
Metabolomics and the Multi-Omics View of Cancer

Abstract
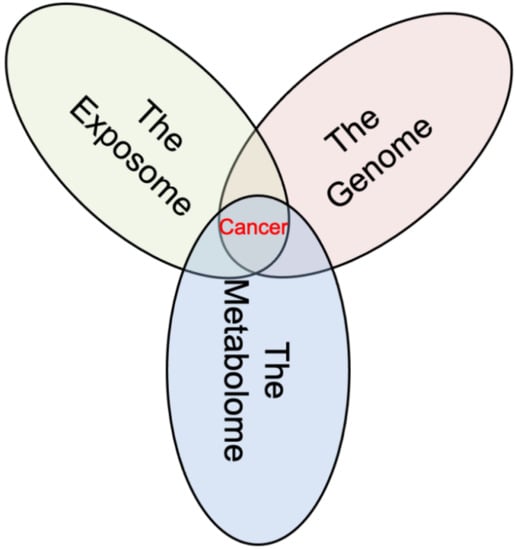
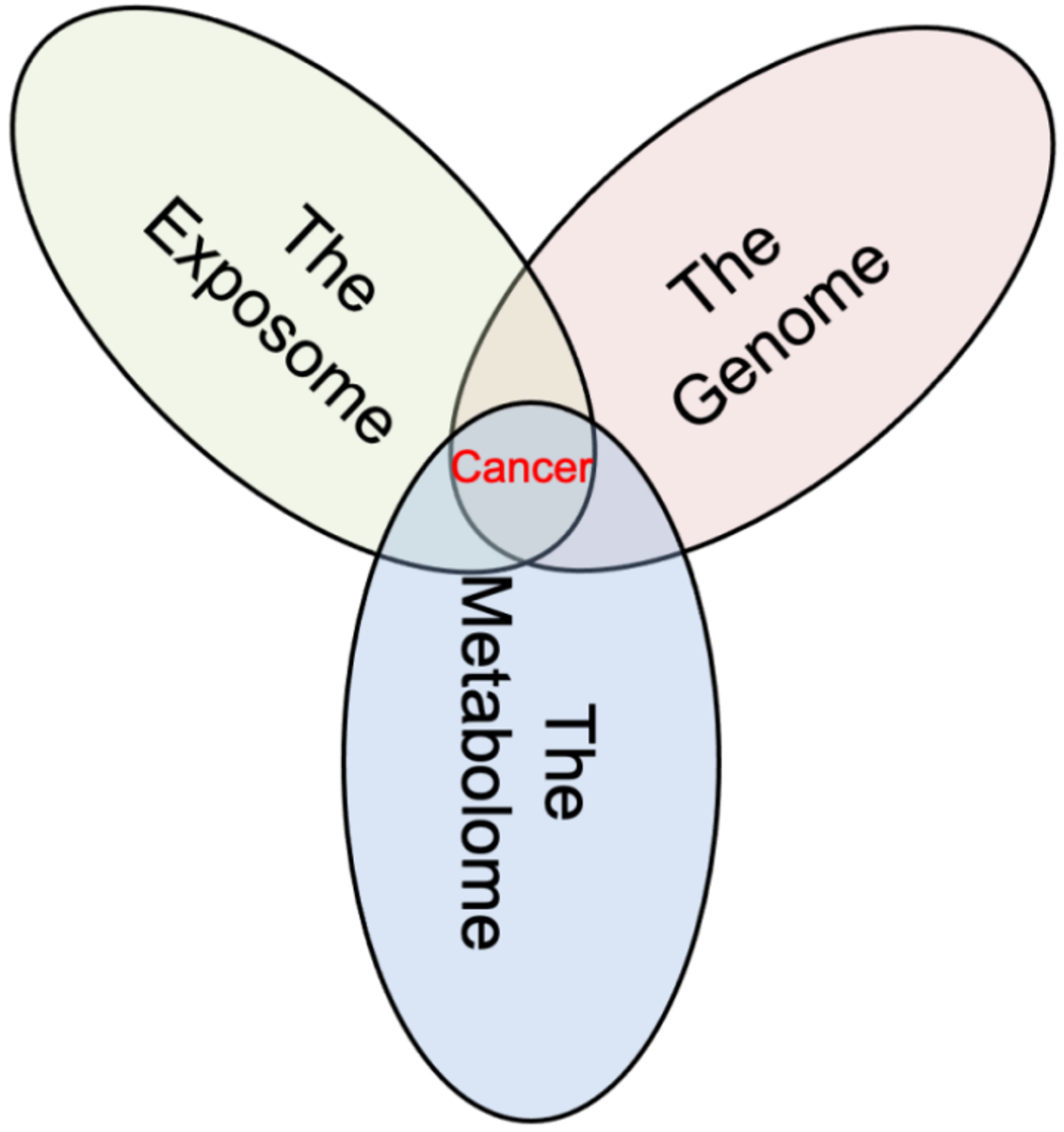
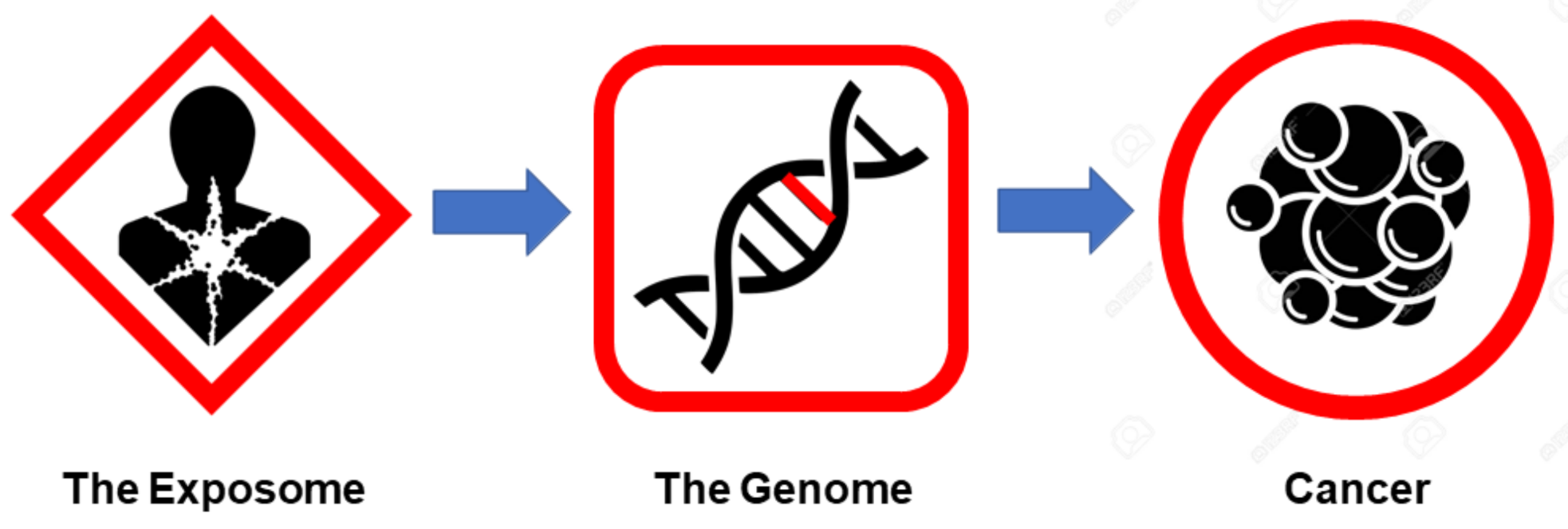
1. Introduction
2. Cancer as a Genetic Disease (the Genome View)
2.1. The Genetics of Familial Cancer
2.2. The Genetics of Sporadic Cancer
3. Cancer as an Environmental Disease (the Exposome View)
4. Cancer as a Metabolic Disease (the Metabolome View)
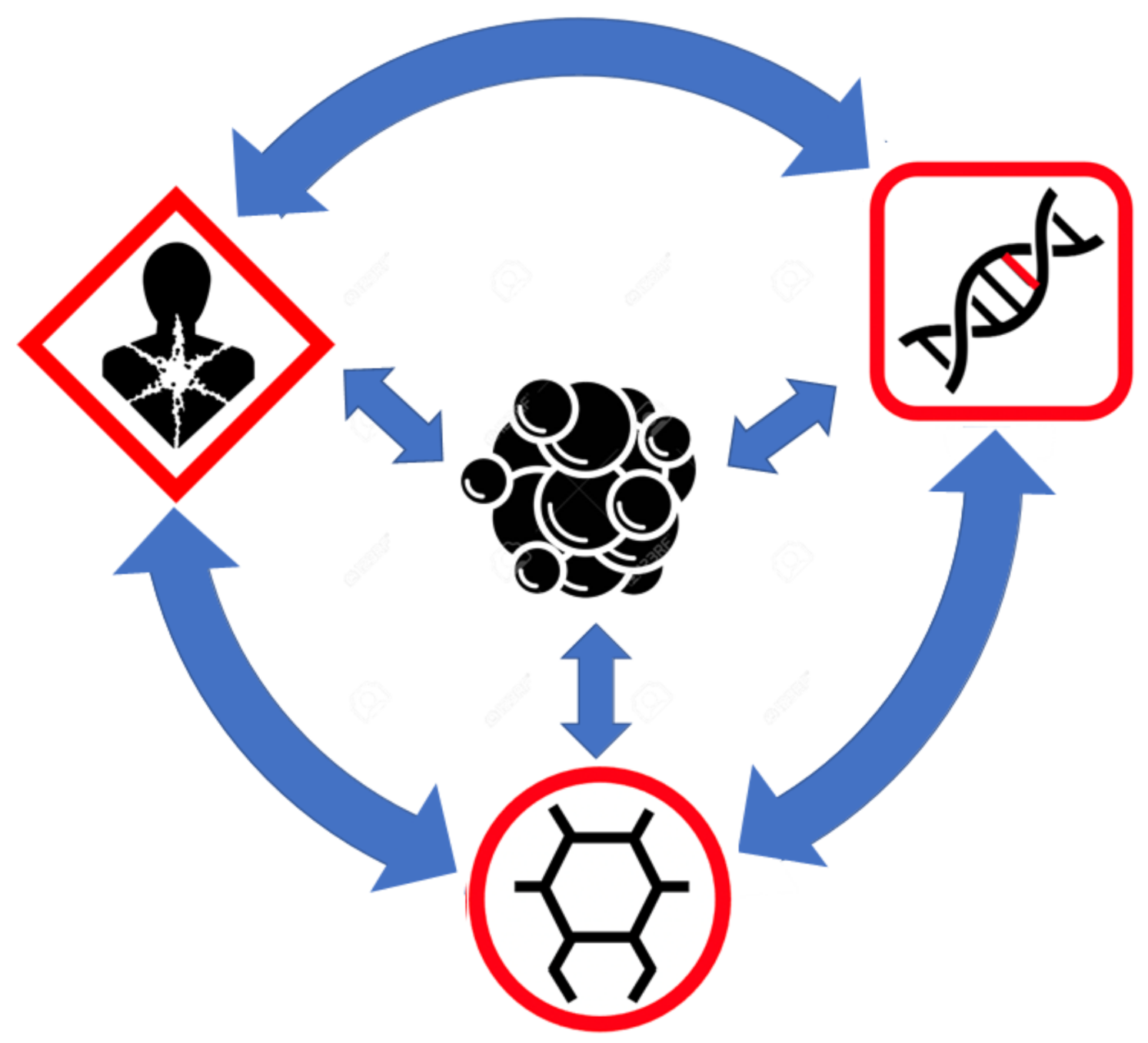
5. Connecting the Multiple Views of Cancer through Metabolomics
6. The Big Picture View of Cancer
Funding
Acknowledgments
Conflicts of Interest
References
- Brown, G. Oncogenes, Proto-oncogenes, and lineage restriction of cancer stem cells. Int. J. Mol. Sci. 2021, 22, 9667. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S. The Emperor of All Maladies; Fourth Estate: London, UK, 2011. [Google Scholar]
- Hadju, S.I. Thoughts about the cause of cancer. Cancer 2006, 106, 1643–1649. [Google Scholar] [CrossRef]
- Faguet, G.B. A brief history of cancer: Age-old milestones underlying our current knowledge database. Int. J. Cancer 2015, 136, 2022–2036. [Google Scholar] [CrossRef]
- Carrel, A.; Ebeling, A.H. The transformation of monocytes into fibroblasts through the action of Rous virus. J. Exp. Med. 1926, 43, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Busk, T.; Clemmesen, J.; Nielsen, A. Twin studies and other genetical investigations in the Danish Cancer Registry. Br. J. Cancer 1948, 2, 156–163. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Comings, D.E. A general theory of carcinogenesis. Proc. Natl. Acad. Sci. USA 1973, 70, 3324–3328. [Google Scholar] [CrossRef] [PubMed]
- Fiala, S. The cancer cell as a stem cell unable to differentiate. A theory of carcinogenesis. Neoplasma 1968, 15, 607–622. [Google Scholar]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Soto, A.M.; Sonnenschein, C. The tissue organization field theory of cancer: A testable replacement for the somatic mutation theory. Bioessays 2011, 33, 332–340. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Chinopoulos, C. Can the mitochondrial metabolic theory explain better the origin and management of cancer than can the somatic mutation theory? Metabolites 2021, 11, 572. [Google Scholar] [CrossRef]
- Wishart, D.S. Is cancer a genetic disease or a metabolic disease? EBioMedicine 2015, 2, 478–479. [Google Scholar] [CrossRef]
- Durham, H.W. Familial cancer of the colon. West. J. Surg. Obstet. Gynecol. 1954, 62, 26–31. [Google Scholar]
- Nagy, R.; Sweet, K.; Eng, C. Highly penetrant hereditary cancer syndromes. Oncogene 2004, 23, 6445–6470. [Google Scholar] [CrossRef]
- Easton, D.; Peto, J. The contribution of inherited predisposition to cancer incidence. Cancer Surv. 1990, 9, 395–416. [Google Scholar]
- Mucci, L.A.; Hjelmborg, J.B.; Harris, J.R.; Czene, K.; Havelick, D.J.; Scheike, T.; Graff, R.E.; Holst, K.; Möller, S.; Unger, R.H.; et al. Nordic twin study of cancer (NorTwinCan) collaboration. Familial risk and heritability of cancer among twins in nordic countries. JAMA 2016, 315, 68–76. [Google Scholar] [CrossRef]
- Clemmensen, S.B.; Harris, J.R.; Mengel-From, J.; Bonat, W.H.; Frederiksen, H.; Kaprio, J.; Hjelmborg, J.V.B. Familial risk and heritability of hematologic malignancies in the nordic twin study of cancer. Cancers 2021, 13, 3023. [Google Scholar] [CrossRef]
- Hemminki, K.; Sundquist, J.; Bermejo, J.L. How common is familial cancer? Ann. Oncol. 2008, 19, 163–167. [Google Scholar] [CrossRef]
- Chen, B.; Zhang, G.; Li, X.; Ren, C.; Wang, Y.; Li, K.; Mok, H.; Cao, L.; Wen, L.; Jia, M.; et al. Comparison of BRCA versus non-BRCA germline mutations and associated somatic mutation profiles in patients with unselected breast cancer. Aging 2020, 12, 3140–3155. [Google Scholar] [CrossRef]
- Momozawa, Y.; Iwasaki, Y.; Parsons, M.T.; Kamatani, Y.; Takahashi, A.; Tamura, C.; Katagiri, T.; Yoshida, T.; Nakamura, S.; Sugano, K.; et al. Germline pathogenic variants of 11 breast cancer genes in 7051 Japanese patients and 11,241 controls. Nat. Commun. 2018, 9, 4083. [Google Scholar] [CrossRef]
- Sun, J.; Meng, H.; Yao, L.; Lv, M.; Bai, J.; Zhang, J.; Wang, L.; Ouyang, T.; Li, J.; Wang, T.; et al. Germline mutations in cancer susceptibility genes in a large series of unselected breast cancer patients. Clin. Cancer Res. 2017, 23, 6113–6119. [Google Scholar] [CrossRef]
- Momozawa, Y.; Iwasaki, Y.; Hirata, M.; Liu, X.; Kamatani, Y.; Takahashi, A.; Sugano, K.; Yoshida, T.; Murakami, Y.; Matsuda, K.; et al. Germline pathogenic variants in 7636 Japanese patients with prostate cancer and 12,366 controls. J. Natl. Cancer Inst. 2020, 112, 369–376. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- Nicolosi, P.; Ledet, E.; Yang, S.; Michalski, S.; Freschi, B.; O’Leary, E.; Esplin, E.D.; Nussbaum, R.L.; Sartor, O. Prevalence of germline variants in prostate cancer and implications for current genetic testing guidelines. JAMA Oncol. 2019, 5, 523–528. [Google Scholar] [CrossRef]
- Hu, X.; Yang, D.; Li, Y.; Li, L.; Wang, Y.; Chen, P.; Xu, S.; Pu, X.; Zhu, W.; Deng, P.; et al. Prevalence and clinical significance of pathogenic germline BRCA1/2 mutations in Chinese non-small cell lung cancer patients. Cancer Biol. Med. 2019, 16, 556–564. [Google Scholar] [CrossRef]
- Liu, M.; Liu, X.; Suo, P.; Gong, Y.; Qu, B.; Peng, X.; Xiao, W.; Li, Y.; Chen, Y.; Zeng, Z.; et al. The contribution of hereditary cancer-related germline mutations to lung cancer susceptibility. Transl. Lung Cancer Res. 2020, 9, 646–658. [Google Scholar] [CrossRef]
