
Development of SFC-MS Method for Quantification of Eicosanoids Biosynthesized in Primary Human Blood Cells

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. SFC-MS Method Development
2.3. Method Validation
2.4. Purification of Primary Human Blood Cells
2.5. Cell Assays
3. Results
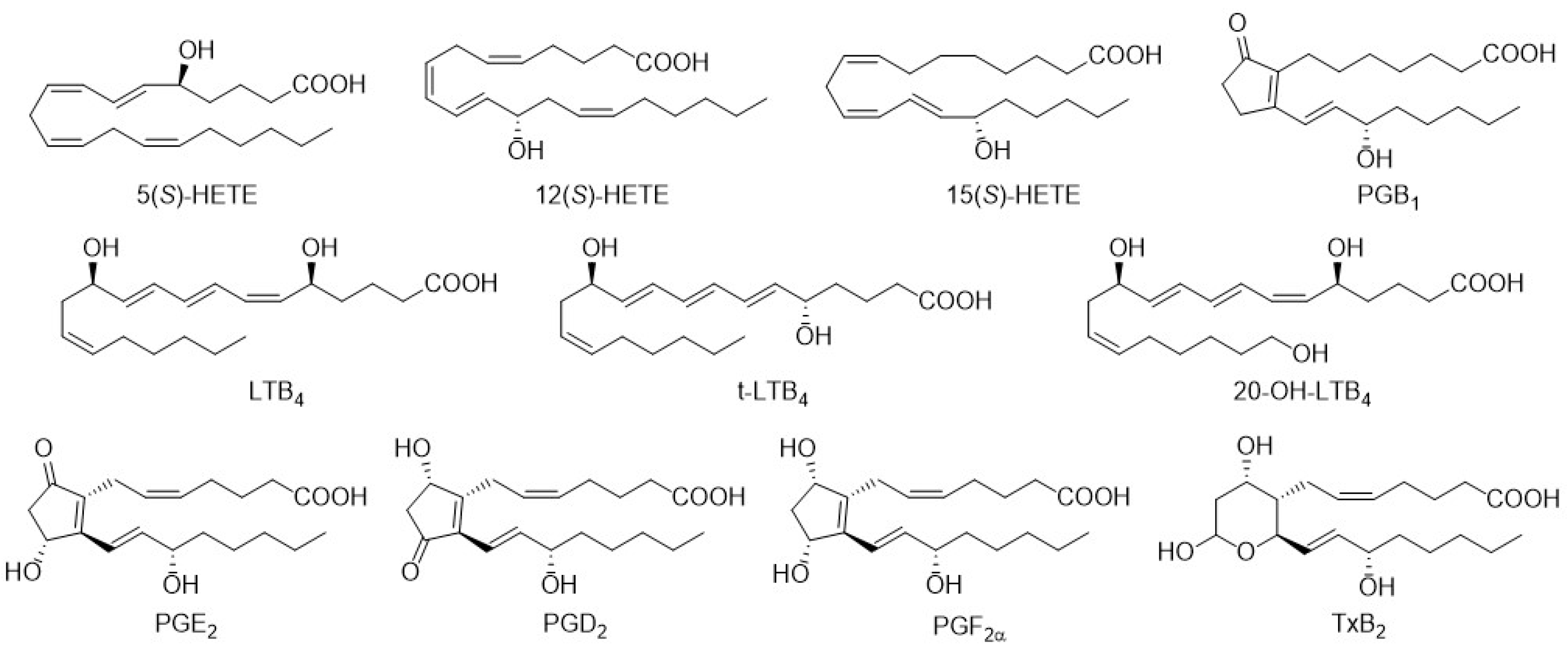
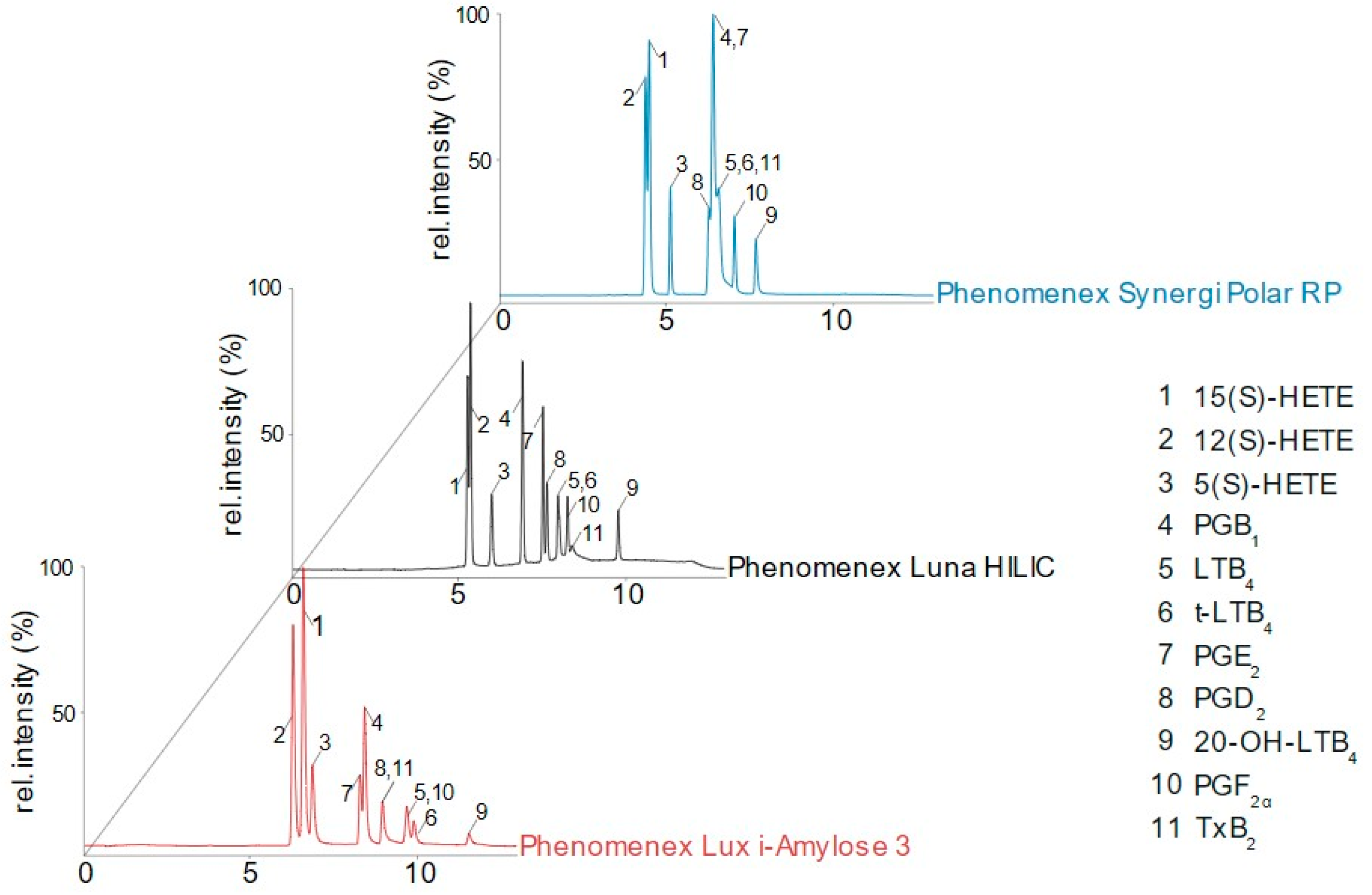
3.1. Column Screening
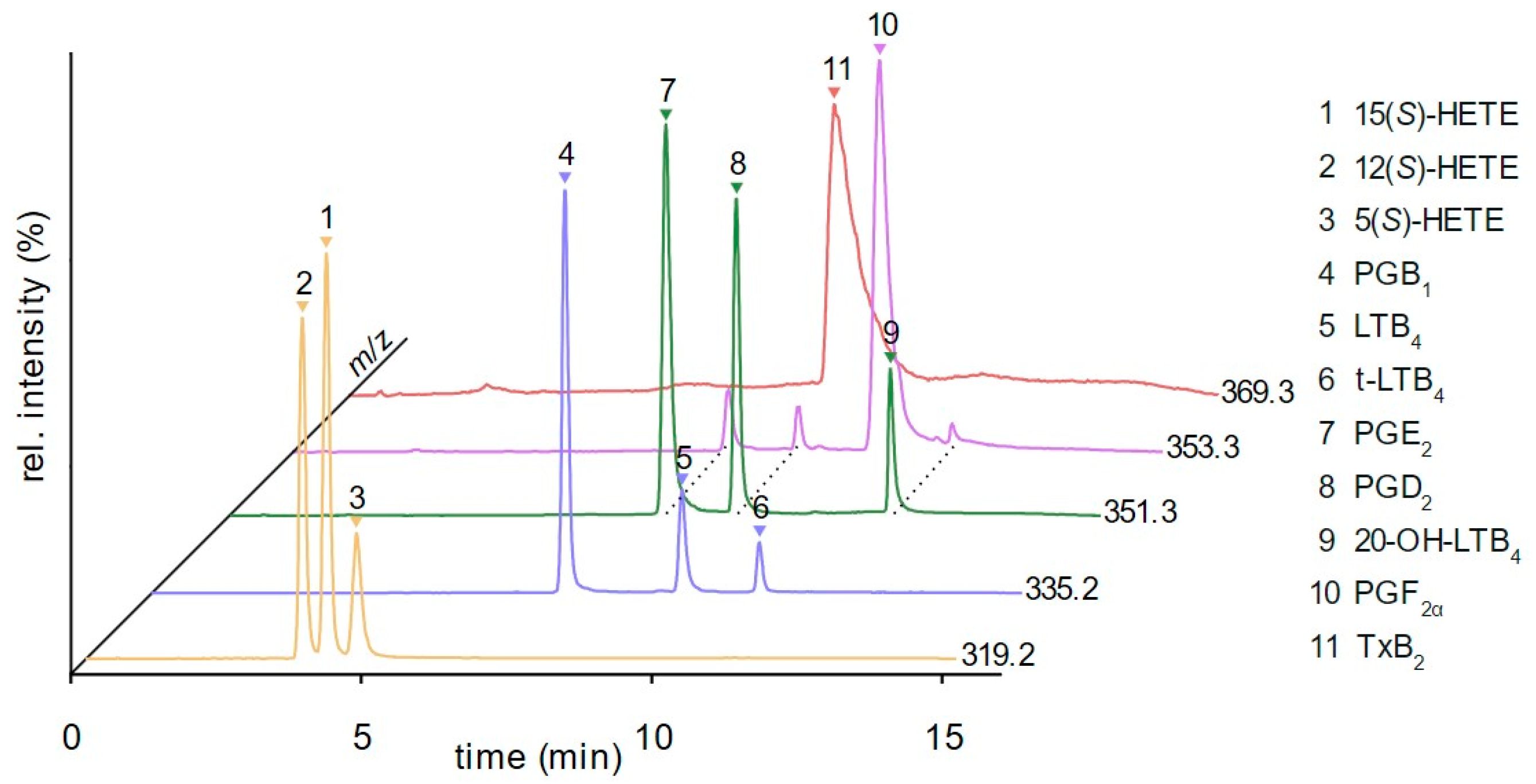
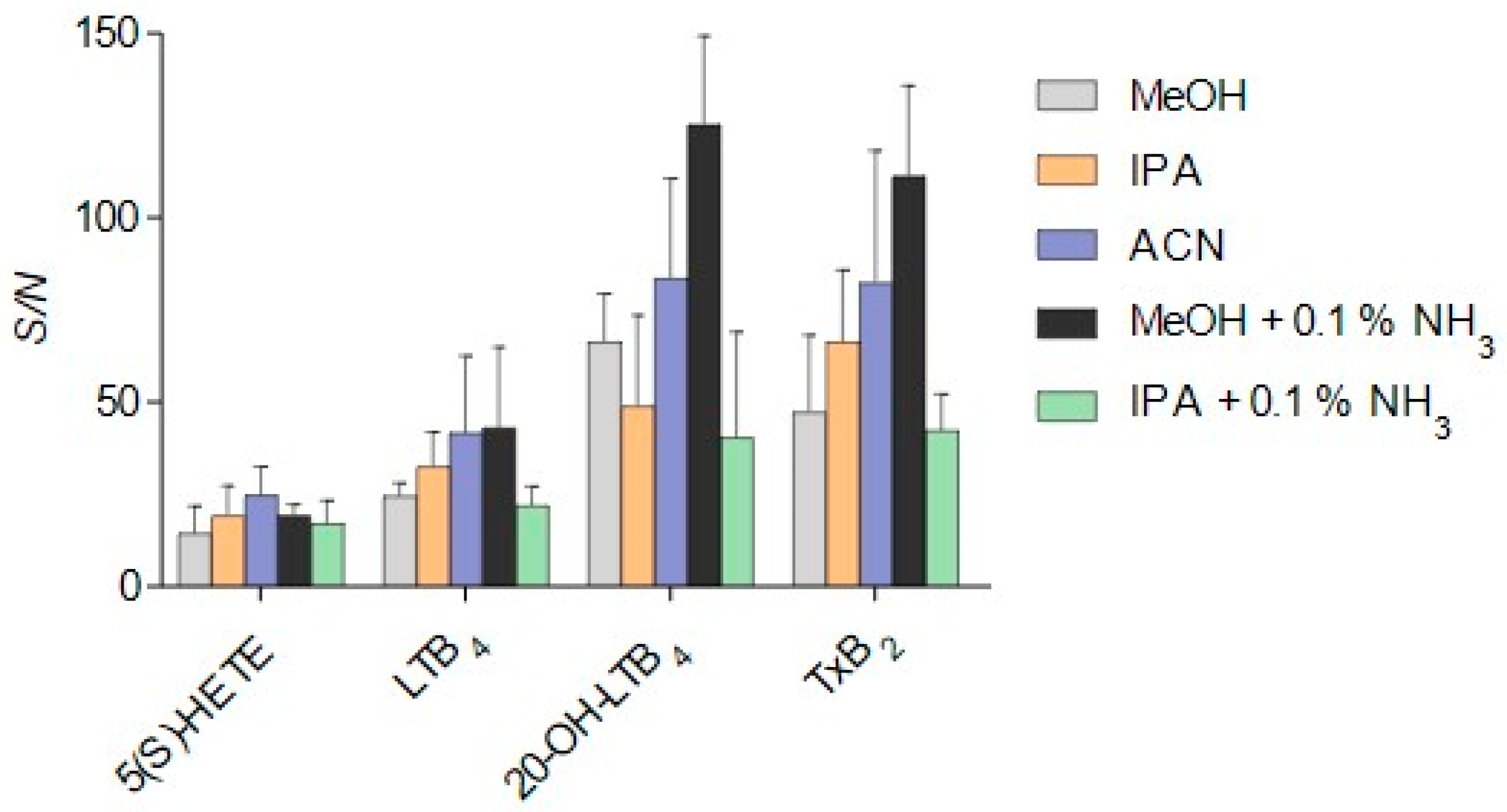
3.2. Method Development
3.3. Method Validation
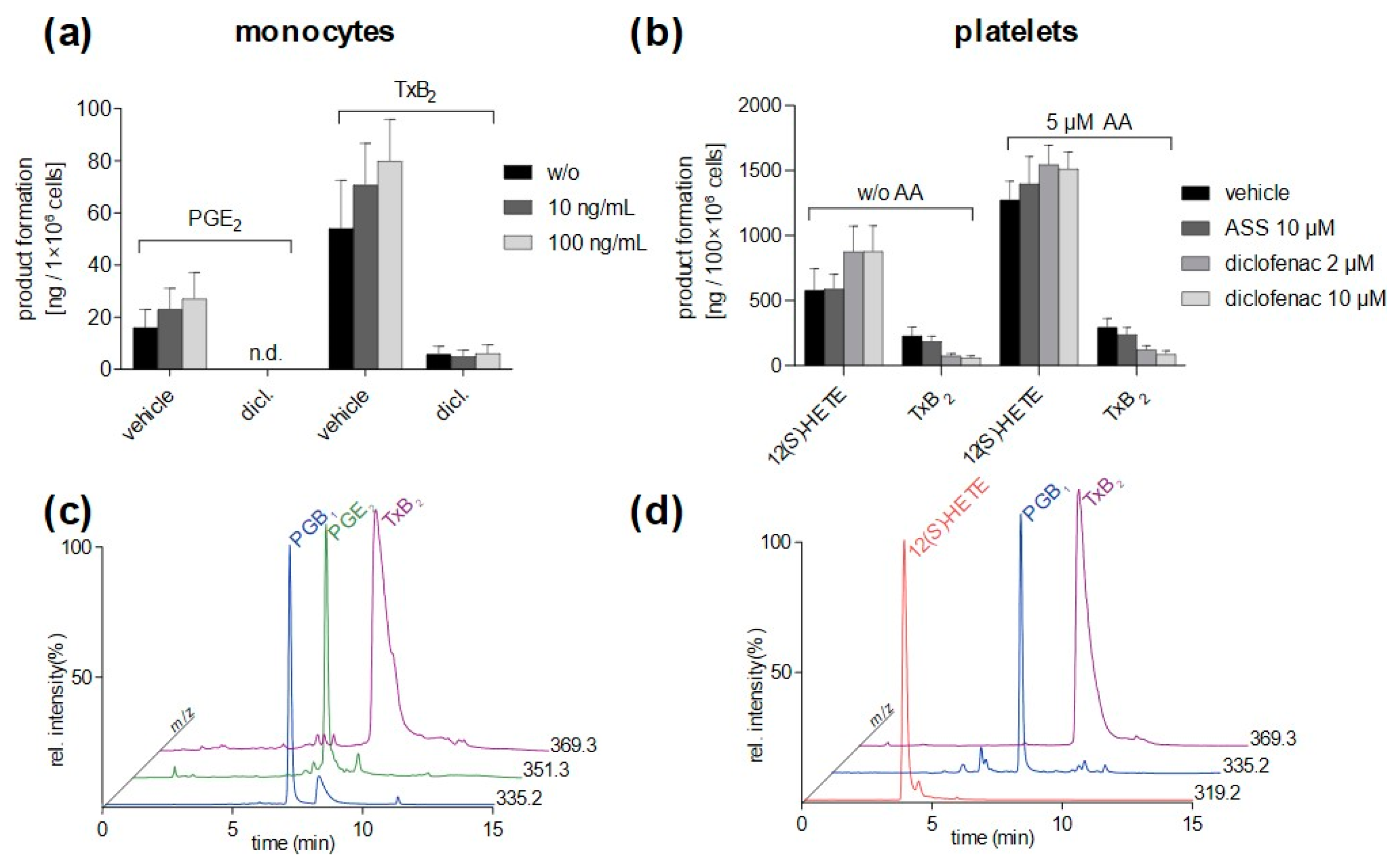
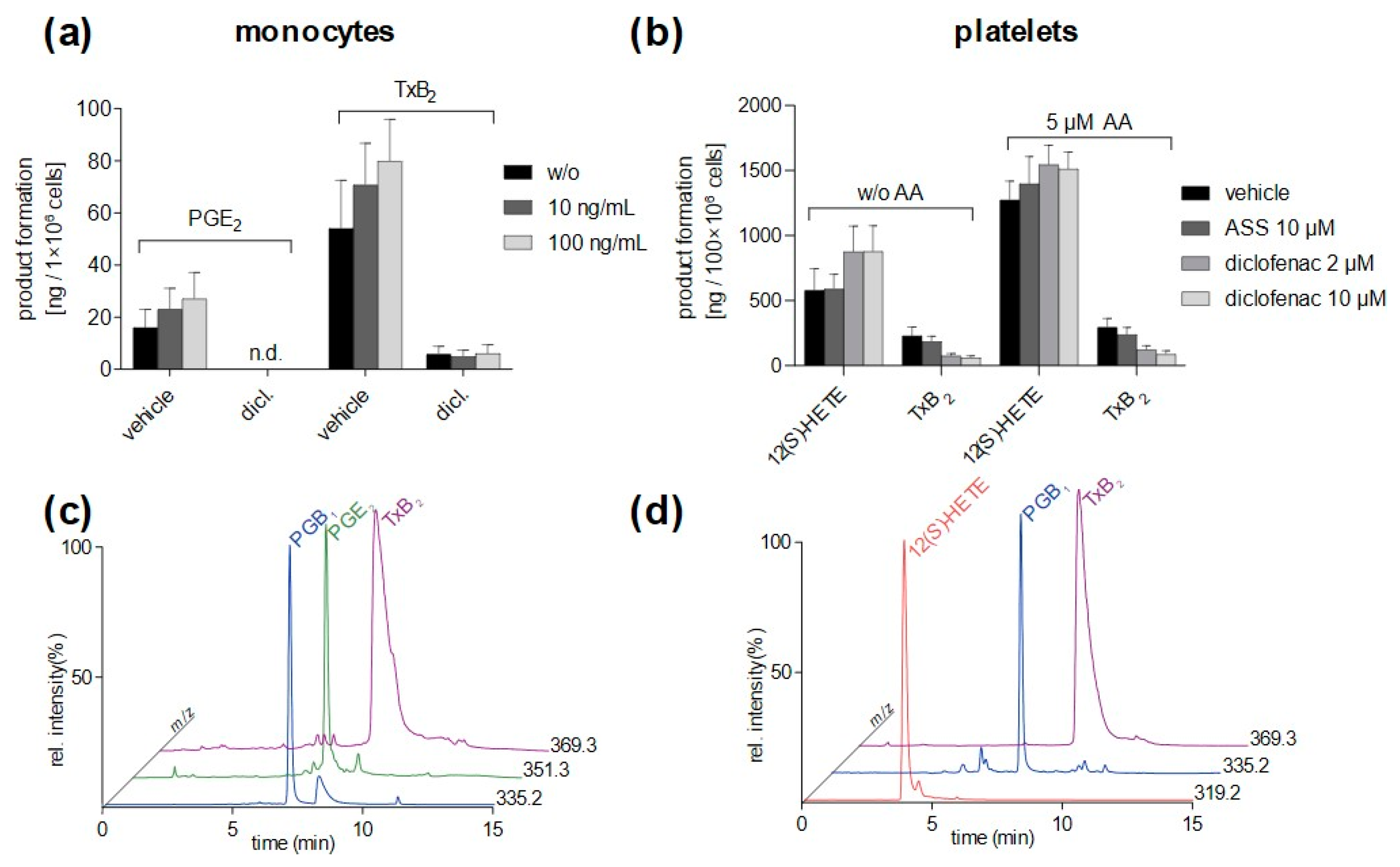
3.4. Method Application
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haeggstrom, J.Z.; Funk, C.D. Lipoxygenase and leukotriene pathways: Biochemistry, biology, and roles in disease. Chem. Rev. 2011, 111, 5866–5898. [Google Scholar] [CrossRef] [PubMed]
- Capdevila, J.H.; Falck, J.R. Biochemical and molecular properties of the cytochrome P450 arachidonic acid monooxygenases. Prostaglandins Other Lipid Mediat. 2002, 68–69, 325–344. [Google Scholar] [CrossRef]
- Oliw, E.H. Oxygenation of polyunsaturated fatty acids by cytochrome P450 monooxygenases. Prog. Lipid Res. 1994, 33, 329–354. [Google Scholar] [CrossRef]
- Nakanishi, M.; Rosenberg, D.W. Multifaceted roles of PGE2 in inflammation and cancer. Semin. Immunopathol. 2013, 35, 123–137. [Google Scholar] [CrossRef]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Urade, Y.; Hayaishi, O. Prostaglandin D-2 and sleep regulation. BBA-Mol. Cell Biol. Lipid 1999, 1436, 606–615. [Google Scholar] [CrossRef]
