
Microbial Interactions in Kombucha through the Lens of Metabolomics

 ,
, 
 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Comparison of General Chemical Compositions and Data Visualization
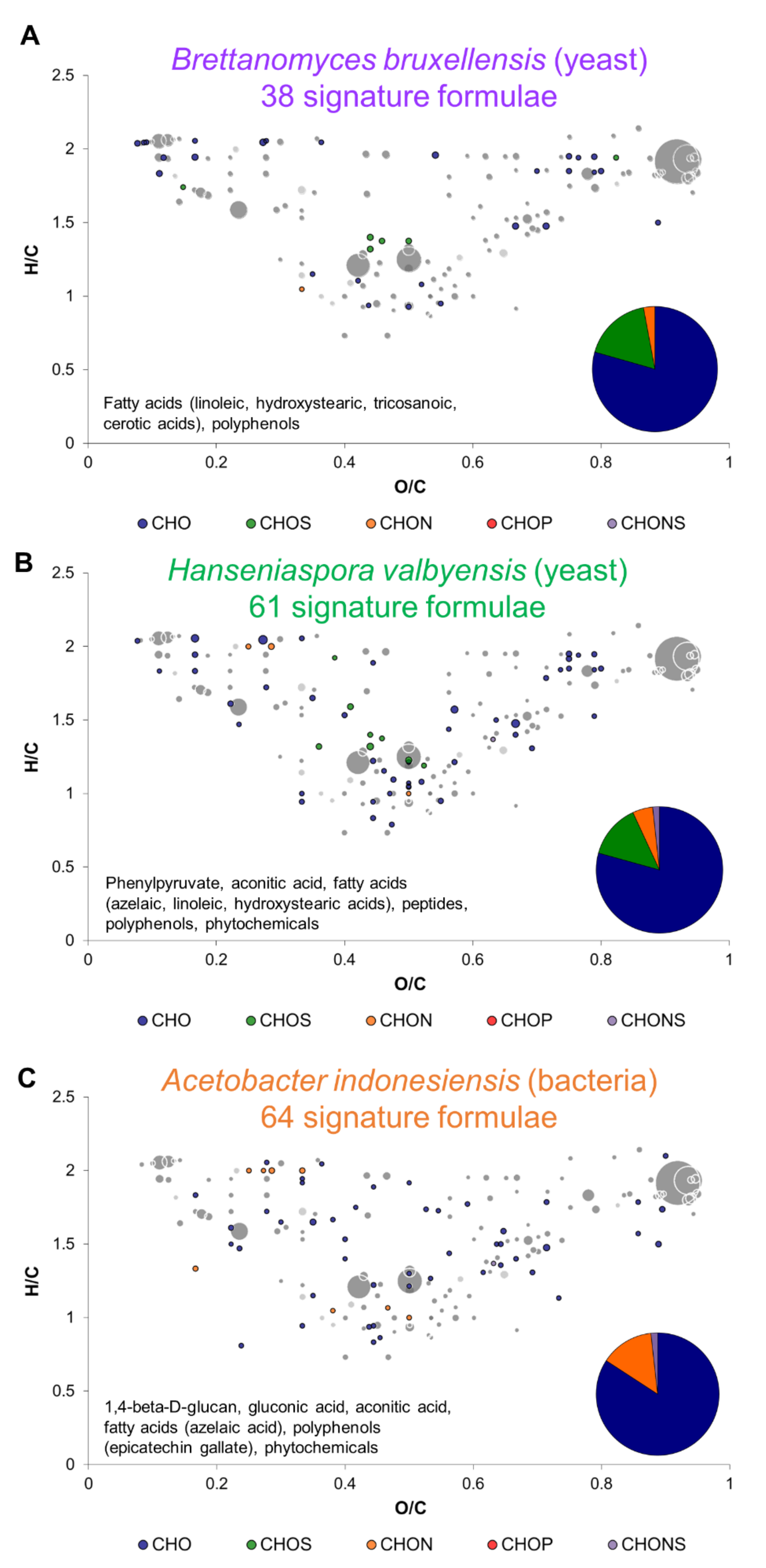
2.2. Metabolic Signature of Individual Microorganisms
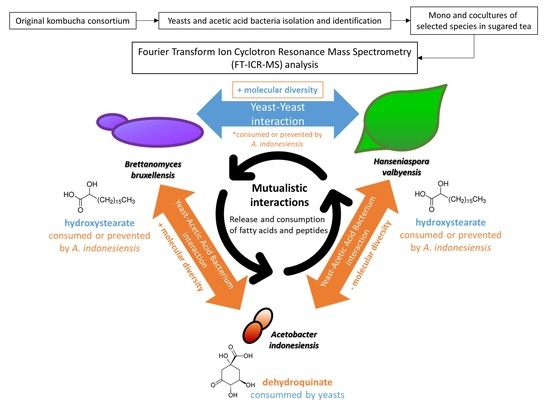
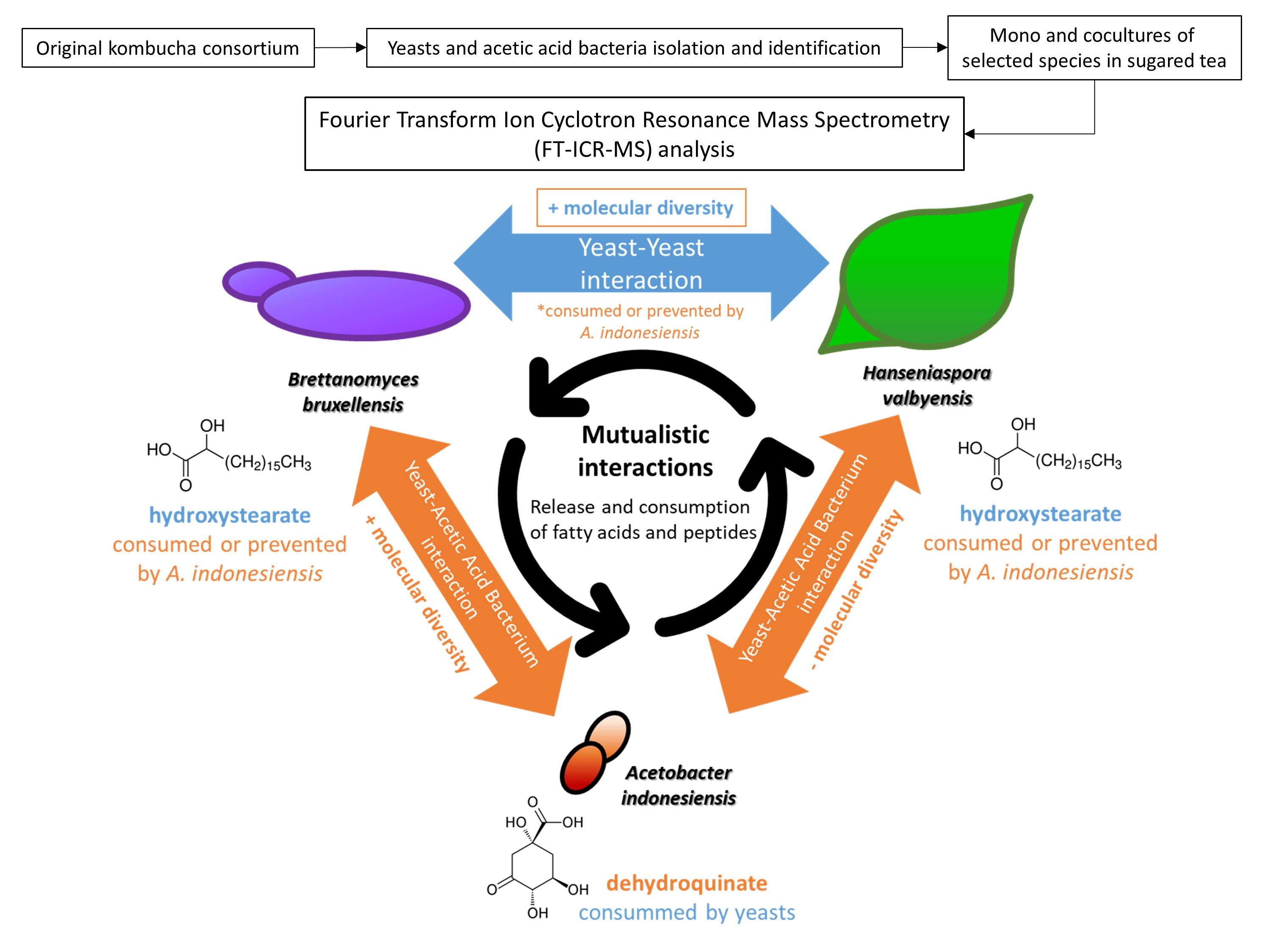
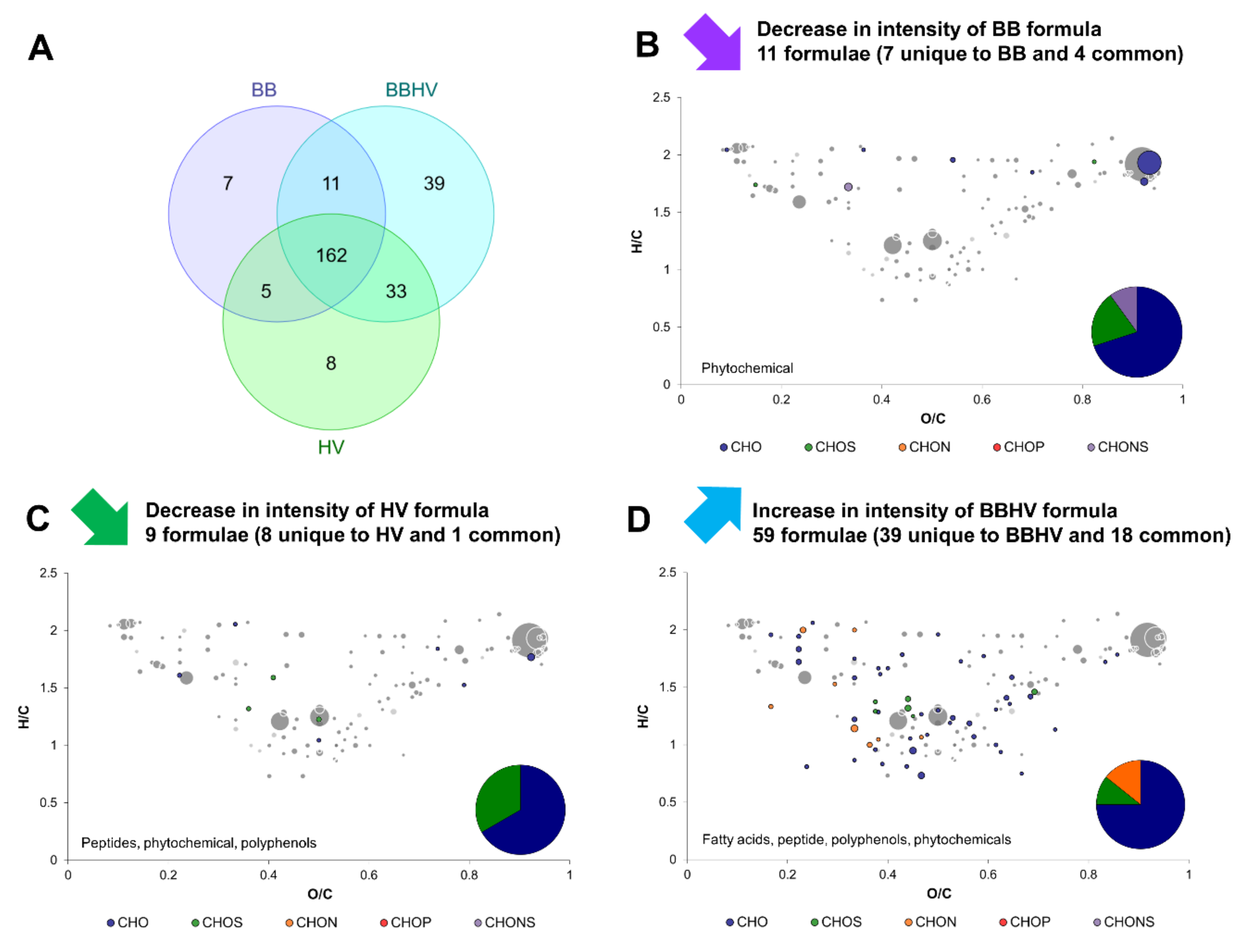
2.3. Yeast–Yeast Interspecies Interactions
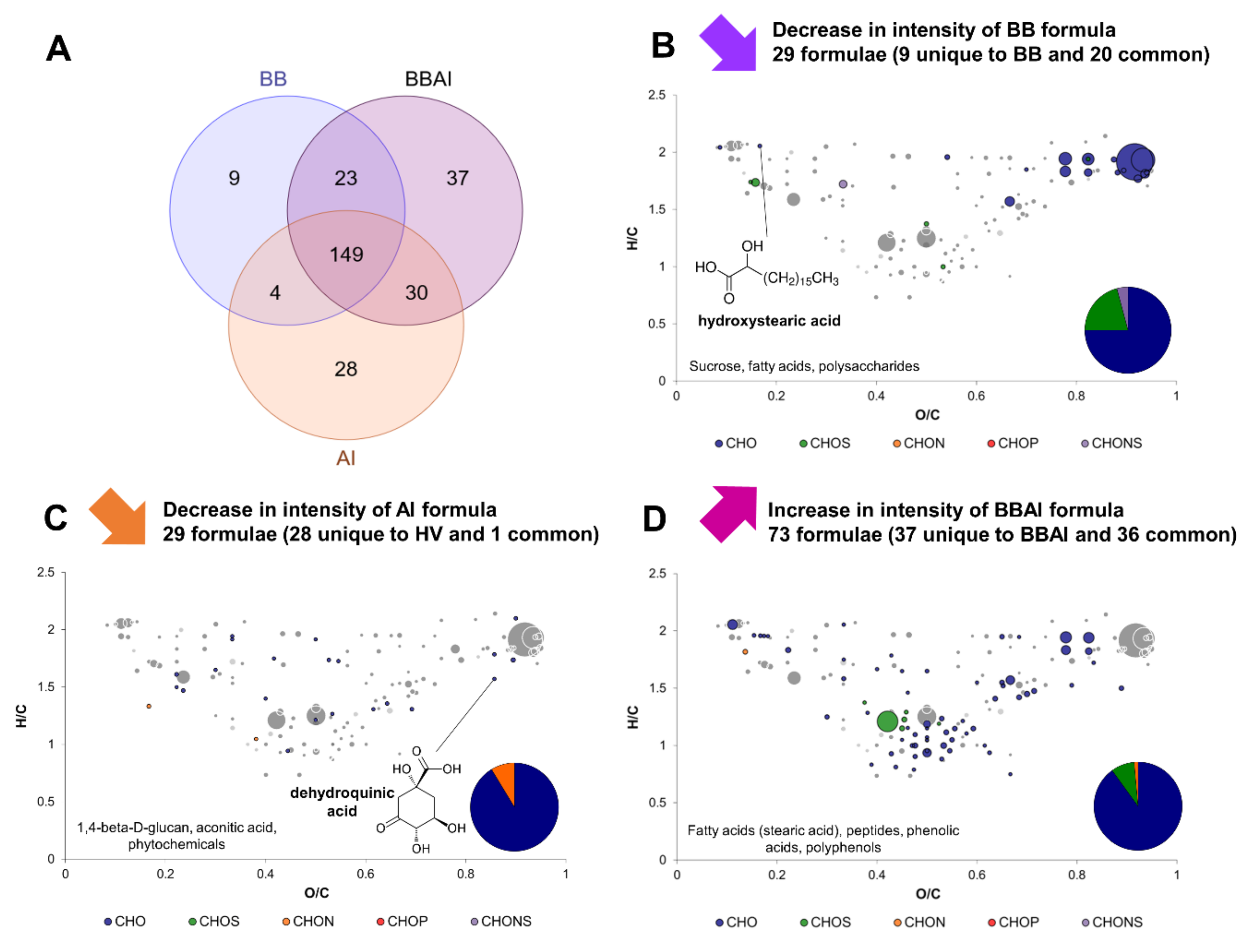
2.4. Yeast–Acetic Acid Bacterium Interactions
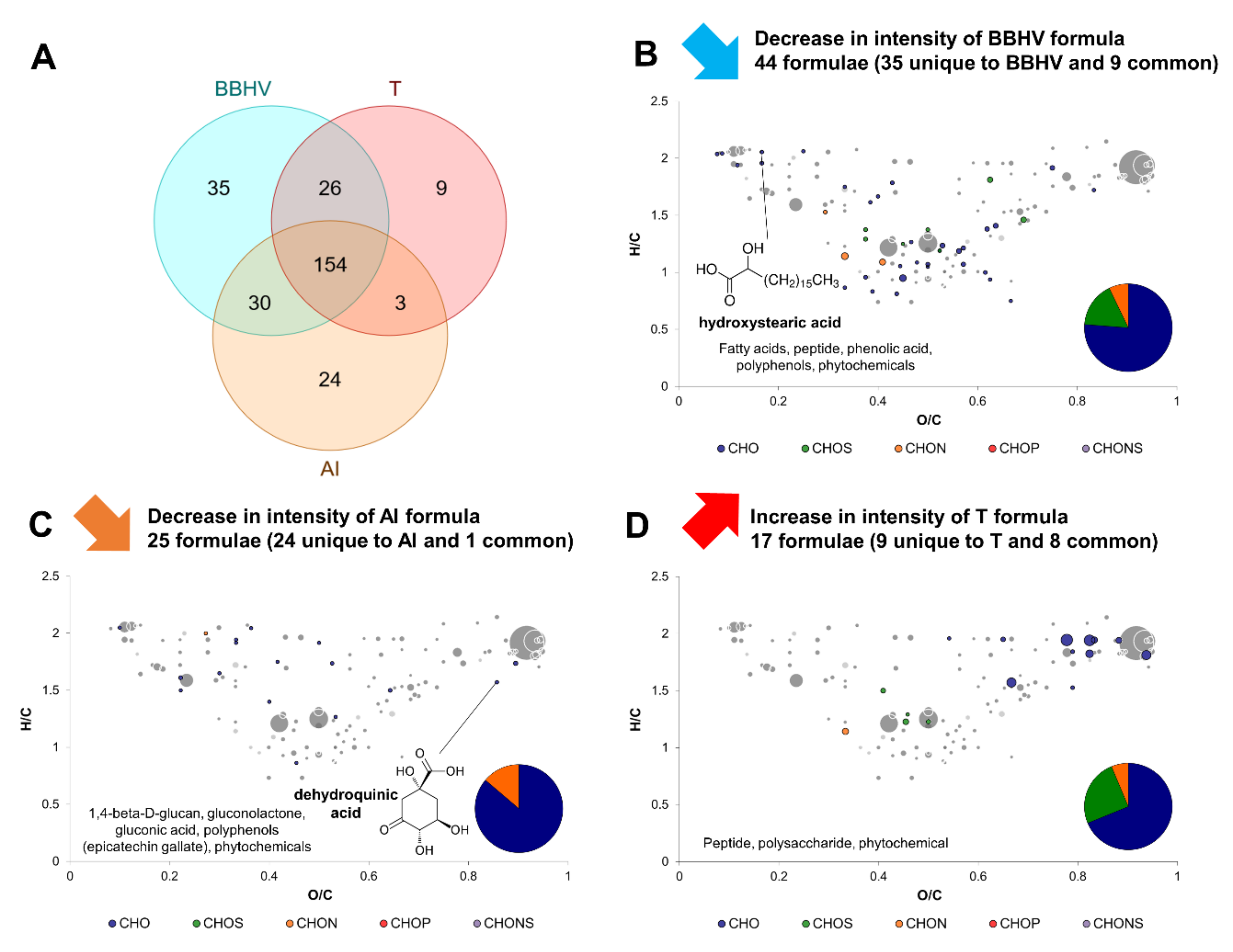
2.5. Complex Interactions Involving Yeast–Yeast and Yeast–Acetic Acid Bacterium Interaction
3. Materials and Methods
3.1. Generation of Biological Samples
3.1.1. Production of Original Black Tea Kombucha
3.1.2. Isolation, Identification, and Selection of Strains
3.1.3. Cultures Conditions and Monitoring
3.2. Sample Preparation
3.3. Fourier Transform-Ion Cyclotron Resonance-Mass Spectrometry (FT-ICR-MS)
3.4. Processing of FT-ICR-MS Data
3.5. Repeatability of FT-ICR-MS Measurements
3.6. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bell, V.; Ferrão, J.; Pimentel, L.; Pintado, M.; Fernandes, T. One Health, Fermented Foods, and Gut Microbiota. Foods 2018, 7, 195. [Google Scholar] [CrossRef] [Green Version]
- Marco, M.L.; Heeney, D.; Binda, S.; Cifelli, C.J.; Cotter, P.D.; Foligné, B.; Gänzle, M.; Kort, R.; Pasin, G.; Pihlanto, A.; et al. Health Benefits of Fermented Foods: Microbiota and Beyond. Curr. Opin. Biotechnol. 