


The Discovery of Potential SARS-CoV-2 Natural Inhibitors among 4924 African Metabolites Targeting the Papain-like Protease: A Multi-Phase In Silico Approach

 ,
,  , , ,
, , ,  and
and 
Abstract

1. Introduction
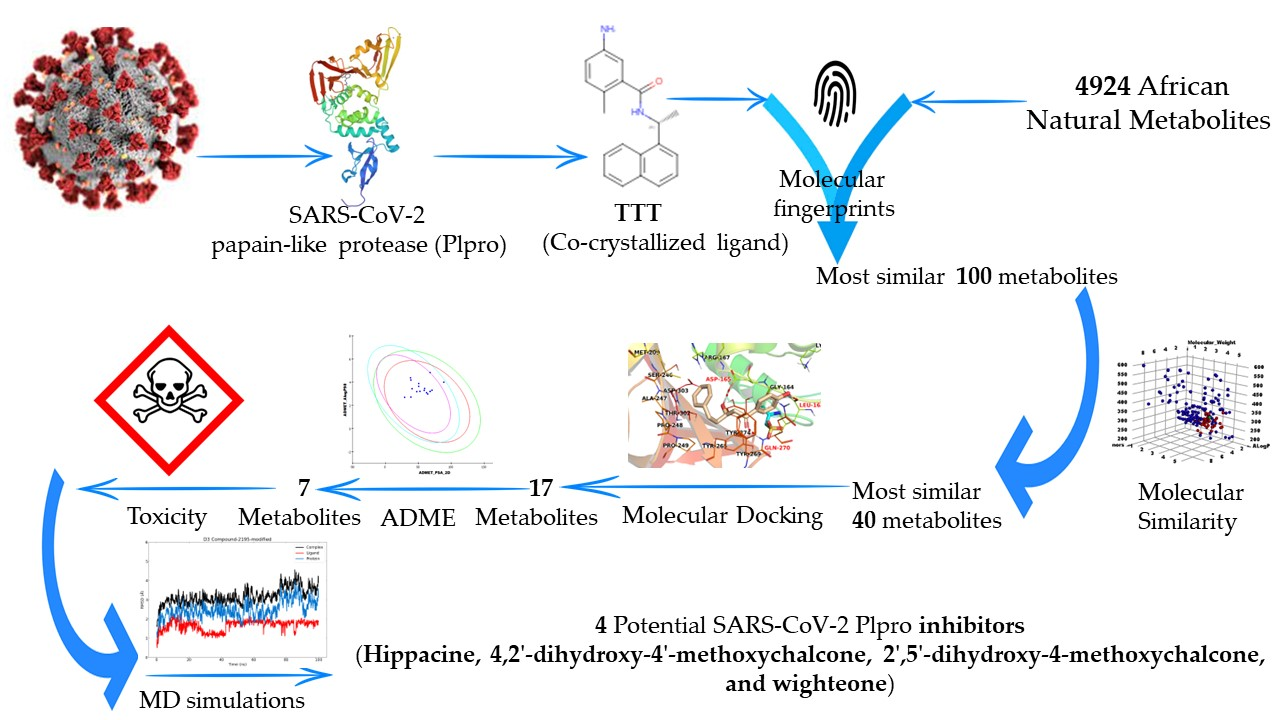
2. Method