- Yang, J.; Li, H.; Lim, B.; Li, W.; Guo, Q.; Hu, L.; Song, Z.; Zhou, B. Profiling oncogenic germline mutations in unselected Chinese lung cancer patients. Front. Oncol. 2021, 11, 647598. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Liu, X.; Iwasaki, Y.; Terao, C.; Mizukami, K.; Kawakami, E.; Takata, S.; Inai, C.; Aoi, T.; Mizukoshi, M.; et al. Population-based Screening for hereditary colorectal cancer variants in Japan. Clin. Gastroenterol. Hepatol. 2020, S1542–3565, 31664–31665. [Google Scholar] [CrossRef]
- DeRycke, M.S.; Gunawardena, S.; Balcom, J.R.; Pickart, A.M.; Waltman, L.A.; French, A.J.; McDonnell, S.; Riska, S.M.; Fogarty, Z.C.; Larson, M.C.; et al. Targeted sequencing of 36 known or putative colorectal cancer susceptibility genes. Mol. Genet. Genomic Med. 2017, 5, 553–569. [Google Scholar] [CrossRef]
- Aoude, L.G.; Gartside, M.; Johansson, P.; Palmer, J.M.; Symmons, J.; Martin, N.G.; Montgomery, G.W.; Hayward, N.K. Prevalence of germline BAP1, CDKN2A and CDK4 mutations in an Australian population-based sample of cutaneous melanoma cases. Twin Res. Hum. Genet. 2015, 18, 126–233. [Google Scholar] [CrossRef] [PubMed]
- Casula, M.; Colombino, M.; Satta, M.P.; Cossu, A.; Lissia, A.; Budroni, M.; Simeone, E.; Calemma, R.; Loddo, C.; Caracò, C.; et al. Factors predicting the occurrence of germline mutations in candidate genes among patients with cutaneous malignant melanoma from South Italy. Eur. J. Cancer. 2007, 43, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Nassar, A.H.; Abou Alaiwi, S.; AlDubayan, S.H.; Moore, N.; Mouw, K.W.; Kwiatkowski, D.J.; Choueiri, T.K.; Curran, C.; Berchuck, J.E.; Harshman, L.C.; et al. Prevalence of pathogenic germline cancer risk variants in high-risk urothelial carcinoma. Genet. Med. 2020, 22, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.J.; Tokaz, M.C.; Jacobs, M.F.; Chinnaiyan, A.M.; Phillips, T.J.; Wilcox, R.A. Germline variants discovered in lymphoma patients undergoing tumor profiling: A case series. Fam. Cancer 2021, 20, 61–65. [Google Scholar] [CrossRef]
- Abou Alaiwi, S.; Nassar, A.H.; Adib, E.; Groha, S.M.; Akl, E.W.; McGregor, B.A.; Esplin, E.D.; Yang, S.; Hatchell, K.; Fusaro, V.; et al. Trans-ethnic variation in germline variants of patients with renal cell carcinoma. Cell Rep. 2021, 34, 108926. [Google Scholar] [CrossRef]
- Long, B.; Lilyquist, J.; Weaver, A.; Hu, C.; Gnanaolivu, R.; Lee, K.Y.; Hart, S.N.; Polley, E.C.; Bakkum-Gamez, J.N.; Couch, F.J.; et al. Cancer susceptibility gene mutations in type I and II endometrial cancer. Gynecol. Oncol. 2019, 152, 20–25. [Google Scholar] [CrossRef]
- Shindo, K.; Yu, J.; Suenaga, M.; Fesharakizadeh, S.; Cho, C.; Macgregor-Das, A.; Siddiqui, A.; Witmer, P.D.; Tamura, K.; Song, T.J.; et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J. Clin. Oncol. 2017, 35, 3382–3390. [Google Scholar] [CrossRef]
- Mezina, A.; Philips, N.; Bogus, Z.; Erez, N.; Xiao, R.; Fan, R.; Olthoff, K.M.; Reddy, K.R.; Samadder, N.J.; Nielsen, S.M.; et al. Multigene panel testing in individuals with hepatocellular carcinoma identifies pathogenic germline variants. JCO Precis. Oncol. 2021, 5, 988–1000. [Google Scholar] [CrossRef]
- Michailidou, K.; Beesley, J.; Lindstrom, S.; Canisius, S.; Dennis, J.; Lush, M.J.; Maranian, M.J.; Bolla, M.K.; Wang, Q.; Shah, M.; et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat. Genet. 2015, 47, 373–380. [Google Scholar] [CrossRef]
- Teerlink, C.C.; Leongamornlert, D.; Dadaev, T.; Thomas, A.; Farnham, J.; Stephenson, R.A.; Riska, S.; McDonnell, S.K.; Schaid, D.J.; Catalona, W.J.; et al. Genome-wide association of familial prostate cancer cases identifies evidence for a rare segregating haplotype at 8q24.21. Hum. Genet. 2016, 135, 923–938. [Google Scholar] [CrossRef]
- Hu, Z.; Wu, C.; Shi, Y.; Guo, H.; Zhao, X.; Yin, Z.; Yang, L.; Dai, J.; Hu, L.; Tan, W.; et al. A genome-wide association study identifies two new lung cancer susceptibility loci at 13q12.12 and 22q12.2 in Han Chinese. Nat. Genet. 2011, 43, 792–796. [Google Scholar] [CrossRef]
- Zhang, B.; Jia, W.H.; Matsuda, K.; Kweon, S.S.; Matsuo, K.; Xiang, Y.B.; Shin, A.; Jee, S.H.; Kim, D.H.; Cai, Q.; et al. Large-scale genetic study in East Asians identifies six new loci associated with colorectal cancer risk. Nat. Genet. 2014, 46, 533–542. [Google Scholar] [CrossRef]
- Bishop, D.T.; Demenais, F.; Iles, M.M.; Harland, M.; Taylor, J.C.; Corda, E.; Randerson-Moor, J.; Aitken, J.F.; Avril, M.F.; Azizi, E.; et al. Genome-wide association study identifies three loci associated with melanoma risk. Nat. Genet. 2009, 41, 920–925. [Google Scholar] [CrossRef]
- Figueroa, J.D.; Ye, Y.; Siddiq, A.; Garcia-Closas, M.; Chatterjee, N.; Prokunina-Olsson, L.; Cortessis, V.K.; Kooperberg, C.; Cussenot, O.; Benhamou, S.; et al. Genome-wide association study identifies multiple loci associated with bladder cancer risk. Hum. Mol. Genet. 2014, 23, 1387–1398. [Google Scholar] [CrossRef]
- Tan, D.E.; Foo, J.N.; Bei, J.X.; Chang, J.; Peng, R.; Zheng, X.; Wei, L.; Huang, Y.; Lim, W.Y.; Li, J.; et al. Genome-wide association study of B cell non-Hodgkin lymphoma identifies 3q27 as a susceptibility locus in the Chinese population. Nat. Genet. 2013, 45, 804–807. [Google Scholar] [CrossRef] [PubMed]
- Purdue, M.P.; Johansson, M.; Zelenika, D.; Toro, J.R.; Scelo, G.; Moore, L.E.; Prokhortchouk, E.; Wu, X.; Kiemeney, L.A.; Gaborieau, V.; et al. Genome-wide association study of renal cell carcinoma identifies two susceptibility loci on 2p21 and 11q13.3. Nat. Genet. 2011, 43, 60–65. [Google Scholar] [CrossRef]
- Cheng, T.H.; Thompson, D.J.; O’Mara, T.A.; Painter, J.N.; Glubb, D.M.; Flach, S.; Lewis, A.; French, J.D.; Freeman-Mills, L.; Church, D.; et al. Five endometrial cancer risk loci identified through genome-wide association analysis. Nat. Genet. 2016, 48, 667–674. [Google Scholar] [CrossRef]