- Reilly, M.; Fitzgerald, G.A. Cellular activation by thromboxane A2 and other eicosanoids. Eur. Heart J. 1993, 14 (Suppl. K), 88–93. [Google Scholar]
- Radmark, O.; Werz, O.; Steinhilber, D.; Samuelsson, B. 5-Lipoxygenase, a key enzyme for leukotriene biosynthesis in health and disease. Biochim. Biophys. Acta 2015, 1851, 331–339. [Google Scholar] [CrossRef]
- Tsikas, D. Application of gas chromatography-mass spectrometry and gas chromatography-tandem mass spectrometry to assess in vivo synthesis of prostaglandins, thromboxane, leukotrienes, isoprostanes and related compounds in humans. J. Chromatogr. B Biomed. Sci. Appl. 1998, 717, 201–245. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.C.; Barkley, R.M.; Zemski Berry, K.; Hankin, J.; Harrison, K.; Johnson, C.; Krank, J.; McAnoy, A.; Uhlson, C.; Zarini, S. Electrospray ionization and tandem mass spectrometry of eicosanoids. Anal. Biochem. 2005, 346, 1–42. [Google Scholar] [CrossRef] [PubMed]
- Klesper, E.; Corwin, A.H.; Turner, D.A. High Pressure Gas Chromatography above Critical Temperatures. J. Org. Chem. 1962, 27, 700–701. [Google Scholar]
- Li, F.; Hsieh, Y. Supercritical fluid chromatography-mass spectrometry for chemical analysis. J. Sep. Sci 2008, 31, 1231–1237. [Google Scholar] [CrossRef]
