
R Shiny App for the Automated Deconvolution of NMR Spectra to Quantify the Solid-State Forms of Pharmaceutical Mixtures

 , , ,
, , , 
{kind=link}
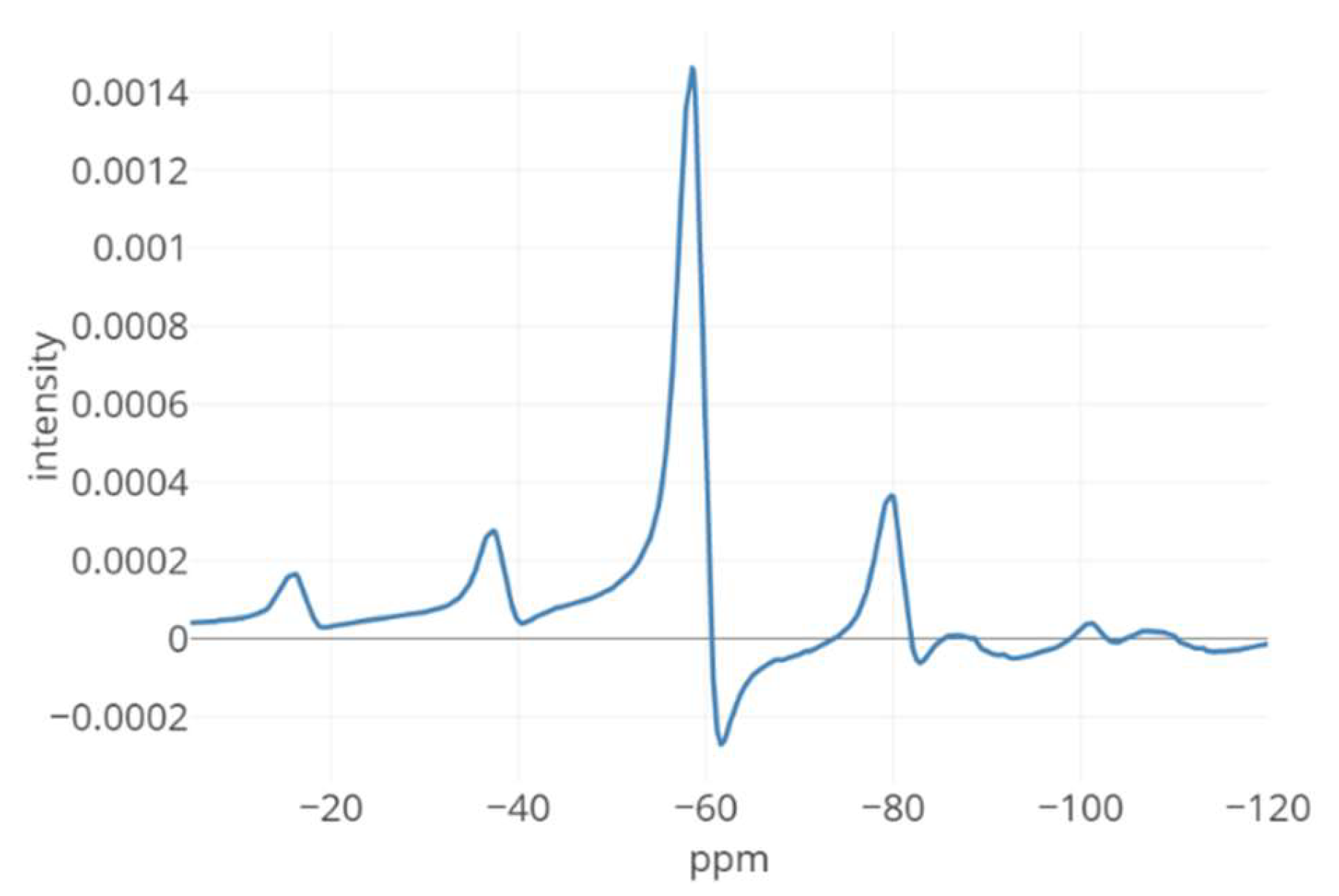{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Algorithm
2.2. R Shiny Application
- “no optimisation, apply the fixed processing values”: No optimisation is performed; the manually specified values in the input fields corresponding to (note that ) are directly applied to process (according to Equation (1)) and visualise the spectra.
- “optimise only proportion”: The same workflow as above, except that now the parameter is estimated, offering a compromise between fully manual and fully automated processing and deconvolution.
- “optimise proportion and other processing parameters”: This entails the fully automated optimisation of the five model parameters.