- Childs, E.J.; Mocci, E.; Campa, D.; Bracci, P.M.; Gallinger, S.; Goggins, M.; Li, D.; Neale, R.E.; Olson, S.H.; Scelo, G.; et al. Common variation at 2p13.3, 3q29, 7p13 and 17q25.1 associated with susceptibility to pancreatic cancer. Nat. Genet. 2015, 47, 911–916. [Google Scholar] [CrossRef]
- Gudmundsson, J.; Sulem, P.; Gudbjartsson, D.F.; Jonasson, J.G.; Masson, G.; He, H.; Jonasdottir, A.; Sigurdsson, A.; Stacey, S.N.; Johannsdottir, H.; et al. Discovery of common variants associated with low TSH levels and thyroid cancer risk. Nat. Genet. 2012, 44, 319–322. [Google Scholar] [CrossRef]
- Li, S.; Qian, J.; Yang, Y.; Zhao, W.; Dai, J.; Bei, J.X.; Foo, J.N.; McLaren, P.J.; Li, Z.; Yang, J.; et al. GWAS identifies novel susceptibility loci on 6p21.32 and 21q21.3 for hepatocellular carcinoma in chronic hepatitis B virus carriers. PLoS Genet. 2012, 8, e1002791. [Google Scholar] [CrossRef]
- Chen, F.; Childs, E.J.; Mocci, E.; Bracci, P.; Gallinger, S.; Li, D.; Neale, R.E.; Olson, S.H.; Scelo, G.; Bamlet, W.R.; et al. Analysis of heritability and genetic architecture of pancreatic cancer: A PanC4 study. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Czene, K.; Lichtenstein, P.; Hemminki, K. Environmental and heritable causes of cancer among 9.6 million individuals in the Swedish family-cancer satabase. Int. J. Cancer 2002, 99, 260–266. [Google Scholar] [CrossRef]
- Turati, F.; Edefonti, V.; Talamini, R.; Ferraroni, M.; Malvezzi, M.; Bravi, F.; Franceschi, S.; Montella, M.; Polesel, J.; Zucchetto, A.; et al. Family history of liver cancer and hepatocellular carcinoma. Hepatology 2012, 55, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Seigel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Anand, P.; Kunnumakkara, A.B.; Sundaram, C.; Harikumar, K.B.; Tharakan, S.T.; Lai, O.S.; Sung, B.; Aggarwal, B.B. Cancer is a preventable disease that requires major lifestyle changes. Pharm. Res. 2008, 25, 2097–2116. [Google Scholar] [CrossRef]
- Freeman, H.J. Colorectal cancer risk in Crohn’s disease. World J. Gastroenterol. 2008, 14, 1810–1811. [Google Scholar] [CrossRef]
- Giovannucci, E.; Harlan, D.M.; Archer, M.C.; Bergenstal, R.M.; Gapstur, S.M.; Habel, L.A.; Pollak, M.; Regensteiner, J.G.; Yee, D. Diabetes and cancer: A consensus report. Diabetes Care 2010, 33, 1674–1685. [Google Scholar] [CrossRef]
- Frank, C.; Sundquist, J.; Yu, H.; Hemminki, A.; Hemminki, K. Concordant and discordant familial cancer: Familial risks, proportions and population impact. Int. J. Cancer 2017, 140, 1510–1516. [Google Scholar] [CrossRef]
- Beck, T.; Shorter, T.; Brookes, A.J. GWAS Central: A comprehensive resource for the discovery and comparison of genotype and phenotype data from genome-wide association studies. Nucleic Acids Res. 2020, 48, D933–D940. [Google Scholar] [CrossRef]
- Patron, J.; Serra-Cayuela, A.; Han, B.; Li, C.; Wishart, D.S. Assessing the performance of genome-wide association studies for predicting disease risk. PLoS ONE 2019, 14, e0220215. [Google Scholar] [CrossRef]
- Segalowitz, S.J. Why twin studies really don’t tell us much about human heritability. Behav. Brain Sci. 1999, 22, 904–905. [Google Scholar] [CrossRef]
- Young, A.I. Solving the missing heritability problem. PLoS Genet. 2019, 15, e1008222. [Google Scholar] [CrossRef] [PubMed]
- Krishna Kumar, S.; Feldman, M.W.; Rehkopf, D.H.; Tuljapurkar, S. Limitations of GCTA as a solution to the missing heritability problem. Proc. Natl. Acad. Sci. USA 2016, 113, E61–E70. [Google Scholar] [CrossRef] [PubMed]
- Hallmayer, J.; Cleveland, S.; Torres, A.; Phillips, J.; Cohen, B.; Torigoe, T.; Miller, J.; Fedele, A.; Collins, J.; Smith, K.; et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 2011, 68, 1095–1102. [Google Scholar] [CrossRef]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Hultman, C.; Larsson, H.; Reichenberg, A. The heritability of autism spectrum disorder. JAMA 2017, 318, 1182–1184. [Google Scholar] [CrossRef]
- De Groot, P.M.; Wu, C.C.; Carter, B.W.; Munden, R.F. The epidemiology of lung cancer. Transl. Lung Cancer Res. 2018, 7, 220–233. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, N.; Yin, C.; Zhu, B.; Li, X. Ultraviolet radiation and melanomagenesis: From mechanism to immunotherapy. Front. Oncol. 2020, 10, 951. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef]
- Wild, C.P. Complementing the genome with an "exposome": The outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1847–1850. [Google Scholar] [CrossRef]
- Shah, D.J.; Sachs, R.K.; Wilson, D.J. Radiation-induced cancer: A modern view. Br. J. Radiol. 2012, 85, e1166–e1173. [Google Scholar] [CrossRef]
- Campbell, T.C. The past, present, and future of nutrition and cancer: Part 1-was a nutritional association acknowledged a century ago? Nutr. Cancer 2017, 69, 811–817. [Google Scholar] [CrossRef]
- Neveu, V.; Moussy, A.; Rouaix, H.; Wedekind, R.; Pon, A.; Knox, C.; Wishart, D.S.; Scalbert, A. Exposome-explorer: A manually-curated database on biomarkers of exposure to dietary and environmental factors. Nucleic Acids Res. 2017, 45, D979–D984. [Google Scholar] [CrossRef]
- Neveu, V.; Nicolas, G.; Salek, R.M.; Wishart, D.S.; Scalbert, A. Exposome-explorer 2.0: An update incorporating candidate dietary biomarkers and dietary associations with cancer risk. Nucleic Acids Res. 2020, 48, D908–D912. [Google Scholar] [CrossRef]
- Liao, J.B. Viruses and human cancer. Yale J. Biol. Med. 2006, 79, 115–122. [Google Scholar]
- Mandong, B.M.; Ngbea, J.A.; Raymond, V. Role of parasites in cancer. Niger. J. Med. 2013, 22, 89–92. [Google Scholar]