2.1. Molecular Similarity Detection
2.2. Fingerprint Studies
2.3. Docking Studies
2.4. ADMET Analysis
2.5. Toxicity Studies
2.6. Molecular Dynamics Simulation
3. Results and Discussion
3.1. Structure Fingerprints Study
3.2. Molecular Similarity
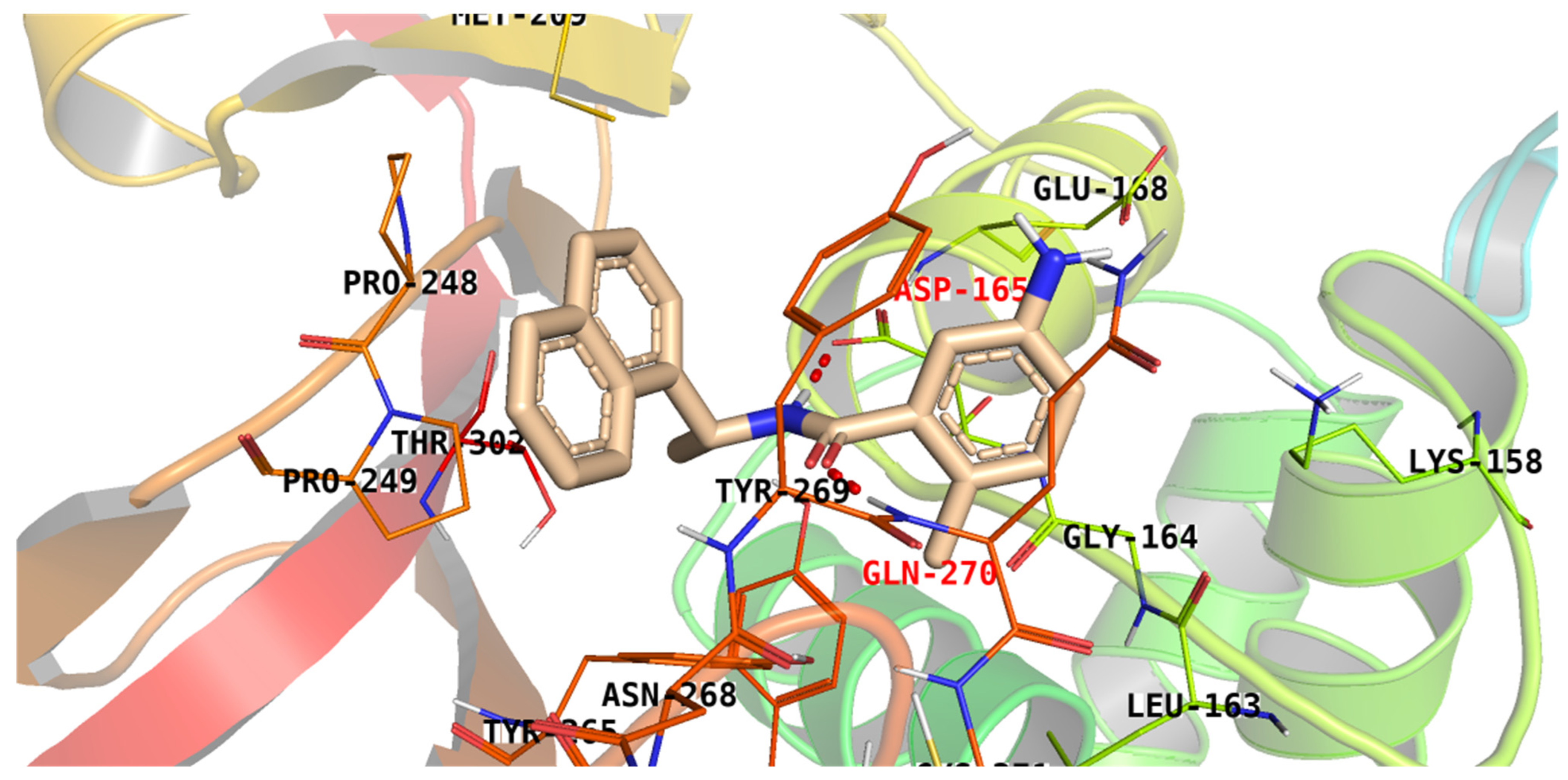