2017, 44, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Marco, M.L.; Sanders, M.E.; Gänzle, M.; Arrieta, M.C.; Cotter, P.D.; De Vuyst, L.; Hill, C.; Holzapfel, W.; Lebeer, S.; Merenstein, D.; et al. The International Scientific Association for Probiotics and Prebiotics (ISAPP) Consensus Statement on Fermented Foods. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 196–208. [Google Scholar] [CrossRef]
- Aldrete-Tapia, J.A.; Miranda-Castilleja, D.E.; Arvizu-Medrano, S.M.; Martínez-Peniche, R.Á.; Soto-Muñoz, L.; Hernández-Iturriaga, M. Pursuing the Perfect Performer of Fermented Beverages: GMMs vs. Microbial Consortium, In Frontiers and New Trends in the Science of Fermented Food and Beverages; Solís-Oviedo, R., de la Cruz Pech-Canul, A., Eds.; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.; Grandvalet, C.; Verdier, F.; Martin, A.; Alexandre, H.; Tourdot-Maréchal, R. Microbiological and Technological Parameters Impacting the Chemical Composition and Sensory Quality of Kombucha. Compr. Rev. Food Sci. Food Saf. 2020, 19, 2050–2070. [Google Scholar] [CrossRef] [PubMed]
- Harrison, K.; Curtin, C. Microbial Composition of SCOBY Starter Cultures Used by Commercial Kombucha Brewers in North America. Microorganisms 2021, 9, 1060. [Google Scholar] [CrossRef] [PubMed]
- Villarreal-Soto, S.A.; Beaufort, S.; Bouajila, J.; Souchard, J.-P.; Taillandier, P. Understanding Kombucha Tea Fermentation: A Review. J. Food Sci. 2018, 83, 580–588. [Google Scholar] [CrossRef]
