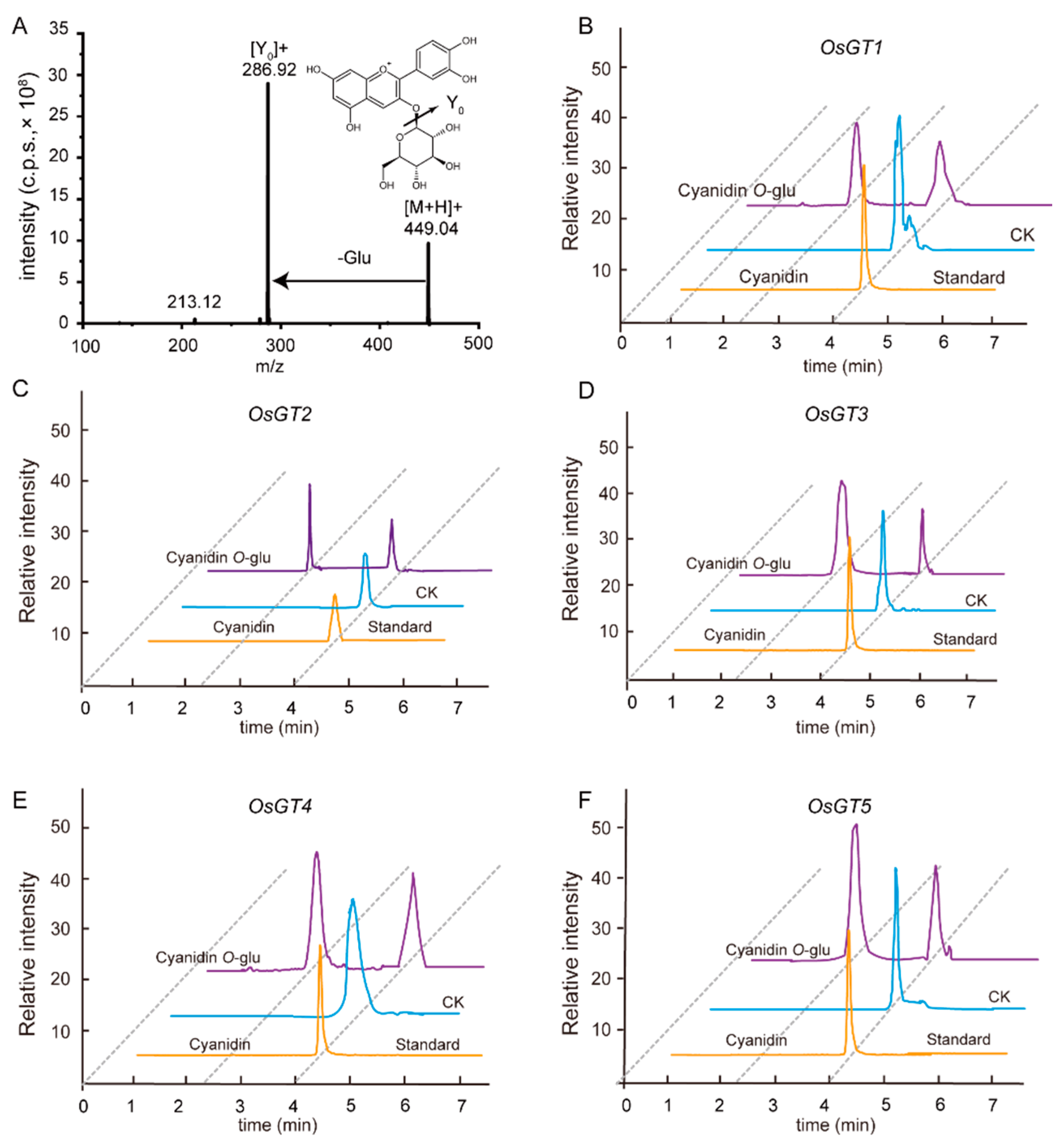
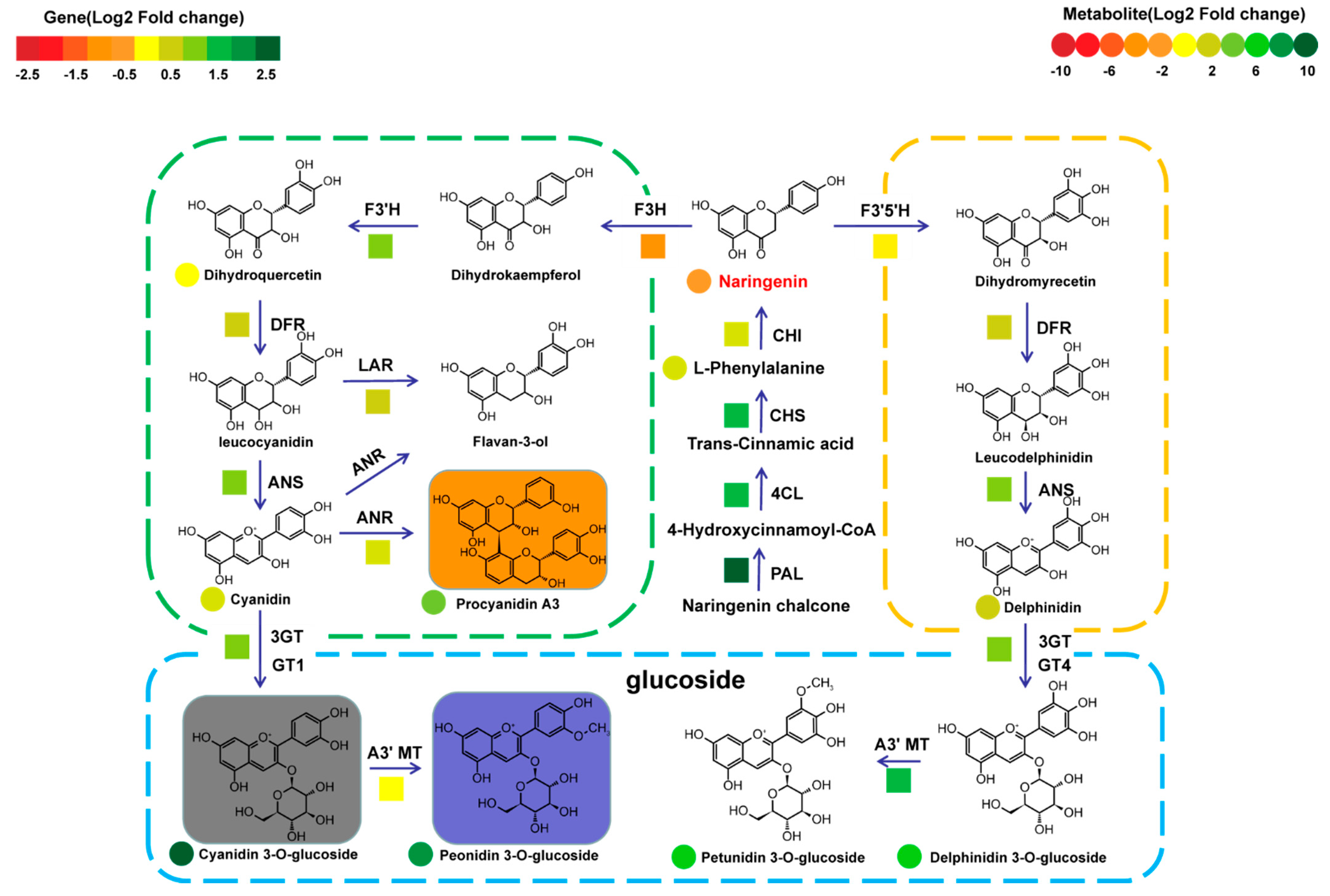
2.4. The Metabolic Diversity of Anthocyanins and Its Molecular Mechanism in Different Types of Rice Seeds
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Metabolic Sample Preparation and Profiling
4.3. Transcriptome Data Analysis
4.4. GO and KEGG Enrichment for Differentially Expressed Genes
4.5. Phylogenetic Analysis and Principal Component Analysis Based on Metabolites
4.6. Co-Expression Network Analysis for the Construction of Modules
4.7. Analysis of Gene Expression Levels by qRT-PCR
4.8. Recombinant Protein Expression and In Vitro Enzyme Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Birla, D.S.; Malik, K.; Sainger, M.; Chaudhary, D.; Jaiwal, R.; Jaiwal, P.K. Progress and challenges in improving the nutritional quality of rice (Oryza sativa L.). Crit. Rev. Food Sci. Nutr. 2017, 57, 2455–2481. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Hu, W.; Xing, Y.Z. The history and prospect of rice genetic breeding in China. Hereditas 2018, 40, 841–857. [Google Scholar] [CrossRef]
- Ke, Y.; Kang, Y.; Wu, M.; Liu, H.; Hui, S.; Zhang, Q.; Li, X.; Xiao, J.; Wang, S. Jasmonic acid-involved OsEDS1 signaling in Rice-bacteria interactions. Rice 2019, 12, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Ning, Y.; Zhang, Y.; Yu, N.; Zhao, C.; Zhan, X.; Wu, W.; Chen, D.; Wei, X.; Wang, G.-L. OsCUL3a negatively regulates cell death and immunity by degrading OsNPR1 in rice. Plant Cell 2017, 29, 345–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anacleto, R.; Cuevas, R.P.; Jimenez, R.; Llorente, C.; Nissila, E.; Henry, R.; Sreenivasulu, N. Prospects of breeding high-quality rice using post-genomic tools. Theor. Appl. Genet. 2015, 128, 1449–1466. [Google Scholar] [CrossRef]
- Bai, S.; Yu, H.; Wang, B.; Li, J. Retrospective and perspective of rice breeding in China. J. Genet. Genom. 2018, 45, 603–612. [Google Scholar] [CrossRef]
- Hour, A.-L.; Hsieh, W.-H.; Chang, S.-H.; Wu, Y.-P.; Chin, H.-S.; Lin, Y.-R. Genetic diversity of landraces and improved varieties of rice (Oryza sativa L.) in Taiwan. Rice 2020, 13, 82. [Google Scholar] [CrossRef]
- Dwivedi, S.L.; Ceccarelli, S.; Blair, M.W.; Upadhyaya, H.D.; Are, A.K.; Ortiz, R. Landrace germplasm for improving yield and abiotic stress adaptation. Trends Plant Sci. 2016, 21, 31–42. [Google Scholar] [CrossRef]