3.3. Docking Studies
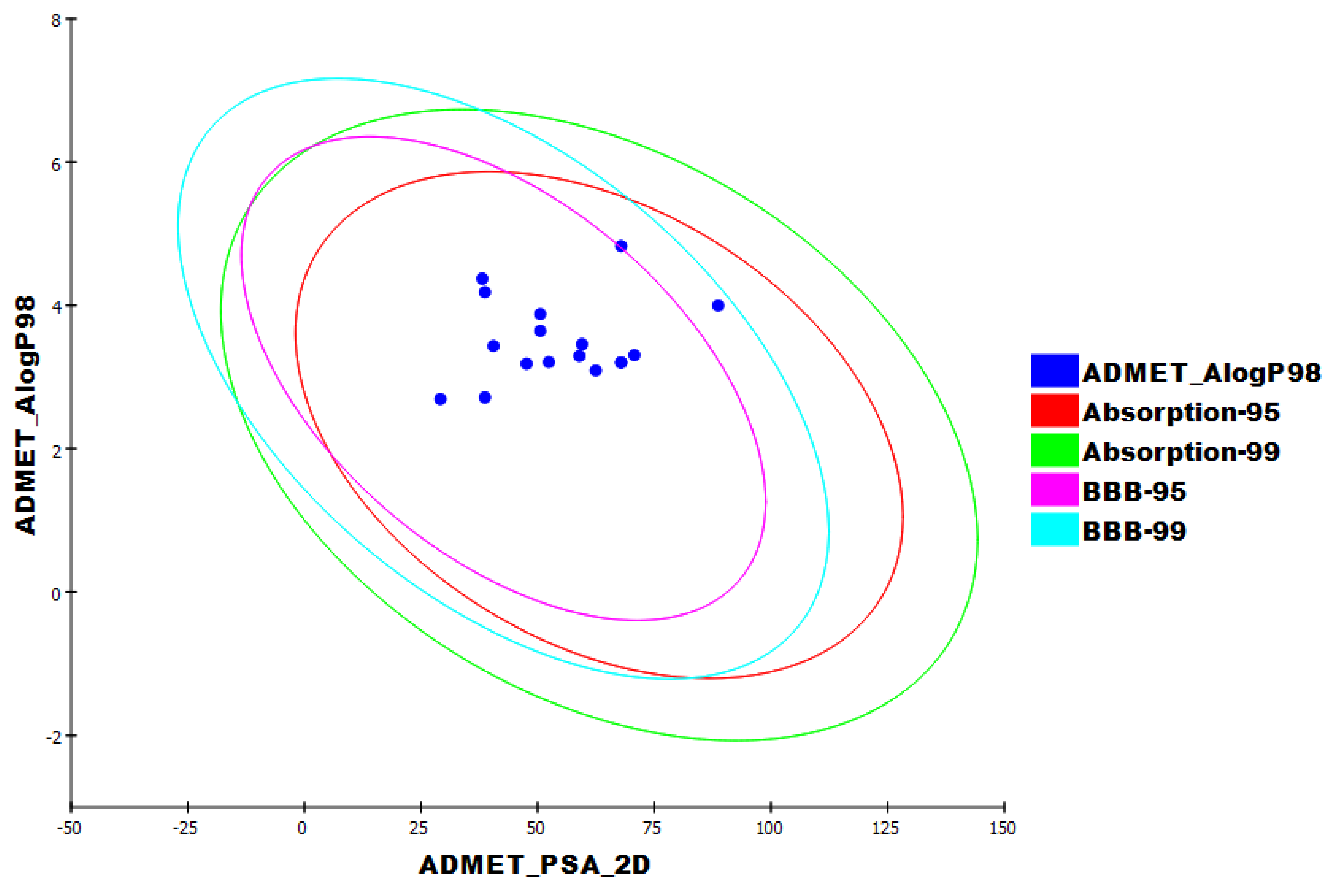
3.4. ADMET Studies
3.5. Toxicity Studies
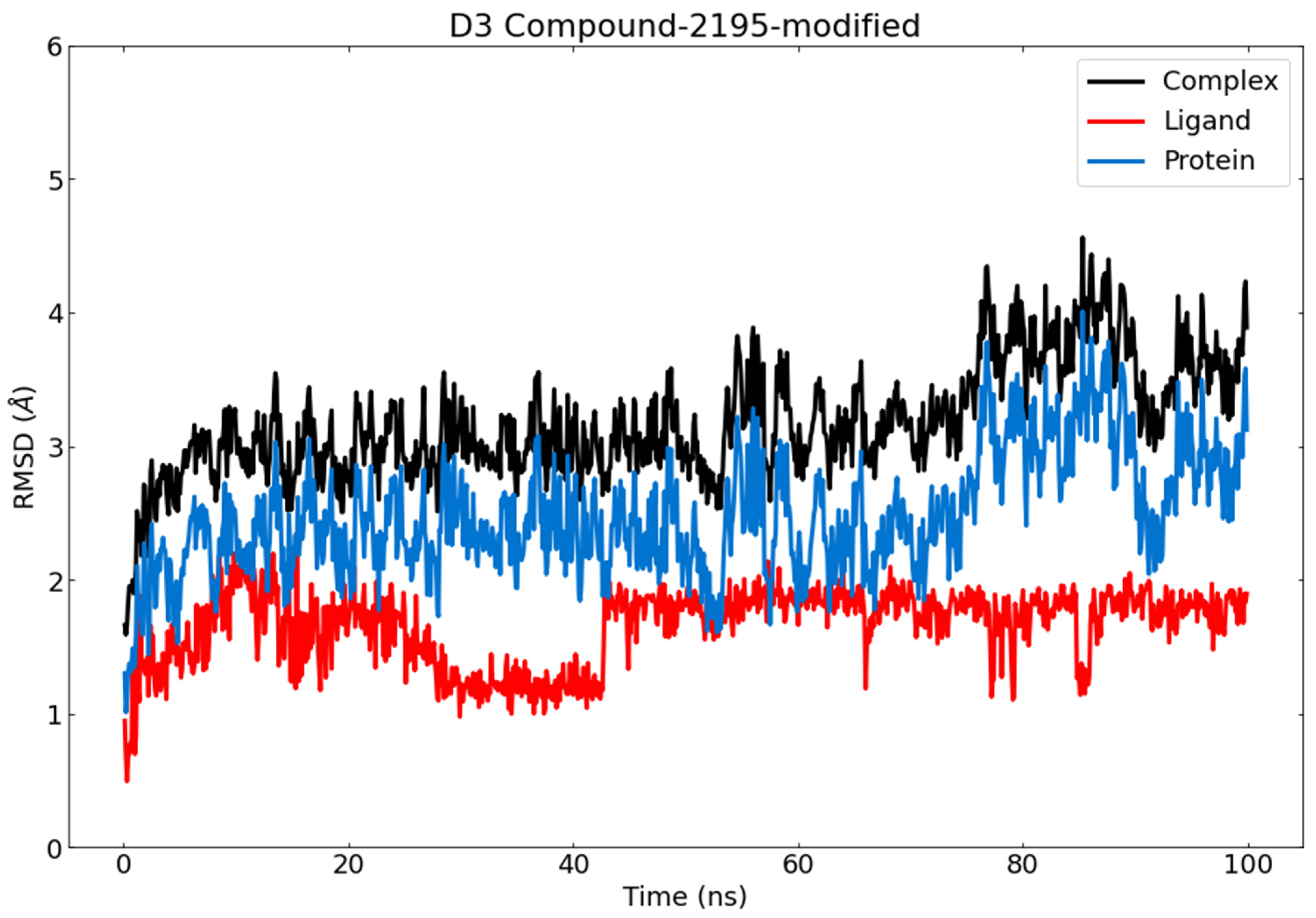
3.6. Molecular Dynamics (MD) Simulations
RMSD and RMSF Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 12 November 2022).