- May, A.; Narayanan, S.; Alcock, J.; Varsani, A.; Maley, C.; Aktipis, A. Kombucha: A Novel Model System for Cooperation and Conflict in a Complex Multi-Species Microbial Ecosystem. PeerJ 2019, 7, e7565. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.; Grandvalet, C.; Verdier, F.; Martin, A.; Alexandre, H.; Tourdot-Maréchal, R. Microbial Dynamics between Yeasts and Acetic Acid Bacteria in Kombucha: Impacts on the Chemical Composition of the Beverage. Foods 2020, 9, 963. [Google Scholar] [CrossRef] [PubMed]
- De Vuyst, L.; Neysens, P. The Sourdough Microflora: Biodiversity and Metabolic Interactions. Trends Food Sci. Technol. 2005, 16, 43–56. [Google Scholar] [CrossRef]
- Leech, J.; Cabrera-Rubio, R.; Walsh, A.M.; Macori, G.; Walsh, C.J.; Barton, W.; Finnegan, L.; Crispie, F.; O’Sullivan, O.; Claesson, M.J.; et al. Fermented-Food Metagenomics Reveals Substrate-Associated Differences in Taxonomy and Health-Associated and Antibiotic Resistance Determinants. mSystems 2020, 5, e00522-20. [Google Scholar] [CrossRef]
- Ivey, M.; Massel, M.; Phister, T.G. Microbial Interactions in Food Fermentations. Annu. Rev. Food Sci. Technol. 2013, 4, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Mashego, M.R.; Rumbold, K.; De Mey, M.; Vandamme, E.; Soetaert, W.; Heijnen, J.J. Microbial Metabolomics: Past, Present and Future Methodologies. Biotechnol. Lett. 2007, 29, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Petitgonnet, C.; Klein, G.L.; Roullier-Gall, C.; Schmitt-Kopplin, P.; Quintanilla-Casas, B.; Vichi, S.; Julien-David, D.; Alexandre, H. Influence of Cell-Cell Contact between L. Thermotolerans and S. Cerevisiae on Yeast Interactions and the Exo-Metabolome. Food Microbiol. 