- Shao, Y.; Hu, Z.; Yu, Y.; Mou, R.; Zhu, Z.; Beta, T. Phenolic acids, anthocyanins, proanthocyanidins, antioxidant activity, minerals and their correlations in non-pigmented, red, and black rice. Food Chem. 2018, 239, 733–741. [Google Scholar] [CrossRef]
- Zhang, Q. Purple tomatoes, black rice and food security. Nat. Rev. Genet. 2021, 22, 414. [Google Scholar] [CrossRef] [PubMed]
- Frank, T.; Reichardt, B.; Shu, Q.; Engel, K.-H. Metabolite profiling of colored rice (Oryza sativa L.) grains. J. Cereal Sci. 2012, 55, 112–119. [Google Scholar] [CrossRef]
- Ito, V.C.; Lacerda, L.G. Black rice (Oryza sativa L.): A review of its historical aspects, chemical composition, nutritional and functional properties, and applications and processing technologies. Food Chem. 2019, 301, 125304. [Google Scholar] [CrossRef] [PubMed]
- Subash, S.; Essa, M.M.; Al-Adawi, S.; Memon, M.A.; Manivasagam, T.; Akbar, M. Neuroprotective effects of berry fruits on neurodegenerative diseases. Neural Regen. Res. 2014, 9, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.C. Anthocyanins in cardiovascular disease. Adv. Nutr. 2011, 2, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Gong, L.; Guo, Z.; Wang, W.; Zhang, H.; Liu, X.; Yu, S.; Xiong, L.; Luo, J. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant 2013, 6, 1769–1780. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Wang, W.; Peng, M.; Gong, L.; Gao, Y.; Wan, J.; Wang, S.; Shi, L.; Zhou, B.; Li, Z. Comparative and parallel genome-wide association studies for metabolic and agronomic traits in cereals. Nat. Commun. 2016, 7, 12767. [Google Scholar] [CrossRef]
- Tieman, D.; Zhu, G.; Resende, M.F.R.; Lin, T.; Nguyen, C.; Bies, D.; Rambla, J.L.; Beltran, K.S.O.; Taylor, M.; Zhang, B.; et al. A chemical genetic roadmap to improved tomato flavor. Science 2017, 355, 391–394. [Google Scholar] [CrossRef]
- Zhu, G.; Wang, S.; Huang, Z.; Zhang, S.; Liao, Q.; Zhang, C.; Lin, T.; Qin, M.; Peng, M.; Yang, C. Rewiring of the fruit metabolome in tomato breeding. Cell 2018, 172, 249–261. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Shen, S.; Zhou, S.; Li, Y.; Mao, Y.; Zhou, J.; Shi, Y.; An, L.; Zhou, Q.; Peng, W. Rice metabolic regulatory network spanning the entire life cycle. Mol. Plant 2021, 15, 258–275. [Google Scholar] [CrossRef]