- Engel, T. Basic overview of chemoinformatics. J. Chem. Inf. Model. 2006, 46, 2267–2277. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Hagler, A. Chemoinformatics and drug discovery. Molecules 2002, 7, 566–600. [Google Scholar] [CrossRef]
- Marrone, T.J.; Briggs, A.; James, M.; McCammon, J.A. Structure-based drug design: Computational advances. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, Y.; Li, W.; Li, H.; Yang, L.; Wang, J.; Mahdy, H.A.; Mehany, A.; Jaiash, D.A.; Santali, E.Y. Screening of Some Sulfonamide and Sulfonylurea Derivatives as Anti-Alzheimer’s Agents Targeting BACE1 and PPARγ. J. Chem. 2020, 2020, 1631243. [Google Scholar] [CrossRef]
- Abdel-Aziz, H.A.; Eldehna, W.M.; Fares, M.; Al-Rashood, S.T.; Al-Rashood, K.A.; Abdel-Aziz, M.M.; Soliman, D.H. Synthesis, biological evaluation and 2D-QSAR study of halophenyl bis-hydrazones as antimicrobial and antitubercular agents. Int. J. Mol. Sci. 2015, 16, 8719–8743. [Google Scholar] [CrossRef]
- March-Vila, E.; Pinzi, L.; Sturm, N.; Tinivella, A.; Engkvist, O.; Chen, H.; Rastelli, G. On the integration of in silico drug design methods for drug repurposing. Front. Pharmacol. 2017, 8, 298. [Google Scholar] [CrossRef]
- Zhang, W.; Pei, J.; Lai, L. Computational multitarget drug design. J. Chem. Inf. Model. 2017, 57, 403–412. [Google Scholar] [CrossRef]
- Youssef, M.I.; Zhou, Y.; Eissa, I.H.; Wang, Y.; Zhang, J.; Jiang, L.; Hu, W.; Qi, J.; Chen, Z. Tetradecyl 2,3-dihydroxybenzoate alleviates oligodendrocyte damage following chronic cerebral hypoperfusion through IGF-1 receptor. Neurochem. Int. 2020, 138, 104749. [Google Scholar] [CrossRef]
- Alsaif, N.A.; Taghour, M.S.; Alanazi, M.M.; Obaidullah, A.J.; Al-Mehizia, A.A.; Alanazi, M.M.; Aldawas, S.; Elwan, A.; Elkady, H. Discovery of new VEGFR-2 inhibitors based on bis ([1, 2, 4] triazolo)[4,3-a:3′,4′-c] quinoxaline derivatives as anticancer agents and apoptosis inducers. J. Enzym. Inhib. Med. Chem. 2021, 36, 1093–1114. [Google Scholar] [CrossRef]
- Alsaif, N.A.; Dahab, M.A.; Alanazi, M.M.; Obaidullah, A.J.; Al-Mehizia, A.A.; Alanazi, M.M.; Aldawas, S.; Mahdy, H.A.; Elkady, H. New quinoxaline derivatives as VEGFR-2 inhibitors with anticancer and apoptotic activity: Design, molecular modeling, and synthesis. Bioorg. Chem. 2021, 110, 104807. [Google Scholar] [CrossRef]
- El-Adl, K.; Ibrahim, M.-K.; Alesawy, M.S.; Eissa, I.H. [1, 2, 4] Triazolo [4, 3-c] quinazoline and bis ([1, 2, 4] triazolo)[4,3-a:4′,3′-c] quinazoline derived DNA intercalators: Design, synthesis, in silico ADMET profile, molecular docking and anti-proliferative evaluation studies. Bioorg. Med. Chem. 2021, 30, 115958. [Google Scholar] [CrossRef] [PubMed]
- El-Metwally, S.A.; Abou-El-Regal, M.M.; Eissa, I.H.; Mehany, A.B.; Mahdy, H.A.; Elkady, H.; Elwan, A.; Elkaeed, E.B. Discovery of thieno [2, 3-d] pyrimidine-based derivatives as potent VEGFR-2 kinase inhibitors and anti-cancer agents. Bioorg. Chem. 2021, 112, 104947. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, M.M.; Eissa, I.H.; Alsaif, N.A.; Obaidullah, A.J.; Alanazi, W.A.; Alasmari, A.F.; Albassam, H.; Elkady, H.; Elwan, A. Design, synthesis, docking, ADMET studies, and anticancer evaluation of new 3-methylquinoxaline derivatives as VEGFR-2 inhibitors and apoptosis inducers. J. Enzym. Inhib. Med. Chem. 2021, 36, 1760–1782. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, M.M.; Alaa, E.; Alsaif, N.A.; Obaidullah, A.J.; Alkahtani, H.M.; Al-Mehizia, A.A.; Alsubaie, S.M.; Taghour, M.S.; Eissa, I.H. Discovery of new 3-methylquinoxalines as potential anti-cancer agents and apoptosis inducers targeting VEGFR-2: Design, synthesis, and in silico studies. J. Enzym. Inhib. Med. Chem. 2021, 36, 1732–1750. [Google Scholar] [CrossRef]
- Elkaeed, E.B.; Yousef, R.G.; Elkady, H.; Alsfouk, A.A.; Husein, D.Z.; Ibrahim, I.M.; Alswah, M.; Elzahabi, H.S.A.; Metwaly, A.M.; Eissa, I.H. A New Theobromine-Based EGFRWT and EGFRT790M Inhibitor and Apoptosis Inducer: Design, Semi-Synthesis, Docking, DFT, MD Simulations, and In Vitro Studies. Processes 2022, 10, 2290. [Google Scholar] [CrossRef]
- Metwaly, A.M.; Ghoneim, M.M.; Eissa, I.H.; Elsehemy, I.A.; Mostafa, A.E.; Hegazy, M.M.; Afifi, W.M.; Dou, D. Traditional ancient Egyptian medicine: A review. Saudi J. Biol. Sci. 2021, 28, 5823–5832. [Google Scholar] [CrossRef]
- Han, X.; Yang, Y.; Metwaly, A.M.; Xue, Y.; Shi, Y.; Dou, D. The Chinese herbal formulae (Yitangkang) exerts an antidiabetic effect through the regulation of substance metabolism and energy metabolism in type 2 diabetic rats. J. Ethnopharmacol. 2019, 239, 111942. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Báez-Santos, Y.M.; John, S.E.S.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef]
- Alesawy, M.S.; Abdallah, A.E.; Taghour, M.S.; Elkaeed, E.B.; Eissa, I.H.; Metwaly, A.M. In Silico Studies of Some Isoflavonoids as Potential Candidates against COVID-19 Targeting Human ACE2 (hACE2) and Viral Main Protease (Mpro). Molecules 2021, 26, 2806. [Google Scholar] [CrossRef]
- El-Demerdash, A.; Metwaly, A.M.; Hassan, A.; El-Aziz, A.; Mohamed, T.; Elkaeed, E.B.; Eissa, I.H.; Arafa, R.K.; Stockand, J.D. Comprehensive virtual screening of the antiviral potentialities of marine polycyclic guanidine alkaloids against SARS-CoV-2 (COVID-19). Biomolecules 2021, 11, 460. [Google Scholar] [CrossRef] [PubMed]
- ANPDB African Natural Products Database. Available online: http://african-compounds.org/anpdb/compounds_list/ (accessed on 11 November 2021).