- Parsonnet, J. Bacterial infection as a cause of cancer. Environ. Health Perspect. 1995, 103, 263–268. [Google Scholar] [CrossRef]
- Maciejewska, A.; Wojtczak, J.; Bielichowska-Cybula, G.; Domańska, A.; Dutkiewicz, J.; Mołocznik, A. Biological effect of wood dust. Med. Pracy 1993, 44, 277–288. [Google Scholar]
- Chen, Y.; Tong, Y.; Yang, C.; Gan, Y.; Sun, H.; Bi, H.; Cao, S.; Yin, X.; Lu, Z. Consumption of hot beverages and foods and the risk of esophageal cancer: A meta-analysis of observational studies. BMC Cancer 2015, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Koritala, B.S.C.; Porter, K.I.; Arshad, O.A.; Gajula, R.P.; Mitchell, H.D.; Arman, T.; Manjanatha, M.G.; Teeguarden, J.; Van Dongen, H.P.A.; McDermott, J.E.; et al. Night shift schedule causes circadian dysregulation of DNA repair genes and elevated DNA damage in humans. J. Pineal Res. 2021, 70, e12726. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative stress in cancer. Cancer Cell. 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Islami, F.; Goding Sauer, A.; Miller, K.D.; Siegel, R.L.; Fedewa, S.A.; Jacobs, E.J.; McCullough, M.L.; Patel, A.V.; Ma, J.; Soerjomataram, I.; et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J. Clin. 2018, 68, 31–54. [Google Scholar] [CrossRef]
- An Update on Cancer Deaths in the United States. Available online: https://www.cdc.gov/cancer/dcpc/research/update-on-cancer-deaths/index.htm (accessed on 31 December 2021).
- Siegel, R.L.; Jacobs, E.J.; Newton, C.C.; Feskanich, D.; Freedman, N.D.; Prentice, R.L.; Jemal, A. Deaths due to cigarette smoking for 12 smoking-related cancers in the United States. JAMA Intern. Med. 2015, 175, 1574–1576. [Google Scholar] [CrossRef]
- Shalo Wilmont, S. Cigarettes still cause a third of U.S. cancer deaths. Am. J. Nurs. 2015, 115, 16. [Google Scholar] [CrossRef]
- Jacobs, E.J.; Newton, C.C.; Carter, B.D.; Feskanich, D.; Freedman, N.D.; Prentice, R.L.; Flanders, W.D. What proportion of cancer deaths in the contemporary United States is attributable to cigarette smoking? Ann. Epidemiol. 2015, 25, 179–182.e1. [Google Scholar] [CrossRef]
- Alvarnas, A.; Alvarnas, J. Obesity and cancer risk: A public health crisis. Am. J. Manag. Care 2019, 25, SP332–SP333. [Google Scholar]
- De Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health. 2020, 8, e180–e190. [Google Scholar] [CrossRef]
- Nelson, D.E.; Jarman, D.W.; Rehm, J.; Greenfield, T.K.; Rey, G.; Kerr, W.C.; Miller, P.; Shield, K.D.; Ye, Y.; Naimi, T.S. Alcohol-attributable cancer deaths and years of potential life lost in the United States. Am. J. Public Health 2013, 103, 641–648. [Google Scholar] [CrossRef]
- Cao, X.; MacNaughton, P.; Laurent, J.C.; Allen, J.G. Radon-induced lung cancer deaths may be overestimated due to failure to account for confounding by exposure to diesel engine exhaust in BEIR VI miner studies. PLoS ONE 2017, 12, e0184298. [Google Scholar] [CrossRef]
- Turner, M.C.; Andersen, Z.J.; Baccarelli, A.; Diver, W.R.; Gapstur, S.M.; Pope, C.A., III; Prada, D.; Samet, J.; Thurston, G.; Cohen, A. Outdoor air pollution and cancer: An overview of the current evidence and public health recommendations. CA Cancer J. Clin. 2020, 25, 460–479. [Google Scholar] [CrossRef]
- Grant, W.B. Air pollution in relation to U.S. cancer mortality rates: An ecological study; likely role of carbonaceous aerosols and polycyclic aromatic hydrocarbons. Anticancer Res. 2009, 29, 3537–3545. [Google Scholar]
- Sunshine, J.E.; Meo, N.; Kassebaum, N.J.; Collison, M.L.; Mokdad, A.H.; Naghavi, M. Association of adverse effects of medical treatment with mortality in the United States: A secondary analysis of the global burden of diseases, injuries, and risk factors study. JAMA Netw. Open 2019, 2, e187041. [Google Scholar] [CrossRef]
- Harding, J.L.; Andes, L.J.; Gregg, E.W.; Cheng, Y.J.; Weir, H.K.; Bullard, K.M.; Burrows, N.R.; Imperatore, G. Trends in cancer mortality among people with vs without diabetes in the USA, 1988–2015. Diabetologia 2020, 63, 75–84. [Google Scholar] [CrossRef]
- Guy, G.P., Jr.; Thomas, C.C.; Thompson, T.; Watson, M.; Massetti, G.M.; Richardson, L.C. Centers for disease control and prevention (CDC). Vital signs: Melanoma incidence and mortality trends and projections—United States, 1982–2030. MMWR Morb. Mortal. Wkly. Rep. 2015, 64, 591–596. [Google Scholar]
- Sinha, R.; Cross, A.J.; Graubard, B.I.; Leitzmann, M.F.; Schatzkin, A. Meat intake and mortality: A prospective study of over half a million people. Arch. Intern. Med. 2009, 169, 562–571. [Google Scholar] [CrossRef]
- Vermeulen, R.; Silverman, D.T.; Garshick, E.; Vlaanderen, J.; Portengen, L.; Steenland, K. Exposure-response estimates for diesel engine exhaust and lung cancer mortality based on data from three occupational cohorts. Environ. Health Perspect. 2014, 122, 172–177. [Google Scholar] [CrossRef]
- Naeem, Z. Second-hand smoke—Ignored implications. Int. J. Health Sci. 2015, 9, V–VI. [Google Scholar] [CrossRef]
- Furuya, S.; Chimed-Ochir, O.; Takahashi, K.; David, A.; Takala, J. Global asbestos disaster. Int. J. Environ. Res. Public Health 2018, 15, 1000. [Google Scholar] [CrossRef]
- Nicholson, W.J.; Perkel, G.; Selikoff, I.J. Occupational exposure to asbestos: Population at risk and projected mortality—1980–2030. Am. J. Ind. Med. 1982, 3, 259–311. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.B.; Galetti, V. Iodine intake as a risk factor for thyroid cancer: A comprehensive review of animal and human studies. Thyroid Res. 2015, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Attributable Fraction: Example Cancers Due to Occupation in the US. Available online: http://www.occupationalcancer.ca/wp-content/uploads/2011/03/Steenland.pdf (accessed on 31 December 2021).