2019, 83, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Roullier-Gall, C.; David, V.; Hemmler, D.; Schmitt-Kopplin, P.; Alexandre, H. Exploring Yeast Interactions through Metabolic Profiling. Sci. Rep. 2020, 10, 6073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Forcisi, S.; Harir, M.; Deleris-Bou, M.; Krieger-Weber, S.; Lucio, M.; Longin, C.; Degueurce, C.; Gougeon, R.D.; Schmitt-Kopplin, P.; et al. New Molecular Evidence of Wine Yeast-Bacteria Interaction Unraveled by Non-Targeted Exometabolomic Profiling. Metabolomics 2016, 12, 69. [Google Scholar] [CrossRef]
- Rivas-Ubach, A.; Liu, Y.; Bianchi, T.S.; Tolić, N.; Jansson, C.; Paša-Tolić, L. Moving beyond the van Krevelen Diagram: A New Stoichiometric Approach for Compound Classification in Organisms. Anal. Chem. 2018, 90, 6152–6160. [Google Scholar] [CrossRef]
- Roullier-Gall, C.; Witting, M.; Gougeon, R.D.; Schmitt-Kopplin, P. High Precision Mass Measurements for Wine Metabolomics. Front. Chem. 2014, 2, 102. [Google Scholar] [CrossRef] [Green Version]
- Roullier-Gall, C.; Witting, M.; Tziotis, D.; Ruf, A.; Gougeon, R.D.; Schmitt-Kopplin, P. Integrating Analytical Resolutions in Non-Targeted Wine Metabolomics. Tetrahedron 2015, 71, 2983–2990. [Google Scholar] [CrossRef]
- Wägele, B.; Witting, M.; Schmitt-Kopplin, P.; Suhre, K. MassTRIX Reloaded: Combined Analysis and Visualization of Transcriptome and Metabolome Data. PLoS ONE 2012, 7, e39860. [Google Scholar] [CrossRef] [Green Version]
- Jayabalan, R.; Marimuthu, S.; Swaminathan, K. Changes in Content of Organic Acids and Tea Polyphenols during Kombucha Tea Fermentation. Food Chem. 2007, 102, 392–398. [Google Scholar] [CrossRef]