- Dong, X.; Chen, W.; Wang, W.; Zhang, H.; Liu, X.; Luo, J. Comprehensive profiling and natural variation of flavonoids in rice. J. Integr. Plant Biol. 2014, 56, 876–886. [Google Scholar] [CrossRef]
- Dong, X.; Gao, Y.; Chen, W.; Wang, W.; Gong, L.; Liu, X.; Luo, J. Spatiotemporal distribution of phenolamides and the genetics of natural variation of hydroxycinnamoyl spermidine in rice. Mol. Plant 2015, 8, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Itkin, M.; Rogachev, I.; Alkan, N.; Rosenberg, T.; Malitsky, S.; Masini, L.; Meir, S.; Iijima, Y.; Aoki, K.; de Vos, R. GLYCOALKALOID METABOLISM1 is required for steroidal alkaloid glycosylation and prevention of phytotoxicity in tomato. Plant Cell 2011, 23, 4507–4525. [Google Scholar] [CrossRef] [Green Version]
- Itkin, M.; Heinig, U.; Tzfadia, O.; Bhide, A.J.; Shinde, B.; Cardenas, P.D.; Bocobza, S.E.; Unger, T.; Malitsky, S.; Finkers, R. Biosynthesis of antinutritional alkaloids in solanaceous crops is mediated by clustered genes. Science 2013, 341, 175–179. [Google Scholar] [CrossRef]
- Polturak, G.; Heinig, U.; Grossman, N.; Battat, M.; Leshkowitz, D.; Malitsky, S.; Rogachev, I.; Aharoni, A. Transcriptome and metabolic profiling provides insights into betalain biosynthesis and evolution in Mirabilis jalapa. Mol. Plant 2018, 11, 189–204. [Google Scholar] [CrossRef] [Green Version]
- Nett, R.S.; Lau, W.; Sattely, E.S. Discovery and engineering of colchicine alkaloid biosynthesis. Nature 2020, 584, 148–153. [Google Scholar] [CrossRef]
- Jeon, J.E.; Kim, J.-G.; Fischer, C.R.; Mehta, N.; Dufour-Schroif, C.; Wemmer, K.; Mudgett, M.B.; Sattely, E. A pathogen-responsive gene cluster for highly modified fatty acids in tomato. Cell 2020, 180, 176–187. [Google Scholar] [CrossRef]
- Cárdenas, P.D.; Sonawane, P.D.; Pollier, J.; Vanden Bossche, R.; Dewangan, V.; Weithorn, E.; Tal, L.; Meir, S.; Rogachev, I.; Malitsky, S. GAME9 regulates the biosynthesis of steroidal alkaloids and upstream isoprenoids in the plant mevalonate pathway. Nat. Commun. 2016, 7, 10654. [Google Scholar] [CrossRef] [Green Version]
- Slate, J.O.N. INVITED REVIEW: Quantitative trait locus mapping in natural populations: Progress, caveats and future directions. Mol. Ecol. 2005, 14, 363–379. [Google Scholar] [CrossRef]
- Angelovici, R.; Fait, A.; Zhu, X.; Szymanski, J.; Feldmesser, E.; Fernie, A.R.; Galili, G. Deciphering transcriptional and metabolic networks associated with lysine metabolism during Arabidopsis seed development. Plant Physiol. 2009, 151, 2058–2072. [Google Scholar] [CrossRef] [Green Version]