- Zallman, L.; Woolhandler, S.; Himmelstein, D.; Bor, D.H.; McCormick, D. Computed tomography associated cancers and cancer deaths following visits to U.S. emergency departments. Int. J. Health Serv. 2012, 42, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Aunan, J.R.; Cho, W.C.; Søreide, K. The biology of aging and cancer: A brief overview of shared and divergent molecular hallmarks. Aging Dis. 2017, 8, 628–642. [Google Scholar] [CrossRef] [PubMed]
- Støer, N.C.; Botteri, E.; Thoresen, G.H.; Karlstad, Ø.; Weiderpass, E.; Friis, S.; Pottegård, A.; Andreassen, B.K. Drug use and cancer risk: A drug-wide association study (DWAS) in Norway. Cancer Epidemiol. Biomark. Prev. 2021, 30, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Wen, C.P.; Tsai, S.P.; Chow, W.H.; Wen, C.; Ye, Y.; Zhao, H.; Tsai, M.K.; Huang, M.; Dinney, C.P.; et al. Cancer risk associated with chronic diseases and disease markers: Prospective cohort study. BMJ 2018, 360, k134. [Google Scholar] [CrossRef] [PubMed]
- Ong, J.S.; An, J.; Law, M.H.; Whiteman, D.C.; Neale, R.E.; Gharahkhani, P.; MacGregor, S. Height and overall cancer risk and mortality: Evidence from a Mendelian randomisation study on 310,000 UK Biobank participants. Br. J. Cancer 2018, 118, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef]
- Emmons, K.M.; Colditz, G.A. Realizing the potential of cancer prevention—The role of implementation science. N. Engl. J. Med. 2017, 376, 986–990. [Google Scholar] [CrossRef]
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Transl. Med. 2018, 7, 11. [Google Scholar] [CrossRef]
- DeVita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 2016, 15, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.B. Metabolic enzymes as oncogenes or tumor suppressors. N. Engl. J. Med. 2009, 360, 813–815. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020, 5, 124. [Google Scholar] [CrossRef]
- Wishart, D.S. Metabolomics for investigating physiological and pathophysiological processes. Physiol. Rev. 2019, 99, 1819–1875. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Ann. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Altenberg, B.; Greulich, K.O. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics 2004, 84, 1014–1020. [Google Scholar] [CrossRef]
- Shuch, B.; Linehan, W.M.; Srinivasan, R. Aerobic glycolysis: A novel target in kidney cancer. Expert Rev. Anticancer Ther. 2013, 13, 711–719. [Google Scholar] [CrossRef]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene. 2016, 35, 3619–3625. [Google Scholar] [CrossRef]
- Goetzman, E.S.; Prochownik, E.V. The role for Myc in coordinating glycolysis, oxidative phosphorylation, glutaminolysis, and fatty acid metabolism in normal and neoplastic tissues. Front. Endocrinol. 2018, 9, 129. [Google Scholar] [CrossRef]
- Li, A.M.; Ye, J. Reprogramming of serine, glycine and one-carbon metabolism in cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165841. [Google Scholar] [CrossRef]
- Porporato, P.E. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis 2016, 5, e200. [Google Scholar] [CrossRef]
- Argiles, J.; Costelli, P.; Carbo, N.; Lopezsoriano, F. Branched-chain amino acid catabolism and cancer cachexia (review). Oncol. Rep. 1996, 3, 687–690. [Google Scholar] [CrossRef]
- Aoyagi, T.; Terracina, K.P.; Raza, A.; Matsubara, H.; Takabe, K. Cancer cachexia, mechanism and treatment. World J. Gastrointest. Oncol. 2015, 7, 17–29. [Google Scholar] [CrossRef]
- Lieu, E.L.; Nguyen, T.; Rhyne, S.; Kim, J. Amino acids in cancer. Exp. Mol. Med. 2020, 52, 15–30. [Google Scholar] [CrossRef]
- Yang, M.; Soga, T.; Pollard, P. J. Oncometabolites: Linking altered metabolism with cancer. J. Clin. Investig. 2013, 123, 3652–3658. [Google Scholar] [CrossRef]
- Seok, J.; Yoon, S.H.; Lee, S.H.; Jung, J.H.; Lee, Y.M. The oncometabolite d-2-hydroxyglutarate induces angiogenic activity through the vascular endothelial growth factor receptor 2 signaling pathway. Int. J. Oncol. 2019, 54, 753–763. [Google Scholar] [CrossRef]
- Yang, Z.; Jiang, B.; Wang, Y.; Ni, H.; Zhang, J.; Xia, J.; Shi, M.; Hung, L.M.; Ruan, J.; Mak, T.W.; et al. 2-HG inhibits necroptosis by stimulating DNMT1-dependent hypermethylation of the RIP3 promoter. Cell Rep. 2017, 19, 1846–1857. [Google Scholar] [CrossRef]
- Richardson, L.G.; Choi, B.D.; Curry, W.T. (R)-2-hydroxyglutarate drives immune quiescence in the tumor microenvironment of IDH-mutant gliomas. Transl. Cancer Res. 2019, 8, S167–S170. [Google Scholar] [CrossRef]
- Carbonneau, M.; Gagné, L.; Lalonde, M.E.; Germain, M.A.; Motorina, A.; Guiot, M.C.; Secco, B.; Vincent, E.E.; Tumber, A.; Hulea, L.; et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat. Commun. 2016, 7, 12700. [Google Scholar] [CrossRef]
- Al-Koussa, H.; El Mais, N.; Maalouf, H.; Abi-Habib, R.; El-Sibai, M. Arginine deprivation: A potential therapeutic for cancer cell metastasis? A review. Cancer Cell Int. 2020, 20, 150. [Google Scholar] [CrossRef]
- Jiang, J.; Batra, S.; Zhang, J. Asparagine: A metabolite to be targeted in cancers. Metabolites 2021, 11, 402. [Google Scholar] [CrossRef]
- Glunde, K.; Bhujwalla, Z.M.; Ronen, S.M. Choline metabolism in malignant transformation. Nat. Rev. Cancer. 2011, 11, 835–848. [Google Scholar] [CrossRef]
- Sen, S.; Kawahara, B.; Mahata, S.K.; Tsai, R.; Yoon, A.; Hwang, L.; Hu-Moore, K.; Villanueva, C.; Vajihuddin, A.; Parameshwar, P.; et al. Cystathionine: A novel oncometabolite in human breast cancer. Arch. Biochem. Biophys. 2016, 604, 95–102. [Google Scholar] [CrossRef]
- Ajouz, H.; Mukherji, D.; Shamseddine, A. Secondary bile acids: An underrecognized cause of colon cancer. World J. Surg. Oncol. 2014, 12, 164. [Google Scholar] [CrossRef]
- Paz, E.A.; LaFleur, B.; Gerner, E.W. Polyamines are oncometabolites that regulate the LIN28/let-7 pathway in colorectal cancer cells. Mol. Carcinog. 2014, 53, E96–E106. [Google Scholar] [CrossRef]
- Rodriguez, A.C.; Blanchard, Z.; Maurer, K.A.; Gertz, J. Estrogen signaling in endometrial cancer: A key oncogenic pathway with several open questions. Horm. Cancer 2019, 10, 51–63. [Google Scholar] [CrossRef]
- Yang, M.; Soga, T.; Pollard, P.J.; Adam, J. The emerging role of fumarate as an oncometabolite. Front. Oncol. 2012, 2, 85. [Google Scholar] [CrossRef] [PubMed]
- Agopian, J.; Da Costa, Q.; Nguyen, Q.V.; Scorrano, G.; Kousteridou, P.; Yuan, M.; Chelbi, R.; Goubard, A.; Castellano, R.; Maurizio, J.; et al. GlcNAc is a mast-cell chromatin-remodeling oncometabolite that promotes systemic mastocytosis aggressiveness. Blood 2021, 138, 1590–1602. [Google Scholar] [CrossRef] [PubMed]
- Hochwald, J.S.; Zhang, J. Glucose oncometabolism of esophageal cancer. Anticancer Agents Med. Chem. 2017, 17, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Park, K.G. Targeting glutamine metabolism for cancer treatment. Biomol. Ther. 2018, 26, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Shim, E.H.; Livi, C.B.; Rakheja, D.; Tan, J.; Benson, D.; Parekh, V.; Kho, E.Y.; Ghosh, A.P.; Kirkman, R.; Velu, S.; et al. L-2-Hydroxyglutarate: An epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov. 2014, 4, 1290–1298. [Google Scholar] [CrossRef]
- Beyoğlu, D.; Idle, J.R. Metabolic rewiring and the characterization of oncometabolites. Cancers 2021, 13, 2900. [Google Scholar] [CrossRef]
- Hasan, T.; Arora, R.; Bansal, A.K.; Bhattacharya, R.; Sharma, G.S.; Singh, L.R. Disturbed homocysteine metabolism is associated with cancer. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Gao, P.; Yang, C.; Nesvick, C.L.; Feldman, M.J.; Sizdahkhani, S.; Liu, H.; Chu, H.; Yang, F.; Tang, L.; Tian, J.; et al. Hypotaurine evokes a malignant phenotype in glioma through aberrant hypoxic signaling. Oncotarget 2016, 7, 15200–15214. [Google Scholar] [CrossRef]
- Sivanand, S.; Vander Heiden, M.G. Emerging roles for branched-chain amino acid metabolism in cancer. Cancer Cell. 2020, 37, 147–156. [Google Scholar] [CrossRef]
- Venkateswaran, N.; Conacci-Sorrell, M. Kynurenine: An oncometabolite in colon cancer. Cell Stress 2020, 4, 24–26. [Google Scholar] [CrossRef]
- De la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef]
- Wanders, D.; Hobson, K.; Ji, X. Methionine restriction and cancer biology. Nutrients 2020, 12, 684. [Google Scholar] [CrossRef]
- Nokin, M.J.; Bellier, J.; Durieux, F.; Peulen, O.; Rademaker, G.; Gabriel, M.; Monseur, C.; Charloteaux, B.; Verbeke, L.; van Laere, S.; et al. Methylglyoxal, a glycolysis metabolite, triggers metastasis through MEK/ERK/SMAD1 pathway activation in breast cancer. Breast Cancer Res. 2019, 21, 11. [Google Scholar] [CrossRef] [PubMed]