- Villarreal-Soto, S.A.; Beaufort, S.; Bouajila, J.; Souchard, J.-P.; Renard, T.; Rollan, S.; Taillandier, P. Impact of Fermentation Conditions on the Production of Bioactive Compounds with Anticancer, Anti-Inflammatory and Antioxidant Properties in Kombucha Tea Extracts. Process Biochem. 2019, 83, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, A.; le Quéré, J.M.; Drilleau, J.F.; Wosiacki, G. Fermentation Behavior of Apiculate Yeast Hanseniaspora Valbyensis in Cider Processing Conditions; International Commission of Agricultural Engineering (CIGR); Institut fur Landtechnik: Bonn, Germany, 2008; p. 20. [Google Scholar]
- Steenwyk, J.L.; Opulente, D.A.; Kominek, J.; Shen, X.-X.; Zhou, X.; Labella, A.L.; Bradley, N.P.; Eichman, B.F.; Čadež, N.; Libkind, D.; et al. Extensive Loss of Cell-Cycle and DNA Repair Genes in an Ancient Lineage of Bipolar Budding Yeasts. PLoS Biol. 2019, 17, e3000255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, K.M.; Zannini, E.; Wilkinson, S.; Daenen, L.; Arendt, E.K. Physiology of Acetic Acid Bacteria and Their Role in Vinegar and Fermented Beverages. Compr. Rev. Food Sci. Food Saf. 2019, 18, 587–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, T.; Grandvalet, C.; Winckler, P.; Verdier, F.; Martin, A.; Alexandre, H.; Tourdot-Maréchal, R. Shedding Light on the Formation and Structure of Kombucha Biofilm Using Two-Photon Fluorescence Microscopy. Front. Microbiol. 2021, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- Braus, G.H. Aromatic Amino Acid Biosynthesis in the Yeast Saccharomyces Cerevisiae: A Model System for the Regulation of a Eukaryotic Biosynthetic Pathway. Microbiol. Rev. 1991, 55, 22. [Google Scholar] [CrossRef] [PubMed]