- Atwell, S.; Huang, Y.S.; Vilhjálmsson, B.J.; Willems, G.; Horton, M.; Li, Y.; Meng, D.; Platt, A.; Tarone, A.M.; Hu, T.T. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 2010, 465, 627–631. [Google Scholar] [CrossRef]
- Chan, E.K.F.; Rowe, H.C.; Corwin, J.A.; Joseph, B.; Kliebenstein, D.J. Combining genome-wide association mapping and transcriptional networks to identify novel genes controlling glucosinolates in Arabidopsis thaliana. PLoS Biol. 2011, 9, e1001125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Gao, Y.; Xie, W.; Gong, L.; Lu, K.; Wang, W.; Li, Y.; Liu, X.; Zhang, H.; Dong, H. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat. Genet. 2014, 46, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Riedelsheimer, C.; Czedik-Eysenberg, A.; Grieder, C.; Lisec, J.; Technow, F.; Sulpice, R.; Altmann, T.; Stitt, M.; Willmitzer, L.; Melchinger, A.E. Genomic and metabolic prediction of complex heterotic traits in hybrid maize. Nat. Genet. 2012, 44, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Kearsey, M.J.; Pooni, H.S.; Syed, N.H. Genetics of quantitative traits in Arabidopsis thaliana. Heredity 2003, 91, 456–464. [Google Scholar] [CrossRef] [Green Version]
- Morgante, M.; Salamini, F. From plant genomics to breeding practice. Curr. Opin. Biotechnol. 2003, 14, 214–219. [Google Scholar] [CrossRef]
- Wambugu, P.W.; Ndjiondjop, M.N.; Henry, R. Genetics and Genomics of African Rice (Oryza glaberrima Steud) Domestication. Rice 2021, 14, 1–14. [Google Scholar] [CrossRef]
- Mbanjo, E.; Kretzschmar, T.; Jones, H.; Ereful, N.; Sreenivasulu, N. The Genetic Basis and Nutritional Benefits of Pigmented Rice Grain. Front. Genet. 2020, 11, 229. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; Deb, D.; Ray, R.; Chattopadhayay, B. Phenotypic characters of rice landraces reveal independent lineages of short-grain aromatic indica rice. AOB Plants 2013, 5, plt032. [Google Scholar] [CrossRef]
- Fukuoka, S.; Suu, T.D.; Ebana, K.; Trinh, L.N.; Nagamine, T.; Okuno, K. Diversity in Phenotypic Profiles in Landrace Populations of Vietnamese Rice: A Case Study of Agronomic Characters for Conserving Crop Genetic Diversity on Farm. Genet. Resour. Crop Evol. 2006, 53, 753–761. [Google Scholar] [CrossRef]