- Ubhayasekera, S.J.K.A.; Acharya, S.R.; Bergquist, J. A novel, fast and sensitive supercritical fluid chromatography-tandem mass spectrometry (SFC-MS/MS) method for analysis of arachidonic acid metabolites. Analyst 2018, 143, 3661–3669. [Google Scholar] [CrossRef] [Green Version]
- Berkecz, R.; Lisa, M.; Holcapek, M. Analysis of oxylipins in human plasma: Comparison of ultrahigh-performance liquid chromatography and ultrahigh-performance supercritical fluid chromatography coupled to mass spectrometry. J. Chromatogr. A 2017, 1511, 107–121. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.; Zhong, Q.; Liang, Y.; Zhou, T. Determination of lipid mediators in breast cancer cells using lyophilization-supercritical fluid extraction online coupled with supercritical fluid chromatography-quadrupole tandem mass spectrometry. J. Sep. Sci. 2020, 43, 1800–1807. [Google Scholar] [CrossRef]
- Quaranta, A.; Zohrer, B.; Revol-Cavalier, J.; Benkestock, K.; Balas, L.; Oger, C.; Keyes, G.S.; Wheelock, A.M.; Durand, T.; Galano, J.M.; et al. Development of a Chiral Supercritical Fluid Chromatography-Tandem Mass Spectrometry and Reversed-Phase Liquid Chromatography-Tandem Mass Spectrometry Platform for the Quantitative Metabolic Profiling of Octadecanoid Oxylipins. Anal. Chem. 2022, 94, 14618–14626. [Google Scholar] [CrossRef]
- Boyum, A. Isolation of mononuclear cells and granulocytes from human blood. Isolation of monuclear cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand. J. Clin. Lab. Investig. Suppl. 1968, 97, 77–89. [Google Scholar]
- Funk, C.D. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef] [Green Version]