- Forny, P.; Hochuli, M.; Rahman, Y.; Deheragoda, M.; Weber, A.; Baruteau, J.; Grunewald, S. Liver neoplasms in methylmalonic aciduria: An emerging complication. J. Inherit. Metab Dis. 2019, 42, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Lala, P.K.; Chakraborty, C. Role of nitric oxide in carcinogenesis and tumour progression. Lancet Oncol. 2001, 2, 149–156. [Google Scholar] [CrossRef]
- Mierzejewska, P.; Kunc, M.; Zabielska-Kaczorowska, M.A.; Kutryb-Zajac, B.; Pelikant-Malecka, I.; Braczko, A.; Jablonska, P.; Romaszko, P.; Koszalka, P.; Szade, J.; et al. An unusual nicotinamide derivative, 4-pyridone-3-carboxamide ribonucleoside (4PYR), is a novel endothelial toxin and oncometabolite. Exp. Mol. Med. 2021, 53, 1402–1412. [Google Scholar] [CrossRef]
- Patel, R.; Raj, A.K.; Lokhande, K.B.; Almasri, M.A.; Alzahrani, K.J.; Almeslet, A.S.; Swamy, K.V.; Sarode, G.S.; Sarode, S.C.; Patil, S.; et al. Detection of nail oncometabolite SAICAR in oral cancer patients and its molecular interactions with PKM2 enzyme. Int. J. Environ. Res. Public Health 2021, 18, 11225. [Google Scholar] [CrossRef]
- Kanaan, Y.M.; Sampey, B.P.; Beyene, D.; Esnakula, A.K.; Naab, T.J.; Ricks-Santi, L.J.; Dasi, S.; Day, A.; Blackman, K.W.; Frederick, W.; et al. Metabolic profile of triple-negative breast cancer in African-American women reveals potential biomarkers of aggressive disease. Cancer Genom. Proteom. 2014, 11, 279–294. [Google Scholar]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef]
- Nowicki, S.; Gottlieb, E. Oncometabolites: Tailoring our genes. FEBS J. 2015, 282, 2796–2805. [Google Scholar] [CrossRef]
- Yang, F.; Li, J.; Deng, H.; Wang, Y.; Lei, C.; Wang, Q.; Xiang, J.; Liang, L.; Xia, J.; Pan, X.; et al. GSTZ1-1 Deficiency activates NRF2/IGF1R axis in HCC via accumulation of oncometabolite succinylacetone. EMBO J. 2019, 38, e101964. [Google Scholar] [CrossRef]
- Mi, S.; Gong, L.; Sui, Z. Friend or foe? An unrecognized role of uric acid in cancer development and the potential anticancer effects of uric acid-lowering drugs. J. Cancer 2020, 11, 5236–5244. [Google Scholar] [CrossRef]
- Croteau, E.; Renaud, J.M.; Richard, M.A.; Ruddy, T.D.; Bénard, F.; deKemp, R.A. PET metabolic biomarkers for cancer. Biomark. Cancer 2016, 8, 61–69. [Google Scholar] [CrossRef]
- Bamji-Stocke, S.; van Berkel, V.; Miller, D.M.; Frieboes, H.B. A review of metabolism-associated biomarkers in lung cancer diagnosis and treatment. Metabolomics 2018, 14, 81. [Google Scholar] [CrossRef]
- Erben, V.; Bhardwaj, M.; Schrotz-King, P.; Brenner, H. Metabolomics biomarkers for detection of colorectal neoplasms: A systematic review. Cancers 2018, 10, 246. [Google Scholar] [CrossRef]
- Dinges, S.S.; Hohm, A.; Vandergrift, L.A.; Nowak, J.; Habbel, P.; Kaltashov, I.A.; Cheng, L.L. Cancer metabolomic markers in urine: Evidence, techniques and recommendations. Nat. Rev. Urol. 2019, 16, 339–362. [Google Scholar] [CrossRef]
- Simińska, E.; Koba, M. Amino acid profiling as a method of discovering biomarkers for early diagnosis of cancer. Amino Acids. 2016, 48, 1339–1345. [Google Scholar] [CrossRef]
- Lee, S.H.; Mahendran, R.; Tham, S.M.; Thamboo, T.P.; Chionh, B.J.; Lim, Y.X.; Tsang, W.C.; Wu, Q.H.; Chia, J.Y.; Tay, M.H.W.; et al. Tryptophan-kynurenine ratio as a biomarker of bladder cancer. BJU Int. 2021, 127, 445–453. [Google Scholar] [CrossRef]
- Wang, W.; Tian, S.L.; Jin, D.; Liu, B.; Wang, W.; Chang, H.; Chen, C.; Yu, Z.; Wang, Y.Z.; Li, Y.L. The role of bile acid subtypes in the diagnosis of cholangiocarcinoma. Asia Pac. J. Clin. Oncol. 2021. [Google Scholar] [CrossRef]
- Hu, X.; Walker, D.I.; Liang, Y.; Smith, M.R.; Orr, M.L.; Juran, B.D.; Ma, C.; Uppal, K.; Koval, M.; Martin, G.S.; et al. A scalable workflow to characterize the human exposome. Nat. Commun. 2021, 12, 5575. [Google Scholar] [CrossRef]
- Chung, M.K.; Kannan, K.; Louis, G.M.; Patel, C.J. Toward capturing the exposome: Exposure biomarker variability and coexposure patterns in the shared environment. Environ. Sci. Technol. 2018, 52, 8801–8810. [Google Scholar] [CrossRef]
- Sexton, K.; Adgate, J.L.; Fredrickson, A.L.; Ryan, A.D.; Needham, L.L.; Ashley, D.L. Using biologic markers in blood to assess exposure to multiple environmental chemicals for inner-city children 3–6 years of age. Environ. Health Perspect. 2006, 114, 453–459. [Google Scholar] [CrossRef]
- Kirman, C.R.; Aylward, L.L.; Blount, B.C.; Pyatt, D.W.; Hays, S.M. Evaluation of NHANES biomonitoring data for volatile organic chemicals in blood: Application of chemical-specific screening criteria. J. Expo. Sci. Environ. Epidemiol. 2012, 22, 24–34. [Google Scholar] [CrossRef]
- Dresen, S.; Ferreirós, N.; Gnann, H.; Zimmermann, R.; Weinmann, W. Detection and identification of 700 drugs by multi-target screening with a 3200 Q TRAP LC-MS/MS system and library searching. Anal. Bioanal. Chem. 2010, 396, 2425–2434. [Google Scholar] [CrossRef]
- Rappaport, S.M.; Barupal, D.K.; Wishart, D.; Vineis, P.; Scalbert, A. The blood exposome and its role in discovering causes of disease. Environ. Health Perspect. 2014, 122, 769–774. [Google Scholar] [CrossRef]
- Wang, A.; Gerona, R.R.; Schwartz, J.M.; Lin, T.; Sirota, M.; Morello-Frosch, R.; Woodruff, T.J. A suspect screening method for characterizing multiple chemical exposures among a demographically diverse population of pregnant women in San Francisco. Environ. Health Perspect. 2018, 126, 077009. [Google Scholar] [CrossRef]
- Walker, D.I.; Juran, B.D.; Cheung, A.C.; Schlicht, E.M.; Liang, Y.; Niedzwiecki, M.; LaRusso, N.F.; Gores, G.J.; Jones, D.P.; Miller, G.W.; et al. High-resolution exposomics and metabolomics reveals specific associations in cholestatic liver diseases. Hepatol. Commun. 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Kowalska, G. The safety assessment of toxic metals in commonly used herbs, spices, tea, and coffee in Poland. Int. J. Environ. Res. Public Health. 2021, 18, 5779. [Google Scholar] [CrossRef]
- Li, X.; Tian, T.; Shang, X.; Zhang, R.; Xie, H.; Wang, X.; Wang, H.; Xie, Q.; Chen, J.; Kadokami, K. Occurrence and health risks of organic micro-pollutants and metals in groundwater of Chinese rural areas. Environ. Health Perspect. 2020, 128, 107010. [Google Scholar] [CrossRef]
- Maruvada, P.; Lampe, J.W.; Wishart, D.S.; Barupal, D.; Chester, D.N.; Dodd, D.; Djoumbou-Feunang, Y.; Dorrestein, P.C.; Dragsted, L.O.; Draper, J.; et al. Perspective: Dietary biomarkers of intake and exposure-exploration with omics approaches. Adv. Nutr. 2020, 11, 200–215. [Google Scholar] [CrossRef]
- Dragsted, L.O.; Gao, Q.; Scalbert, A.; Vergères, G.; Kolehmainen, M.; Manach, C.; Brennan, L.; Afman, L.A.; Wishart, D.S.; Andres Lacueva, C.; et al. Validation of biomarkers of food intake-critical assessment of candidate biomarkers. Genes Nutr. 2018, 13, 14. [Google Scholar] [CrossRef] [PubMed]
- Loftfield, E.; Stepien, M.; Viallon, V.; Trijsburg, L.; Rothwell, J.A.; Robinot, N.; Biessy, C.; Bergdahl, I.A.; Bodén, S.; Schulze, M.B.; et al. Novel biomarkers of habitual alcohol intake and associations with risk of pancreatic and liver cancers and liver disease mortality. J. Natl. Cancer Inst. 2021, 113, 1542–1550. [Google Scholar] [CrossRef]
- Schmidt, J.A.; Fensom, G.K.; Rinaldi, S.; Scalbert, A.; Gunter, M.J.; Holmes, M.V.; Key, T.J.; Travis, R.C. NMR metabolite profiles in male meat-eaters, fish-eaters, vegetarians and vegans, and comparison with MS metabolite profiles. Metabolites 2021, 11, 121. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, J.A.; Murphy, N.; Bešević, J.; Kliemann, N.; Jenab, M.; Ferrari, P.; Achaintre, D.; Gicquiau, A.; Vozar, B.; Scalbert, A.; et al. Metabolic signatures of healthy lifestyle patterns and colorectal cancer risk in a european cohort. Clin. Gastroenterol. Hepatol. 2020, S1542–3565, 31635–31639. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Kuo, T.C.; Kuo, H.C.; Tseng, Y.J.; Kuo, C.H.; Yuan, T.H.; Chan, C.C. Metabolomics of children and adolescents exposed to industrial carcinogenic pollutants. Environ. Sci. Technol. 2019, 53, 5454–5465. [Google Scholar] [CrossRef]
- Chen, C.S.; Kuo, T.C.; Kuo, H.C.; Tseng, Y.J.; Kuo, C.H.; Yuan, T.H.; Chan, C.C. Lipidomics of children and adolescents exposed to multiple industrial pollutants. Environ. Res. 2021, 201, 111448. [Google Scholar] [CrossRef]
- Wang, Z.; Zheng, Y.; Zhao, B.; Zhang, Y.; Liu, Z.; Xu, J.; Chen, Y.; Yang, Z.; Wang, F.; Wang, H.; et al. Human metabolic responses to chronic environmental polycyclic aromatic hydrocarbon exposure by a metabolomic approach. J. Proteome Res. 2015, 14, 2583–2593. [Google Scholar] [CrossRef]