- Luong, N.H.; Linh, L.-H.; Shim, K.-C.; Adeva, C.; Lee, H.-S.; Ahn, S.-N. Genetic Structure and Geographical Differentiation of Traditional Rice (Oryza sativa L.) from Northern Vietnam. Plants 2021, 10, 2094. [Google Scholar] [CrossRef]
- Koes, R.E.; Quattrocchio, F.; Mol, J. The flavonoid biosynthetic pathway in plants: Function and evolution. BioEssays 1994, 16, 123–132. [Google Scholar] [CrossRef]
- Petroni, K.; Pilu, R.; Tonelli, C. Anthocyanins in corn: A wealth of genes for human health. Planta 2014, 240, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Ns, A.; Ak, A.; Vc, A.; Sk, A.; Jb, C.; Ag, B.; Mg, A. Anthocyanin biofortified black, blue and purple wheat exhibited lower amino acid cooking losses than white wheat. LWT 2021, 154, 112802. [Google Scholar] [CrossRef]
- Abdel-Aal, E.; Young, J.C.; Rabalski, I. Anthocyanin Composition in Black, Blue, Pink, Purple, and Red Cereal Grains. J. Agric. Food Chem. 2006, 54, 4696–4704. [Google Scholar] [CrossRef] [PubMed]
- Thanyacharoen, T.; Chuysinuan, P.; Techasakul, S.; Noenplab, A.L.; Ummartyotin, S. The chemical composition and antioxidant and release properties of a black rice (Oryza sativa L.)-loaded chitosan and polyvinyl alcohol composite. J. Mol. Liq. 2017, 248, 1065–1070. [Google Scholar] [CrossRef]
- Vargas, C.G.; da Silva Junior, J.D.; Rabelo, T.K.; Moreira, J.C.F.; Gelain, D.P.; Rodrigues, E.; Augusti, P.R.; de Oliveira Rios, A.; Flôres, S.H. Bioactive compounds and protective effect of red and black rice brans extracts in human neuron-like cells (SH-SY5Y). Food Res. Int. 2018, 113, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Luo, J. Metabolite-based genome-wide association studies in plants. Curr. Opin. Plant Biol. 2015, 24, 31–38. [Google Scholar] [CrossRef]
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Leung, H. Genomic variation in 3010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef]
- Peng, M.; Shahzad, R.; Gul, A.; Subthain, H.; Shen, S.; Lei, L.; Zheng, Z.; Zhou, J.; Lu, D.; Wang, S. Differentially evolved glucosyltransferases determine natural variation of rice flavone accumulation and UV-tolerance. Nat. Commun. 2017, 8, 1975. [Google Scholar] [CrossRef]
- Giovannoni, J. Tomato multiomics reveals consequences of crop domestication and improvement. Cell 2018, 172, 6–8. [Google Scholar] [CrossRef]