- Hansson, G.; Lindgren, J.A.; Dahlen, S.E.; Hedqvist, P.; Samuelsson, B. Identification and biological activity of novel omega-oxidized metabolites of leukotriene B4 from human leukocytes. FEBS Lett. 1981, 130, 107–112. [Google Scholar] [CrossRef] [Green Version]
- Haeggstrom, J.Z.; Rinaldo-Matthis, A.; Wheelock, C.E.; Wetterholm, A. Advances in eicosanoid research, novel therapeutic implications. Biochem. Biophys. Res. Commun. 2010, 396, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Chhonker, Y.S.; Bala, V.; Murry, D.J. Quantification of eicosanoids and their metabolites in biological matrices: A review. Bioanalysis 2018, 10, 2027–2046. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, R.K.; Hasan, M.; Eckert, C.; Link, A. Supercritical fluid chromatography: From science fiction to scientific fact. Chemtexts 2019, 5, 1–12. [Google Scholar] [CrossRef]
- Kostiainen, R.; Kauppila, T.J. Effect of eluent on the ionization process in liquid chromatography-mass spectrometry. J. Chromatogr. A 2009, 1216, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Ferreiros, N.; Homann, J.; Labocha, S.; Grossmann, N.; Hahn, J.S.; Brune, B.; Geisslinger, G. Lipoxin A(4): Problems with its determination using reversed phase chromatography-tandem mass spectrometry and confirmation with chiral chromatography. Talanta 2014, 127, 82–87. [Google Scholar] [CrossRef]