- Fu, Z.; Huang, B.; Tang, J.; Liu, S.; Liu, M.; Ye, Y.; Liu, Z.; Xiong, Y.; Zhu, W.; Cao, D. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 2021, 12, 488. [Google Scholar] [CrossRef]
- R.P.D. Bank. X-ray Structural and Biological Evaluation of a Series of Potent and Highly Selective Inhibitors of Human Coronavirus Papain-Like Proteases. Available online: https://www.rcsb.org/structure/4OW0 (accessed on 14 August 2022).
- Parmar, D.R.; Soni, J.Y.; Guduru, R.; Rayani, R.H.; Kusurkar, R.V.; Vala, A.G.; Talukdar, S.N.; Eissa, I.H.; Metwaly, A.M.; Khalil, A. Discovery of new anticancer thiourea-azetidine hybrids: Design, synthesis, in vitro antiproliferative, SAR, in silico molecular docking against VEGFR-2, ADMET, toxicity, and DFT studies. Bioorg. Chem. 2021, 115, 105206. [Google Scholar] [CrossRef] [PubMed]
- Yousef, R.; Sakr, H.; Eissa, I.; Mehany, A.; Metwaly, A.; Elhendawy, M.A.; Radwan, M.; ElSohly, M.A.; Abulkhair, H.S.; El-Adl, K. New quinoxaline-2 (1H)-ones as potential VEGFR-2 inhibitors: Design, synthesis, molecular docking, ADMET profile and anti-proliferative evaluations. New J. Chem. 2021, 45, 16949–16964. [Google Scholar] [CrossRef]
- Amer, H.H.; Alotaibi, S.H.; Trawneh, A.H.; Metwaly, A.M.; Eissa, I.H. Anticancer activity, spectroscopic and molecular docking of some new synthesized sugar hydrazones, Arylidene and α-Aminophosphonate derivatives. Arab. J. Chem. 2021, 14, 103348. [Google Scholar] [CrossRef]
- Alesawy, M.S.; Al-Karmalawy, A.A.; Elkaeed, E.B.; Alswah, M.; Belal, A.; Taghour, M.S.; Eissa, I.H. Design and discovery of new 1,2,4-triazolo[4,3-c] quinazolines as potential DNA intercalators and topoisomerase II inhibitors. Arch. Der Pharm. 2021, 354, 2000237. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Wawer, M.; Peltason, L.; Weskamp, N.; Teckentrup, A.; Bajorath, J. Structure–activity relationship anatomy by network-like similarity graphs and local structure–activity relationship indices. J. Med. Chem. 2008, 51, 6075–6084. [Google Scholar] [CrossRef] [PubMed]
- Farhadi, F.; Khameneh, B.; Iranshahi, M.; Iranshahy, M. Antibacterial activity of flavonoids and their structure–activity relationship: An update review. Phytother. Res. 2019, 33, 13–40. [Google Scholar] [CrossRef] [PubMed]
- Burke, B.J. Developments in Molecular Shape Analysis to Establish Spatial Similarity among Flexible Molecules; University of Illinois at Chicago, Health Sciences Center: Chicago, IL, USA, 1993. [Google Scholar]
- Willett, P. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Briem, H.; Kuntz, I.D. Molecular similarity based on DOCK-generated fingerprints. J. Med. Chem. 1996, 39, 3401–3408. [Google Scholar] [CrossRef] [PubMed]
- Willett, P. Similarity searching using 2D structural fingerprints. Chemoinform. Comput. Chem. Biol. 2010, 672, 133–158. [Google Scholar]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Chu, H.; He, Q.-X.; Wang, J.; Hu, Y.; Wang, Y.-Q.; Lin, Z.-H. In silico design of novel benzohydroxamate-based compounds as inhibitors of histone deacetylase 6 based on 3D-QSAR, molecular docking, and molecular dynamics simulations. New J. Chem. 2020, 44, 21201–21210. [Google Scholar] [CrossRef]
- Ieritano, C.; Campbell, J.L.; Hopkins, W.S. Predicting differential ion mobility behaviour in silico using machine learning. Analyst 2021, 146, 4737–4743. [Google Scholar] [CrossRef]
- Taha, M.; Ismail, N.H.; Ali, M.; Rashid, U.; Imran, S.; Uddin, N.; Khan, K.M. Molecular hybridization conceded exceptionally potent quinolinyl-oxadiazole hybrids through phenyl linked thiosemicarbazide antileishmanial scaffolds: In silico validation and SAR studies. Bioorg. Chem. 2017, 71, 192–200. [Google Scholar] [CrossRef]
- Heikamp, K.; Bajorath, J. How do 2D fingerprints detect structurally diverse active compounds? Revealing compound subset-specific fingerprint features through systematic selection. J. Chem. Inf. Model. 2011, 51, 2254–2265. [Google Scholar] [CrossRef] [PubMed]