- Schilmiller, A.L.; Charbonneau, A.L.; Last, R.L. Identification of a BAHD acetyltransferase that produces protective acyl sugars in tomato trichomes. Proc. Natl. Acad. Sci. USA 2012, 109, 16377–16382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, L.; Chen, W.; Gao, Y.; Liu, X.; Zhang, H.; Xu, C.; Yu, S.; Zhang, Q.; Luo, J. Genetic analysis of the metabolome exemplified using a rice population. Proc. Natl. Acad. Sci. USA 2013, 110, 20320–20325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Yu, S.; Zeng, D.; Liu, H.; Wang, H.; Yang, Z.; Xie, X.; Shen, R.; Tan, J.; Li, H.; et al. Development of “Purple Endosperm Rice” by Engineering Anthocyanin Biosynthesis in the Endosperm with a High-Efficiency Transgene Stacking System. Mol. Plant 2017, 10, 918–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, D.; Zhou, H.; Wang, Y.; Li, P.; Fu, P.; Wu, B.; He, Y. How rice organs are colored: The genetic basis of anthocyanin biosynthesis in rice. Crop J. 2021, 9, 598–608. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Zhang, F.; Deng, Y.; Sun, L.; Mao, M.; Chen, R.; Qiang, Q.; Zhou, J.; Long, T.; Zhao, X.; et al. Integrated Metabolomics and Transcriptomics Analyses Reveal the Metabolic Differences and Molecular Basis of Nutritional Quality in Landraces and Cultivated Rice. Metabolites 2022, 12, 384. https://doi.org/10.3390/metabo12050384
Zhang Z, Zhang F, Deng Y, Sun L, Mao M, Chen R, Qiang Q, Zhou J, Long T, Zhao X, et al. Integrated Metabolomics and Transcriptomics Analyses Reveal the Metabolic Differences and Molecular Basis of Nutritional Quality in Landraces and Cultivated Rice. Metabolites. 2022; 12(5):384. https://doi.org/10.3390/metabo12050384
Chicago/Turabian StyleZhang, Zhonghui, Feng Zhang, Yuan Deng, Lisong Sun, Mengdi Mao, Ridong Chen, Qi Qiang, Junjie Zhou, Tuan Long, Xuecheng Zhao, and et al. 2022. "Integrated Metabolomics and Transcriptomics Analyses Reveal the Metabolic Differences and Molecular Basis of Nutritional Quality in Landraces and Cultivated Rice" Metabolites 12, no. 5: 384. https://doi.org/10.3390/metabo12050384
APA StyleZhang, Z., Zhang, F., Deng, Y., Sun, L., Mao, M., Chen, R., Qiang, Q., Zhou, J., Long, T., Zhao, X., Liu, X., Wang, S., Yang, J., & Luo, J. (2022). Integrated Metabolomics and Transcriptomics Analyses Reveal the Metabolic Differences and Molecular Basis of Nutritional Quality in Landraces and Cultivated Rice. Metabolites, 12(5), 384. https://doi.org/10.3390/metabo12050384