- van Roermund, C.W.T.; Waterham, H.R.; Ijlst, L.; Wanders, R.J.A. Fatty Acid Metabolism in Saccharomyces Cerevisiae. Cell. Mol. Life Sci. CMLS 2003, 60, 1838–1851. [Google Scholar] [CrossRef]
- Yamada, Y.; Nunoda, M.; Ishikawa, T.; Tahara, Y. The Cellular Fatty Acid Composition in Acetic Acid Bacteria. J. Gen. Appl. Microbiol. 1981, 27, 405–417. [Google Scholar] [CrossRef]
- Cardoso, R.R.; Neto, R.O.; Dos Santos D’Almeida, C.T.; do Nascimento, T.P.; Pressete, C.G.; Azevedo, L.; Martino, H.S.D.; Cameron, L.C.; Ferreira, M.S.L.; Barros, F.A.R. de Kombuchas from Green and Black Teas Have Different Phenolic Profile, Which Impacts Their Antioxidant Capacities, Antibacterial and Antiproliferative Activities. Food Res. Int. 2020, 128, 108782. [Google Scholar] [CrossRef]
- Dueñas, M.; Cueva, C.; Muñoz-González, I.; Jiménez-Girón, A.; Sánchez-Patán, F.; Santos-Buelga, C.; Moreno-Arribas, M.; Bartolomé, B. Studies on Modulation of Gut Microbiota by Wine Polyphenols: From Isolated Cultures to Omic Approaches. Antioxidants 2015, 4, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Serra, S.; De Simeis, D. New Insights on the Baker’s Yeast-Mediated Hydration of Oleic Acid: The Bacterial Contaminants of Yeast Are Responsible for the Stereoselective Formation of ( R )-10-Hydroxystearic Acid. J. Appl. Microbiol. 2018, 124, 719–729. [Google Scholar] [CrossRef]