- Opo, F.A.D.M.; Rahman, M.M.; Ahammad, F.; Ahmed, I.; Bhuiyan, M.A.; Asiri, A.M. Structure based pharmacophore modeling, virtual screening, molecular docking and ADMET approaches for identification of natural anti-cancer agents targeting XIAP protein. Sci. Rep. 2021, 11, 4049. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Dixon, S.L.; Lowrie, J.F.; Sherman, W. Analysis and comparison of 2D fingerprints: Insights into database screening performance using eight fingerprint methods. J. Mol. Graph. Model. 2010, 29, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Sastry, M.; Lowrie, J.F.; Dixon, S.L.; Sherman, W. Large-scale systematic analysis of 2D fingerprint methods and parameters to improve virtual screening enrichments. J. Chem. Inf. Model. 2010, 50, 771–784. [Google Scholar] [CrossRef]
- Kogej, T.; Engkvist, O.; Blomberg, N.; Muresan, S. Multifingerprint based similarity searches for targeted class compound selection. J. Chem. Inf. Model. 2006, 46, 1201–1213. [Google Scholar] [CrossRef]
- Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular similarity in medicinal chemistry: Miniperspective. J. Med. Chem. 2014, 57, 3186–3204. [Google Scholar] [CrossRef]
- Muegge, I.; Mukherjee, P. An overview of molecular fingerprint similarity search in virtual screening. Expert Opin. Drug Discov. 2016, 11, 137–148. [Google Scholar] [CrossRef]
- Turchi, M.; Cai, Q.; Lian, G. An evaluation of in-silico methods for predicting solute partition in multiphase complex fluids—A case study of octanol/water partition coefficient. Chem. Eng. Sci. 2019, 197, 150–158. [Google Scholar] [CrossRef]
- Sullivan, K.M.; Enoch, S.J.; Ezendam, J.; Sewald, K.; Roggen, E.L.; Cochrane, S. An adverse outcome pathway for sensitization of the respiratory tract by low-molecular-weight chemicals: Building evidence to support the utility of in vitro and in silico methods in a regulatory context. Appl. Vitr. Toxicol. 2017, 3, 213–226. [Google Scholar] [CrossRef]
- Altamash, T.; Amhamed, A.; Aparicio, S.; Atilhan, M. Effect of hydrogen bond donors and acceptors on CO2 absorption by deep eutectic solvents. Processes 2020, 8, 1533. [Google Scholar] [CrossRef]
- Wan, Y.; Tian, Y.; Wang, W.; Gu, S.; Ju, X.; Liu, G. In silico studies of diarylpyridine derivatives as novel HIV-1 NNRTIs using docking-based 3D-QSAR, molecular dynamics, and pharmacophore modeling approaches. RSC Adv. 2018, 8, 40529–40543. [Google Scholar] [CrossRef] [PubMed]
- Escamilla-Gutiérrez, A.; Ribas-Aparicio, R.M.; Córdova-Espinoza, M.G.; Castelán-Vega, J.A. In silico strategies for modeling RNA aptamers and predicting binding sites of their molecular targets. Nucleosides Nucleotides Nucleic Acids 2021, 40, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.C.; Kumar, A.; Bharadwaj, S.; Chaudhary, R.; Sahi, S. Ligand-Based Approach for In-silico Drug Designing. In Bioinformatics Techniques for Drug Discovery; Springer: Berlin/Heidelberg, Germany, 2018; pp. 11–19. [Google Scholar]
- Jain, A.N. Morphological similarity: A 3D molecular similarity method correlated with protein-ligand recognition. J. Comput.-Aided Mol. Des. 2000, 14, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ren, J.-X.; Ma, J.-X.; Ding, L. Development of an in silico prediction model for chemical-induced urinary tract toxicity by using naïve Bayes classifier. Mol. Divers. 2019, 23, 381–392. [Google Scholar] [CrossRef]
- Norinder, U.; Bergström, C.A. Prediction of ADMET properties. ChemMedChem Chem. Enabling Drug Discov. 2006, 1, 920–937. [Google Scholar]
- Ferreira, L.L.; Andricopulo, A.D. ADMET modeling approaches in drug discovery. Drug Discov. Today 2019, 24, 1157–1165. [Google Scholar] [CrossRef]
- Idakwo, G.; Luttrell, J.; Chen, M.; Hong, H.; Zhou, Z.; Gong, P.; Zhang, C. A review on machine learning methods for in silico toxicity prediction. J. Environ. Sci. Health Part C 2018, 36, 169–191. [Google Scholar] [CrossRef]
- Raies, A.B.; Bajic, V.B. In silico toxicology: Computational methods for the prediction of chemical toxicity. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2016, 6, 147–172. [Google Scholar] [CrossRef]
- Xia, X.; Maliski, E.G.; Gallant, P.; Rogers, D. Classification of kinase inhibitors using a Bayesian model. J. Med. Chem. 2004, 47, 4463–4470. [Google Scholar] [CrossRef]
- BIOVIA QSAR, ADMET and Predictive Toxicology. Available online: https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/qsar-admet-and-predictive-toxicology.html (accessed on 18 August 2022).