- Goloshchapova, K.; Stehling, S.; Heydeck, D.; Blum, M.; Kuhn, H. Functional characterization of a novel arachidonic acid 12S-lipoxygenase in the halotolerant bacterium Myxococcus fulvus exhibiting complex social living patterns. Microbiologyopen 2018, 8, e775. [Google Scholar] [CrossRef]
- Cebo, M.; Fu, X.; Gawaz, M.; Chatterjee, M.; Lammerhofer, M. Enantioselective ultra-high performance liquid chromatography-tandem mass spectrometry method based on sub-2microm particle polysaccharide column for chiral separation of oxylipins and its application for the analysis of autoxidized fatty acids and platelet releasates. J. Chromatogr. A 2020, 1624, 461206. [Google Scholar] [CrossRef]
- Homann, J.; Lehmann, C.; Kahnt, A.S.; Steinhilber, D.; Parnham, M.J.; Geisslinger, G.; Ferreiros, N. Chiral chromatography-tandem mass spectrometry applied to the determination of pro-resolving lipid mediators. J. Chromatogr. A 2014, 1360, 150–163. [Google Scholar] [CrossRef]
- Garscha, U.; Romp, E.; Pace, S.; Rossi, A.; Temml, V.; Schuster, D.; Konig, S.; Gerstmeier, J.; Liening, S.; Werner, M.; et al. Pharmacological profile and efficiency in vivo of diflapolin, the first dual inhibitor of 5-lipoxygenase-activating protein and soluble epoxide hydrolase. Sci. Rep.-UK 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Romp, E.; Arakandy, V.; Fischer, J.; Wolz, C.; Siegmund, A.; Loffler, B.; Tuchscherr, L.; Werz, O.; Garscha, U. Exotoxins from Staphylococcus aureus activate 5-lipoxygenase and induce leukotriene biosynthesis. Cell. Mol. Life Sci. 2020, 77, 3841–3858. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Brune, K.; Pahl, A. Cyclooxygenase-2 expression in lipopolysaccharide-stimulated human monocytes is modulated by cyclic AMP, prostaglandin E-2, and nonsteroidal anti-inflammatory drugs. Biochem. Bioph. Res. Commun. 2000, 278, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Skarke, C.; Reus, M.; Schmidt, R.; Grundei, I.; Schuss, P.; Geisslinger, G.; Lotsch, J. The cyclooxygenase 2 genetic variant -765G>C does not modulate the effects of celecoxib on prostaglandin E2 production. Clin. Pharmacol. Ther. 2006, 80, 621–632. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | m/z | k Screening Condition | k Final Method Condition |
|---|---|---|---|
| 12(S)-HETE | 319.2 | 9.7 | 5.3 |
| 15(S)-HETE | 319.2 | 10.2 | 6.1 |
| 5(S)-HETE | 319.2 | 10.7 | 6.8 |
| PGB1 | 335.2 | 13.3 | 11.1 |
| LTB4 | 335.2 | 15.5 | 14.5 |
| t-LTB4 | 335.2 | 15.8 | 16.7 |
| PGE2 | 351.3 | 13.1 | 11.7 |
| PGD2 | 351.3 | 14.2 | 13.7 |
| 20-OH-LTB4 | 351.3 | 18.6 | 18.3 |
| PGF2α | 353.3 | 15.4 | 16.2 |
| TxB2 | 369.3 | 14.3 | 13.3 |
| Accuracy (%) | Precision (RSD%) | ||||
|---|---|---|---|---|---|
| Compound | QC | Intraday (n = 6) | Interday (n = 3) | Intraday (n = 6) | Interday (n = 3) |
| 12(S)-HETE | LLQC | 109.6 | 109.0 | 5.1 | 0.5 |
| LQC | 91.0 | 98.0 | 6.8 | 6.8 | |
| MQC | 93.0 | 93.5 | 6.6 | 4.5 | |
| HQC | 89.3 | 93.7 | 7.3 | 4.8 | |
| 15(S)-HETE | LLQC | 110.7 | 112.4 | 5.7 | 2.6 |
| LQC | 91.3 | 99.8 | 7.0 | 8.1 | |
| MQC | 94.4 | 95.6 | 7.3 | 3.0 | |