- Gocho, S.; Tabogami, N.; Inagaki, M.; Kawabata, C.; Komai, T. Biotransformation of Oleic Acid to Optically Active γ-Dodecalactone. Biosci. Biotechnol. Biochem. 1995, 59, 1571–1572. [Google Scholar] [CrossRef]
- Wanikawa, A.; Hosoi, K.; Kato, T. Conversion of Unsaturated Fatty Acids to Precursors of γ-Lactones by Lactic Acid Bacteria during the Production of Malt Whisky. J. Am. Soc. Brew. Chem. 2000, 58, 51–56. [Google Scholar] [CrossRef]
- Romano, A.; Gandolfi, R.; Nitti, P.; Rollini, M.; Molinari, F. Acetic Acid Bacteria as Enantioselective Biocatalysts. J. Mol. Catal. B Enzym. 2002, 17, 235–240. [Google Scholar] [CrossRef]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Adachi, O.; Moonmangmee, D.; Toyama, H.; Yamada, M.; Shinagawa, E.; Matsushita, K. New Developments in Oxidative Fermentation. Appl. Microbiol. Biotechnol. 2003, 60, 643–653. [Google Scholar] [CrossRef]
- Tran, T.; Billet, K.; Torres-Cobos, B.; Vichi, S.; Alexandre, H.; Grandvalet, C.; Tourdot-Maréchal, R. Use of a Minimal Microbial Consortium to Determine the Origin of Kombucha Flavor. Front. Microbiol. 2022. Under press. [Google Scholar]
- Pallmann, C.L.; Brown, J.A.; Olineka, T.L.; Cocolin, L.; Mills, D.A.; Bisson, L.F. Use of WL Medium to Profile Native Flora Fermentations. Am. J. Enol. Vitic. 2001, 52, 198–203. [Google Scholar]
- Cvetković, D.; Markov, S.; Djurić, M.; Savić, D.; Velićanski, A. Specific Interfacial Area as a Key Variable in Scaling-up Kombucha Fermentation. J. Food Eng. 2008, 85, 387–392. [Google Scholar] [CrossRef]