- Venkatapathy, R.; Wang, N.C.Y.; Martin, T.M.; Harten, P.F.; Young, D. Structure–Activity Relationships for Carcinogenic Potential. Gen. Appl. Syst. Toxicol. 2009. [Google Scholar] [CrossRef]
- Goodrnan, G.; Wilson, R. Comparison of the dependence of the TD50 on maximum tolerated dose for mutagens and nonmutagens. Risk Anal. 1992, 12, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Council, N.R. Correlation Between Carcinogenic Potency and the Maximum Tolerated Dose: Implications for Risk Assessment. In Issues in Risk Assessment; National Academies Press (US): Washington, DC, USA, 1993. [Google Scholar]
- Gonella Diaza, R.; Manganelli, S.; Esposito, A.; Roncaglioni, A.; Manganaro, A.; Benfenati, E. Comparison of in silico tools for evaluating rat oral acute toxicity. SAR QSAR Environ. Res. 2015, 26, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, F.; Benfenati, E. In silico models for repeated-dose toxicity (RDT): Prediction of the no observed adverse effect level (NOAEL) and lowest observed adverse effect level (LOAEL) for drugs. In In Silico Methods for Predicting Drug Toxicity; Springer: Berlin/Heidelberg, Germany, 2016; pp. 163–176. [Google Scholar]
- Venkatapathy, R.; Moudgal, C.J.; Bruce, R.M. Assessment of the oral rat chronic lowest observed adverse effect level model in TOPKAT, a QSAR software package for toxicity prediction. J. Chem. Inf. Comput. Sci. 2004, 44, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmus, K.R. The Draize eye test. Surv. Ophthalmol. 2001, 45, 493–515. [Google Scholar] [CrossRef]
- Pillay, C.C.; Jäger, A.K.; Mulholland, D.A.; Van Staden, J. Cyclooxygenase inhibiting and anti-bacterial activities of South African Erythrina species. J. Ethnopharmacol. 2001, 74, 231–237. [Google Scholar] [CrossRef]
- Oyama, S.D.O.; Souza, L.A.D.; Baldoqui, D.C.; Sarragiotto, M.H.; Silva, A.A. Prenylated flavonoids from Maclura tinctoria fruits. Química Nova 2013, 36, 800–802. [Google Scholar] [CrossRef][Green Version]
- Delle Monache, G.; De Rosa, M.C.; Scurria, R.; Vitali, A.; Cuteri, A.; Monacelli, B.; Pasqua, G.; Botta, B. Comparison between metabolite productions in cell culture and in whole plant of Maclura pomifera. Phytochemistry 1995, 39, 575–580. [Google Scholar] [CrossRef]
- Atta-ur-Rahman. Studies in Natural Products Chemistry: Indices Part A; Elsevier Science: Amsterdam, The Netherlands, 1997. [Google Scholar]
- Akhtar, A.; Hussain, W.; Rasool, N. Probing the pharmacological binding properties, and reactivity of selective phytochemicals as potential HIV-1 protease inhibitors. Univ. Sci. 2019, 24, 441–464. [Google Scholar] [CrossRef]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Protein–ligand docking: Current status and future challenges. Proteins Struct. Funct. Bioinform. 2006, 65, 15–26. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef]
- Liu, X.; Shi, D.; Zhou, S.; Liu, H.; Liu, H.; Yao, X. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 2018, 13, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Hansson, T.; Oostenbrink, C.; van Gunsteren, W. Molecular dynamics simulations. Curr. Opin. Struct. Biol. 2002, 12, 190–196. [Google Scholar] [CrossRef]
- Kuzmanic, A.; Zagrovic, B. Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys. J. 2010, 98, 861–871. [Google Scholar] [CrossRef]
- Liu, P.; Lu, J.; Yu, H.; Ren, N.; Lockwood, F.E.; Wang, Q.J. Lubricant shear thinning behavior correlated with variation of radius of gyration via molecular dynamics simulations. J. Chem. Phys. 2017, 147, 084904. [Google Scholar] [CrossRef]
- Kumar, K.; Anbarasu, A.; Ramaiah, S. Molecular docking and molecular dynamics studies on β-lactamases and penicillin binding proteins. Mol. Biosyst. 2014, 10, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Mitternacht, S. FreeSASA: An open source C library for solvent accessible surface area calculations. F1000Research 2016, 5, 189. [Google Scholar] [CrossRef] [PubMed]
- Naik, V.R.; Munikumar, M.; Ramakrishna, U.; Srujana, M.; Goudar, G.; Naresh, P.; Kumar, B.N.; Hemalatha, R.J. Dynamics, Remdesivir (GS-5734) as a therapeutic option of 2019-nCOV main protease–in silico approach. J. Biomol. Struct. Dyn. 2021, 39, 4701–4714. [Google Scholar] [CrossRef]
- Magro, P.; Zanella, I.; Pescarolo, M.; Castelli, F.; Quiros-Roldan, E.J.B.J. Lopinavir/ritonavir: Repurposing an old drug for HIV infection in COVID-19 treatment. Biomed. J. 2021, 44, 43–53. [Google Scholar] [CrossRef]