- Orešič, M.; McGlinchey, A.; Wheelock, C.E.; Hyötyläinen, T. Metabolic signatures of the exposome-quantifying the impact of exposure to environmental chemicals on human health. Metabolites 2020, 10, 454. [Google Scholar] [CrossRef]
- Bessonneau, V.; Rudel, R.A. Mapping the human exposome to uncover the causes of breast cancer. Int. J. Environ. Res. Public Health 2019, 17, 189. [Google Scholar] [CrossRef]
- Deng, L.; Chang, D.; Foshaug, R.R.; Eisner, R.; Tso, V.K.; Wishart, D.S.; Fedorak, R.N. Development and validation of a high-throughput mass spectrometry based urine metabolomic test for the detection of colonic adenomatous polyps. Metabolites 2017, 7, 32. [Google Scholar] [CrossRef]
- Tsoli, M.; Daskalakis, K.; Kassi, E.; Kaltsas, G.; Tsolakis, A.V. A critical appraisal of contemporary and novel biomarkers in pheochromocytomas and adrenocortical tumors. Biology 2021, 10, 580. [Google Scholar] [CrossRef]
- Weber, D.D.; Aminzadeh-Gohari, S.; Tulipan, J.; Catalano, L.; Feichtinger, R.G.; Kofler, B. Ketogenic diet in the treatment of cancer—Where do we stand? Mol. Metab. 2020, 33, 102–121. [Google Scholar] [CrossRef]
- Maddocks, O.D.; Berkers, C.R.; Mason, S.M.; Zheng, L.; Blyth, K.; Gottlieb, E.; Vousden, K.H. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013, 493, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Brandhorst, S.; Longo, V.D. Fasting and caloric restriction in cancer prevention and treatment. Recent results. Cancer Res. 2016, 207, 241–266. [Google Scholar] [CrossRef]
- Clifton, K.K.; Ma, C.X.; Fontana, L.; Peterson, L.L. Intermittent fasting in the prevention and treatment of cancer. CA Cancer J. Clin. 2021, 71, 527–546. [Google Scholar] [CrossRef]
- Butler, M.; van der Meer, L.T.; van Leeuwen, F.N. Amino acid depletion therapies: Starving cancer cells to death. Trends Endocrinol. Metab. 2021, 32, 367–381. [Google Scholar] [CrossRef]
- Ribeiro, M.D.; Silva, A.S.; Bailey, K.M.; Kumar, N.B.; Sellers, T.A.; Gatenby, R.A.; Ibrahim-Hashim, A.; Gillies, R.J. Buffer therapy for cancer. J. Nutr. Food Sci. 2012, 2, 6. [Google Scholar] [CrossRef]
- Ibrahim-Hashim, A.; Abrahams, D.; Enriquez-Navas, P.M.; Luddy, K.; Gatenby, R.A.; Gillies, R.J. Tris-base buffer: A promising new inhibitor for cancer progression and metastasis. Cancer Med. 2017, 6, 1720–1729. [Google Scholar] [CrossRef]
- Yang, M.; Zhong, X.; Yuan, Y. Does baking soda function as a magic bullet for patients with cancer? A mini review. Integr. Cancer Ther. 2020, 19, 1534735420922579. [Google Scholar] [CrossRef]
- Gonzalez, C.A.; Riboli, E. Diet and cancer prevention: Contributions from the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Eur. J. Cancer 2010, 46, 2555–2562. [Google Scholar] [CrossRef]
- Geijsen, A.J.M.R.; Brezina, S.; Keski-Rahkonen, P.; Baierl, A.; Bachleitner-Hofmann, T.; Bergmann, M.M.; Boehm, J.; Brenner, H.; Chang-Claude, J.; van Duijnhoven, F.J.B.; et al. Plasma metabolites associated with colorectal cancer: A discovery-replication strategy. Int. J. Cancer 2019, 145, 1221–1231. [Google Scholar] [CrossRef]
- Weir, T.L.; Manter, D.K.; Sheflin, A.M.; Barnett, B.A.; Heuberger, A.L.; Ryan, E.P. Stool microbiome and metabolome differences between colorectal cancer patients and healthy adults. PLoS ONE 2013, 8, e70803. [Google Scholar] [CrossRef]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef]
- Pham, C.H.; Lee, J.E.; Yu, J.; Lee, S.H.; Yu, K.R.; Hong, J.; Cho, N.; Kim, S.; Kang, D.; Lee, S.; et al. Anticancer effects of propionic acid inducing cell death in cervical cancer cells. Molecules 2021, 26, 4951. [Google Scholar] [CrossRef]
- Sreedhar, A.; Zhao, Y. Dysregulated metabolic enzymes and metabolic reprogramming in cancer cells. Biomed. Rep. 2018, 8, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and metabolism. J. Mol. Cell. Biol. 2019, 11, 284–292. [Google Scholar] [CrossRef]
- Huang, R.; Liu, X.; Li, H.; Zhou, Y.; Zhou, P.K. Integrated analysis of transcriptomic and metabolomic profiling reveal the p53 associated pathways underlying the response to ionizing radiation in HBE cells. Cell. Biosci. 2020, 10, 56. [Google Scholar] [CrossRef]
- Zhu, Y.; Piao, C.; Zhang, Z.; Jiang, Y.; Kong, C. The potential role of c-MYC and polyamine metabolism in multiple drug resistance in bladder cancer investigated by metabonomics. Genomics 2021, 114, 125–137. [Google Scholar] [CrossRef]
- Dogra, R.; Bhatia, R.; Shankar, R.; Bansal, P.; Rawal, R.K. Enasidenib: First mutant IDH2 inhibitor for the treatment of refractory and relapsed acute myeloid leukemia. Anticancer Agents Med. Chem. 2018, 18, 1936–1951. [Google Scholar] [CrossRef]
- Roboz, G.J.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Mims, A.S.; Prince, G.T.; Altman, J.K.; Arellano, M.L.; Donnellan, W.; Erba, H.P.; et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood 2020, 135, 463–471. [Google Scholar] [CrossRef]
- Oh, S.; Cho, Y.; Chang, M.; Park, S.; Kwon, H. Metformin decreases 2-HG production through the MYC-PHGDH pathway in suppressing breast cancer cell proliferation. Metabolites 2021, 11, 480. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Number of Cases/Year in USA (2021) | Germline Prevalence (%) | Familial Prevalence (%) | GWAS Heritability (%) | Twin Heritability (%) |
|---|---|---|---|---|---|
| Breast | 281,550 [55] | 5.7–11.1 [21,22,23] Ave. = 8.4 | 13.6 [20] | 9.7 [40] | 31 [18] |
| Prostate | 248,530 [55] | 2.9–17.2 [24,25,26] Ave. = 10.1 | 20.2 [20] | 11.0 [41] | 57 [18] |
| Lung | 235,760 [55] | 0.3–1.4 [27,28,29] Ave. = 0.9 | 8.7 [20] | 0.7 [42] | 18 [18] |
| Colorectal | 149,500 [55] | 3.5–7.5 [30,31] Ave. = 5.5 | 12.8 [20] | 1.2 [43] | 15 [18] |
| Melanoma | 106,110 [55] | 1.9–3.1 [32,33] Ave. = 2.5 | 4.9 [20] | 0.9 [44] | 58 [18] |
| Bladder | 83,730 [55] | 8.9 [34] | 5.4 [20] | 0.9 [45] | 30 [18] |
| Non-Hodgkin Lymphoma | 81,560 [55] | 7.7 [35] | 2.9 [20] | 0.7 [46] | 25 [19] |
| Kidney (RCC) | 76,080 [55] | 7.9 [36] | 3.6 [20] | 0.6 [47] | 38 [18] |
| Endometrial Uterine | 66,570 [55] | 4.6 [37] | 4.1 [20] | 0.6 [48] | 27 [18] |
| Pancreatic | 60,430 [55] | 3.9 [38] | 3.7 [20] | 0.6 [49] | 36 [52] |
| Thyroid | 44,280 [55] | NA | 3.5 [20] | 1.5 [50] | 53 [53] |
| Liver/bile Duct | 42,230 [55] | 5.9 [39] | 2.6 [20] | 1.7 [51] | 30 [54] |
| Range | 0.3–17.2 | 2.6–20.2 | 0.6–11.0 | 15.0–57.0 | |
| Case-weighted average | 6.2 | 10.2 | 4.3 | 34.2 |
| Cause | Percentage of Cancer Deaths in the US (%) | References |
|---|---|---|
| Age (>65) | 72.0 | [86] |
| Smoking | 28.8–31.7 | [87,88,89] |
| Obesity | 7.0 | [90] |
| Germline mutations/heritable cancers | 3.3–5.9 | This paper |
| Infectious agents | 5.9 | [91] |
| Alcohol | 3.5–4.0 | [90,92] |
| Radon exposure | 3.5 | [93] |
| Outdoor air pollution (PM 2.5) | 3.1 | [94,95] |
| Adverse effects of cancer treatment | 2.8 | [96] |
| Low fruit/vegetable diet | 2.7 | [85] |
| Diabetes | 2.5 | [97] |
| Physical inactivity | 2.2 | [85] |
| UV exposure | 1.5 | [85,98] |
| Red meat consumption | 0.5–1.4 | [99] |
| Diesel fumes | 1.3 | [100] |
| Second-hand smoke | 1.2 | [101] |
| Low fiber intake | 0.9 | [85] |
| Processed meat intake | 0.7–0.8 | [85,99] |
| Asbestos exposure | 0.7 | [102,103] |
| Low calcium and iodine intake | 0.5 | [85,104] |
| Miscellaneous occupational chemical exposures | 0.5 | [105] |
| Ionizing radiation (CT scans, radiotherapy) | 0.3 | [106] |
| Total (excluding age) | 73.4–80.4 |
| Oncometabolite | Cancer(s) | Mechanisms | Cancer HallMarks | Reference |
|---|---|---|---|---|
| Arginine | Ovarian cancer, pancreatic cancer, glioma, acute lymphoblastic leukemia (ALL), lung cancer, bladder cancer, colon cancer | Metastasis signaling, cell growth signaling (mTOR), reduced autophagy, DNA instability, mitochondrial dysfunction, Angiogenesis, anti-apoptosis, immune suppression | Evading growth suppressors, sustained proliferative signaling, genome instability, resisting cell death, replicative immortality, evading immune destruction, inducing angiogenesis | [135] |
| Asparagine | Acute lymphoblastic leukemia, breast cancer, lung cancer | Anti-apoptosis, Cell growth signaling, metastasis signaling | Dysregulated metabolism, resisting cell death, sustained proliferative signaling, evading growth suppressors, activating invasion and metastasis | [136] |
| Choline | Prostate cancer, brain cancer, breast cancer | Hypoxic, hyperglycemic growth, epigenetic modifications | Dysregulated metabolism, genome instability, sustained proliferative signaling | [137] |
| Cystathionine | Breast cancer | ROS protection, anti-apoptosis | replicative immortality, resisting cell death | [138] |