| HQC | 90.4 | 95.4 | 9.3 | 5.4 | |
| 5(S)-HETE | LLQC | 123.3 | 113.6 | 4.1 | 8.4 |
| LQC | 87.8 | 96.6 | 6.4 | 8.9 | |
| MQC | 92.9 | 92.6 | 10.8 | 2.7 | |
| HQC | 89.5 | 94.9 | 12.6 | 7.0 | |
| PGB1 | LLQC | 94.1 | 100.0 | 8.8 | 7.3 |
| LQC | 93.1 | 95.5 | 7.8 | 6.7 | |
| MQC | 95.0 | 93.3 | 4.4 | 4.0 | |
| HQC | 102.9 | 97.8 | 6.2 | 5.9 | |
| LTB4 | LLQC | 114.5 | 110.7 | 2.0 | 4.8 |
| LQC | 97.0 | 100.7 | 4.9 | 3.4 | |
| MQC | 102.0 | 98.9 | 2.7 | 2.7 | |
| HQC | 103.6 | 101.1 | 4.5 | 3.4 | |
| t-LTB4 | LLQC | 107.4 | 98.6 | 5.9 | 9.2 |
| LQC | 102.3 | 99.6 | 3.9 | 15.3 | |
| MQC | 101.9 | 98.9 | 5.3 | 5.4 | |
| HQC | 104.7 | 100.5 | 4.4 | 4.4 | |
| PGE2 | LLQC | 119.0 | 104.6 | 4.1 | 12.0 |
| LQC | 94.7 | 92.4 | 4.4 | 3.9 | |
| MQC | 98.4 | 92.6 | 3.2 | 5.6 | |
| HQC | 97.9 | 95.7 | 9.4 | 2.6 | |
| PGD2 | LLQC | 111.9 | 104.4 | 5.5 | 6.2 |
| LQC | 96.7 | 95.9 | 7.1 | 5.0 | |
| MQC | 103.1 | 96.1 | 8.5 | 9.9 | |
| HQC | 102.4 | 99.5 | 8.0 | 4.8 | |
| 20-OH-LTB4 | LLQC | 110.2 | 106.9 | 6.7 | 16.6 |
| LQC | 97.1 | 92.0 | 10.0 | 11.8 | |
| MQC | 95.8 | 88.6 | 9.0 | 12.1 | |
| HQC | 97.9 | 92.0 | 9.9 | 8.7 | |
| PGF2α | LLQC | 112.7 | 107.7 | 7.2 | 5.0 |
| LQC | 96.3 | 96.7 | 5.4 | 3.8 | |
| MQC | 114.0 | 102.3 | 10.9 | 11.1 | |
| HQC | 114.0 | 110.0 | 3.2 | 5.0 | |
| TxB2 | LLQC | 117.2 | 106.3 | 7.2 | 9.1 |
| LQC | 99.4 | 97.8 | 5.5 | 2.7 | |
| MQC | 105.0 | 96.4 | 8.8 | 10.4 | |
| HQC | 113.2 | 103.8 | 5.4 | 9.7 | |
| Recovery (%) | ||
| I | II | |