- Tziotis, D.; Hertkorn, N.; Schmitt-Kopplin, P. Kendrick-Analogous Network Visualisation of Ion Cyclotron Resonance Fourier Transform Mass Spectra: Improved Options for the Assignment of Elemental Compositions and the Classification of Organic Molecular Complexity. Eur. J. Mass Spectrom. 2011, 17, 415–421. [Google Scholar] [CrossRef]
- Mazauric, J.-P.; Salmon, J.-M. Interactions between Yeast Lees and Wine Polyphenols during Simulation of Wine Aging: I. Analysis of Remnant Polyphenolic Compounds in the Resulting Wines. J. Agric. Food Chem. 2005, 53, 5647–5653. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, T.; Roullier-Gall, C.; Verdier, F.; Martin, A.; Schmitt-Kopplin, P.; Alexandre, H.; Grandvalet, C.; Tourdot-Maréchal, R. Microbial Interactions in Kombucha through the Lens of Metabolomics. Metabolites 2022, 12, 235. https://doi.org/10.3390/metabo12030235
Tran T, Roullier-Gall C, Verdier F, Martin A, Schmitt-Kopplin P, Alexandre H, Grandvalet C, Tourdot-Maréchal R. Microbial Interactions in Kombucha through the Lens of Metabolomics. Metabolites. 2022; 12(3):235. https://doi.org/10.3390/metabo12030235
Chicago/Turabian StyleTran, Thierry, Chloé Roullier-Gall, François Verdier, Antoine Martin, Philippe Schmitt-Kopplin, Hervé Alexandre, Cosette Grandvalet, and Raphaëlle Tourdot-Maréchal. 2022. "Microbial Interactions in Kombucha through the Lens of Metabolomics" Metabolites 12, no. 3: 235. https://doi.org/10.3390/metabo12030235
APA StyleTran, T., Roullier-Gall, C., Verdier, F., Martin, A., Schmitt-Kopplin, P., Alexandre, H., Grandvalet, C., & Tourdot-Maréchal, R. (2022). Microbial Interactions in Kombucha through the Lens of Metabolomics. Metabolites, 12(3), 235. https://doi.org/10.3390/metabo12030235