| Deoxycholic acid | Colon cancer | Mitochondrial dysfunction, ROS production, anti-apoptosis, proinflammation | evading growth suppressors, tumor promoting inflammation, resisting cell death | [139] |
| Diacetylspermine | Neuroblastoma, liver cancer, breast cancer, colon cancer, lung cancer | Anti-apoptosis, cell growth signaling, immune suppression | Resisting cell death, sustained proliferative signaling, evading immune destruction | [140] |
| Estradiol | Ovarian cancer, endometrial cancer breast cancer | Cell growth signaling, metastasis signaling | Sustained proliferative signaling, activating invasion and metastasis | [141] |
| Fumarate | Praganglioma, pheochromocytoma, renal cell carcinoma | Epigenetic modifications, protein modification | Dysregulated metabolism, genome instability, sustained proliferative signaling | [142] |
| N-acetyl-D-glucosamine | Systemic mastocytosis | Cell growth signaling, proinflammation | Sustained proliferative signaling, tumor promoting inflammation | [143] |
| Glucose | Most cancers | Hyperglycemic growth, aerobic glycolysis, protein modification | Dysregulated metabolism, sustained proliferative signaling, replicative immortality | [144] |
| Glutamine | Glioma, acute myeloid leukemia, lung cancer, breast cancer | Glutaminolysis, ROS protection, cell growth signaling (mTOR), reduced autophagy, DNA instability, mitochondrial dysfunction, metastasis signaling | Dysregulated metabolism, replicative immortality, sustained proliferative signaling, evading growth suppressors, genome instability, resisting cell death, activating invasion and metastasis | [145] |
| D-2-hydroxy-glutarate | Glioma, acute myeloid leukemia, prostate cancer, colon cancer | Epigenetic modifications, hypoxic, hyperglycemic growth, cell growth signaling (mTOR), ROS production, angiogenesis, immune suppression | Dysregulated metabolism, genome instability, inducing angiogenesis, resisting cell death, sustained proliferative signaling, evading immune destruction | [10] |
| L-2-hydroxy-glutarate | Renal cell carcinoma | Epigenetic modifications, hypoxic, hyperglycemic growth, cell growth signaling (mTOR), immune suppression, ROS production | Dysregulated metabolism, genome instability, resisting cell death, sustained proliferative signaling, evading immune destruction | [146] |
| Glycine | Lung cancer, glioma | hyperglycemic growth, aerobic glycolysis, epigenetic modifications | Dysregulated metabolism, genome instability | [147] |
| Homocysteine | Most cancers | Reduced DNA repair, proinflammation, epigenetic modifications | Genome instability, tumor promoting inflammation | [148] |
| Hypotaurine | Glioma | Epigenetic modifications, hypoxic, hyperglycemic growth | Dysregulated metabolism, genome instability, sustained proliferative signaling | [149] |
| Isoleucine | Lung cancer, glioma, breast cancer, glioma, endometrial cancer | Cell growth signaling (mTOR), reduced autophagy, DNA instability, mitochondrial dysfunction | Evading growth suppressors, sustained proliferative signaling, genome instability, resisting cell death, replicative immortality | [150] |
| Kynurenine | Colon cancer, lung cancer, prostate cancer, glioma, breast cancer | Cell growth signaling, immune suppression, metastasis signaling, proinflammation | Sustained proliferative signaling, evading immune destruction, tumor promoting inflammation, activating invasion and metastasis | [151] |
| Lactate | Most cancers | Metastasis signaling, immune suppression, angiogenesis, anti-apoptosis, proinflammation | Dysregulated metabolism, activating invasion and metastasis, inducing angiogenesis, evading immune destruction, tumor promoting inflammation | [152] |
| Leucine | Lung cancer, glioma, breast cancer, glioma, endometrial cancer | Cell growth signaling (mTOR), reduced autophagy, DNA instability, mitochondrial dysfunction | Evading growth suppressors, sustained proliferative signaling, genome instability, resisting cell death, replicative immortality | [150] |
| Lithocholic acid | Colon cancer | Mitochondrial dysfunction, ROS production, anti-apoptosis, proinflammation | Evading growth suppressors, tumor promoting inflammation, resisting cell death | [139] |
| Methionine | Colon cancer, pancreatic cancer, glioma, endometrial cancer | Cell growth signaling (mTOR), reduced autophagy, epigenetic modifications, mitochondrial dysfunction, anti-apoptosis, Immune suppression | Evading growth suppressors, sustained proliferative signaling, genome instability, resisting cell death, replicative immortality, evading immune destruction | [153] |
| Methylglyoxal | Breast cancer | Metastasis signaling, protein modification, proinflammation | Dysregulated metabolism, activating invasion and metastasis, tumor promoting inflammation | [154] |
| Methylmalonate | Liver cancer | Mitochondrial dysfunction, ROS production, DNA instability, proinflammation | Dysregulated metabolism, resisting cell death, genome instability, tumor promoting inflammation | [155] |
| Nitric Oxide | Lung cancer, colon cancer, breast cancer, pancreatic cancer, prostate Cancer | Angiogenesis, metastasis signaling, DNA instability, proinflammation | Inducing angiogenesis, activating invasion and metastasis, genome instability, tumor promoting inflammation | [156] |
| Progesterone | Ovarian cancer | Cell growth signaling, metastasis signaling | Sustained proliferative signaling, activating invasion and metastasis | [141] |
| Putrescine | Neuroblastoma, liver cancer, breast cancer, colon cancer, lung cancer | Anti-apoptosis, cell growth signaling, immune suppression | Resisting cell death, sustained proliferative signaling, evading immune destruction | [140] |
| 4-Pyridone-3-carboxamide-1-beta-D-ribonucleoside | Lung cancer, breast cancer | Metastasis signaling | Activating invasion and metastasis | [157] |
| SAICAR | Oral cancer, most cancers | Aerobic glycolysis, PKM2 signaling, cell growth signaling | Dysregulated metabolism, sustained proliferative signaling | [158] |
| Sarcosine | Prostate cancer | Epigenetic modifications, metastasis signaling | Dysregulated metabolism, genome instability, activating invasion and metastasis | [159] |
| Serine | Breast cancer, glioma, cervical cancer | Hyperglycemic growth, aerobic glycolysis, PKM2 signaling | dysregulated metabolism, replicative immortality | [160] |
| Spermidine | Neuroblastoma, liver cancer, breast cancer, colon cancer, lung cancer | Anti-apoptosis, cell growth signaling, immune suppression | resisting cell death, sustained proliferative signaling, evading immune destruction | [140] |
| Spermine | Neuroblastoma, liver cancer, breast cancer, colon cancer, lung cancer | Anti-apoptosis, cell growth signaling, immune suppression | Resisting cell death, sustained proliferative signaling, evading immune destruction | [140] |
| Succinate | Praganglioma, pheochromocytoma, renal cell carcinoma | Epigenetic modifications, hypoxic, hyperglycemic growth, angiogenesis, proinflammation, cell growth signaling | Dysregulated metabolism, genome instability, tumor promoting inflammation, inducing angiogenesis, sustained proliferative signaling | [161] |
| Succinyl-acetoacetate | Liver cancer | Protein modification, cell growth signaling | Dysregulated metabolism, Genome instability, sustained proliferative signaling | [162] |
| Succinyl-acetone | Liver cancer | Protein modification, cell growth signaling | Dysregulated metabolism, genome instability, sustained proliferative signaling | [162] |
| Uric acid | Liver cancer, lung cancer, liver cancer, bladder cancer, prostate cancer | Proinflammation, ROS protection | Tumor promoting inflammation, replicative immortality | [163] |
| Valine | Lung cancer, glioma, breast cancer, glioma, endometrial cancer | Cell growth signaling (mTOR), reduced autophagy, DNA instability, mitochondrial dysfunction | Evading growth suppressors, sustained proliferative signaling, genome instability, resisting cell death, replicative immortality | [150] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wishart, D. Metabolomics and the Multi-Omics View of Cancer. Metabolites 2022, 12, 154. https://doi.org/10.3390/metabo12020154
Wishart D. Metabolomics and the Multi-Omics View of Cancer. Metabolites. 2022; 12(2):154. https://doi.org/10.3390/metabo12020154
Chicago/Turabian StyleWishart, David. 2022. "Metabolomics and the Multi-Omics View of Cancer" Metabolites 12, no. 2: 154. https://doi.org/10.3390/metabo12020154
APA StyleWishart, D. (2022). Metabolomics and the Multi-Omics View of Cancer. Metabolites, 12(2), 154. https://doi.org/10.3390/metabo12020154