| 12(S)-HETE | 63.3 ± 5.2 | 64.2 ± 3.3 |
| 15(S)-HETE | 65.3 ± 3.3 | 66.5 ± 3.0 |
| 5(S)-HETE | 73.0 ± 7.6 | 69.8 ± 8.7 |
| PGB1 | 80.4 ± 5.3 | 81.6 ± 2.3 |
| LTB4 | 61.8 ± 3.5 | 63.6 ± 1.9 |
| t-LTB4 | 64.4 ± 3.1 | 60.9 ± 0.7 |
| PGE2 | 73.0 ± 3.4 | 80.4 ± 2.4 |
| PGD2 | 75.8 ± 4.3 | 74.9 ± 1.5 |
| 20-OH-LTB4 | 60.2 ± 4.0 | 58.9 ± 0.7 |
| PGF2α | 69.8 ± 6.0 | 73.0 ± 5.8 |
| TxB2 | 70.3 ± 3.6 | 76.6 ± 3.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, L.; Burmeister, L.S.; Greinacher, A.; König, S.; Garscha, U. Development of SFC-MS Method for Quantification of Eicosanoids Biosynthesized in Primary Human Blood Cells. Metabolites 2022, 12, 1198. https://doi.org/10.3390/metabo12121198
Schmidt L, Burmeister LS, Greinacher A, König S, Garscha U. Development of SFC-MS Method for Quantification of Eicosanoids Biosynthesized in Primary Human Blood Cells. Metabolites. 2022; 12(12):1198. https://doi.org/10.3390/metabo12121198
Chicago/Turabian StyleSchmidt, Louis, Laura Sophie Burmeister, Andreas Greinacher, Stefanie König, and Ulrike Garscha. 2022. "Development of SFC-MS Method for Quantification of Eicosanoids Biosynthesized in Primary Human Blood Cells" Metabolites 12, no. 12: 1198. https://doi.org/10.3390/metabo12121198
APA StyleSchmidt, L., Burmeister, L. S., Greinacher, A., König, S., & Garscha, U. (2022). Development of SFC-MS Method for Quantification of Eicosanoids Biosynthesized in Primary Human Blood Cells. Metabolites, 12(12), 1198. https://doi.org/10.3390/metabo12121198