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haleblian, J.; McCrone, W. Pharmaceutical Applications of Polymorphism. J. Pharm. Sci. 1969, 58, 911–929. [Google Scholar] [CrossRef] [PubMed]
- Hilfiker, R.; von Raumer, M. Polymorphism in the Pharmaceutical Industry: Solid Form and Drug Development; Wiley: Hoboken, NJ, USA, 2019; ISBN 978-3-527-69785-4. [Google Scholar]
- Vanhamme, L.; van den Boogaart, A.; Van Huffel, S. Improved Method for Accurate and Efficient Quantification of MRS Data with Use of Prior Knowledge. J. Magn. Reson. 1997, 129, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Ratiney, H.; Coenradie, Y.; Cavassila, S.; van Ormondt, D.; Graveron-Demilly, D. Time-Domain Quantitation of 1 H Short Echo-Time Signals: Background Accommodation. MAGMA Magn. Reson. Mater. Phys. Biol. Med. 2004, 16, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Poullet, J.-B.; Sima, D.M.; Simonetti, A.W.; De Neuter, B.; Vanhamme, L.; Lemmerling, P.; Van Huffel, S. An Automated Quantitation of Short Echo Time MRS Spectra in an Open Source Software Environment: AQSES. NMR Biomed. 2007, 20, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Stefan, D.; Cesare, F.D.; Andrasescu, A.; Popa, E.; Lazariev, A.; Vescovo, E.; Strbak, O.; Williams, S.; Starcuk, Z.; Cabanas, M.; et al. Quantitation of Magnetic Resonance Spectroscopy Signals: The JMRUI Software Package. Meas. Sci. Technol. 2009, 20, 104035. [Google Scholar] [CrossRef]
- Wilson, M.; Reynolds, G.; Kauppinen, R.A.; Arvanitis, T.N.; Peet, A.C. A Constrained Least-Squares Approach to the Automated Quantitation of in Vivo 1H Magnetic Resonance Spectroscopy Data. Magn. Reson. Med. 2011, 65, 2579. [Google Scholar] [CrossRef] [PubMed]
- Provencher, S.W. Automatic Quantitation of Localized in Vivo 1H Spectra with LCModel. NMR Biomed. 2001, 14, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Young, K.; Soher, B.J.; Maudsley, A.A. Automated Spectral Analysis II: Application of Wavelet Shrinkage for Characterization of Non-Parameterized Signals. Magn. Reson. Med. 1998, 40, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Edden, R.A.E.; Puts, N.A.J.; Harris, A.D.; Barker, P.B.; Evans, C.J. Gannet: A Batch-Processing Tool for the Quantitative Analysis of Gamma-Aminobutyric Acid–Edited MR Spectroscopy Spectra. J. Magn. Reson. Imaging 2014, 40, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Oeltzschner, G.; Zöllner, H.J.; Hui, S.C.N.; Mikkelsen, M.; Saleh, M.G.; Tapper, S.; Edden, R.A.E. Osprey: Open-Source Processing, Reconstruction and Estimation of Magnetic Resonance Spectroscopy Data. J. Neurosci. Methods 2020, 343, 108827. [Google Scholar] [CrossRef] [PubMed]
- Massiot, D.; Fayon, F.; Capron, M.; King, I.; Le Calvé, S.; Alonso, B.; Durand, J.-O.; Bujoli, B.; Gan, Z.; Hoatson, G. Modelling One- and Two-Dimensional Solid-State NMR Spectra. Magn. Reson. Chem. 2002, 40, 70–76. [Google Scholar] [CrossRef]
- Rowan, T.H. Functional Stability Analysis of Numerical Algorithms; University of Texas at Austin: Austin, TX, USA, 1990. [Google Scholar]
- Martin, M.; Legat, B.; Leenders, J.; Vanwinsberghe, J.; Rousseau, R.; Boulanger, B.; Eilers, P.H.C.; De Tullio, P.; Govaerts, B. PepsNMR for 1H NMR Metabolomic Data Pre-Processing. Anal. Chim. Acta 2018, 1019, 67. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2022. Available online: https://www.r-project (accessed on 30 September 2022).
- Nloptr. Available online: https://github.com/astamm/nloptr (accessed on 30 September 2022).
- Johnson, S.G. The NLopt Nonlinear-Optimization Package. Available online: https://nlopt.readthedocs (accessed on 30 September 2022).





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prostko, P.; Pikkemaat, J.; Selter, P.; Lukaschek, M.; Wechselberger, R.; Khamiakova, T.; Valkenborg, D. R Shiny App for the Automated Deconvolution of NMR Spectra to Quantify the Solid-State Forms of Pharmaceutical Mixtures. Metabolites 2022, 12, 1248. https://doi.org/10.3390/metabo12121248
Prostko P, Pikkemaat J, Selter P, Lukaschek M, Wechselberger R, Khamiakova T, Valkenborg D. R Shiny App for the Automated Deconvolution of NMR Spectra to Quantify the Solid-State Forms of Pharmaceutical Mixtures. Metabolites. 2022; 12(12):1248. https://doi.org/10.3390/metabo12121248
Chicago/Turabian StyleProstko, Piotr, Jeroen Pikkemaat, Philipp Selter, Michail Lukaschek, Rainer Wechselberger, Tatsiana Khamiakova, and Dirk Valkenborg. 2022. "R Shiny App for the Automated Deconvolution of NMR Spectra to Quantify the Solid-State Forms of Pharmaceutical Mixtures" Metabolites 12, no. 12: 1248. https://doi.org/10.3390/metabo12121248
APA StyleProstko, P., Pikkemaat, J., Selter, P., Lukaschek, M., Wechselberger, R., Khamiakova, T., & Valkenborg, D. (2022). R Shiny App for the Automated Deconvolution of NMR Spectra to Quantify the Solid-State Forms of Pharmaceutical Mixtures. Metabolites, 12(12), 1248. https://doi.org/10.3390/metabo12121248