- Alexpandi, R.; De Mesquita, J.F.; Pandian, S.K.; Ravi, A.V. Quinolines-based SARS-CoV-2 3CLpro and RdRp inhibitors and Spike-RBD-ACE2 inhibitor for drug-repurposing against COVID-19: An in silico analysis. Front. Microbiol. 2020, 11, 1796. [Google Scholar] [CrossRef]
- Elmezayen, A.D.; Al-Obaidi, A.; Şahin, A.T.; Yelekçi, K. Dynamics, Drug repurposing for coronavirus (COVID-19): In silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. J. Biomol. Struct. Dyn. 2021, 39, 2980–2992. [Google Scholar] [CrossRef]
- Keretsu, S.; Bhujbal, S.P.; Cho, S.J. Rational approach toward COVID-19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Sci. Rep. 2020, 10, 17716. [Google Scholar] [CrossRef] [PubMed]
- Koulgi, S.; Jani, V.; Uppuladinne, M.; Sonavane, U.; Nath, A.K.; Darbari, H.; Joshi, R. Dynamics, Drug repurposing studies targeting SARS-CoV-2: An ensemble docking approach on drug target 3C-like protease (3CLpro). J. Biomol. Struct. Dyn. 2021, 39, 5735–5755. [Google Scholar] [CrossRef] [PubMed]
- Eissa, I.H.; Khalifa, M.M.; Elkaeed, E.B.; Hafez, E.E.; Alsfouk, A.A.; Metwaly, A.M. In Silico Exploration of Potential Natural Inhibitors against SARS-Cov-2 nsp10. Molecules 2021, 26, 6151. [Google Scholar] [CrossRef]
- Elkaeed, E.B.; Youssef, F.S.; Eissa, I.H.; Elkady, H.; Alsfouk, A.A.; Ashour, M.L.; El Hassab, M.A.; Abou-Seri, S.M.; Metwaly, A.M. Multi-Step In Silico Discovery of Natural Drugs against COVID-19 Targeting Main Protease. Int. J. Mol. Sci. 2022, 23, 6912. [Google Scholar] [CrossRef]
- Elkaeed, E.B.; Eissa, I.H.; Elkady, H.; Abdelalim, A.; Alqaisi, A.M.; Alsfouk, A.A.; Elwan, A.; Metwaly, A.M. A Multistage In Silico Study of Natural Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Int. J. Mol. Sci. 2022, 23, 8407. [Google Scholar] [CrossRef] [PubMed]
- Elkaeed, E.B.; Metwaly, A.M.; Alesawy, M.S.; Saleh, A.M.; Alsfouk, A.A.; Eissa, I.H. Discovery of Potential SARS-CoV-2 Papain-like Protease Natural Inhibitors Employing a Multi-Phase In Silico Approach. Life 2022, 12, 1407. [Google Scholar] [CrossRef]
- Eissa, I.H.; Alesawy, M.S.; Saleh, A.M.; Elkaeed, E.B.; Alsfouk, B.A.; El-Attar, A.-A.M.M.; Metwaly, A.M. Ligand and Structure-Based In Silico Determination of the Most Promising SARS-CoV-2 nsp16-nsp10 2′-o-Methyltransferase Complex Inhibitors among 3009 FDA Approved Drugs. Molecules 2022, 27, 2287. [Google Scholar]
- Elkaeed, E.B.; Elkady, H.; Belal, A.; Alsfouk, B.A.; Ibrahim, T.H.; Abdelmoaty, M.; Arafa, R.K.; Metwaly, A.M.; Eissa, I.H. Multi-Phase In Silico Discovery of Potential SARS-CoV-2 RNA-Dependent RNA Polymerase Inhibitors among 3009 Clinical and FDA-Approved Related Drugs. Processes 2022, 10, 530. [Google Scholar] [CrossRef]
- Metwaly, A.M.; Elwan, A.; El-Attar, A.-A.M.M.; Al-Rashood, S.T.; Eissa, I.H. Structure-Based Virtual Screening, Docking, ADMET, Molecular Dynamics, and MM-PBSA Calculations for the Discovery of Potential Natural SARS-CoV-2 Helicase Inhibitors from the Traditional Chinese Medicine. J. Chem. 2022, 2022, 7270094. [Google Scholar] [CrossRef]
- Alesawy, M.S.; Elkaeed, E.B.; Alsfouk, A.A.; Metwaly, A.M.; Eissa, I. In Silico Screening of Semi-Synthesized Compounds as Potential Inhibitors for SARS-CoV-2 Papain-Like Protease: Pharmacophoric Features, Molecular Docking, ADMET, Toxicity and DFT Studies. Molecules 2021, 26, 6593. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Similarity | SA | SB | SC | Compound | Similarity | SA | SB | SC |
|---|---|---|---|---|---|---|---|---|---|
| TTT | 1.000 | 454 | 0 | 0 | 3448 | 0.632653 | 279 | −13 | 175 |
| 2538 | 0.758 | 322 | −29 | 132 | 3647 | 0.632479 | 296 | 14 | 158 |
| 3518 | 0.747 | 324 | −20 | 130 | 292 | 0.632249 | 447 | 253 | 7 |
| 3323 | 0.743 | 324 | −18 | 130 | 1795 | 0.632054 | 280 | −11 | 174 |
| 2982 | 0.743 | 329 | −11 | 125 | 3414 | 0.631699 | 554 | 423 | −100 |
| 2981 | 0.738 | 327 | −11 | 127 | 2259 | 0.631188 | 255 | −50 | 199 |
| 182 | 0.732 | 314 | −25 | 140 | 3040 | 0.63035 | 324 | 60 | 130 |
| 2677 | 0.720 | 317 | −14 | 137 | 1157 | 0.630081 | 310 | 38 | 144 |
| 2558 | 0.712 | 442 | 167 | 12 | 1141 | 0.629797 | 279 | −11 | 175 |
| 2554 | 0.710 | 316 | −9 | 138 | 2180 | 0.62963 | 255 | −49 | 199 |
| 1875 | 0.710 | 320 | −3 | 134 | 203 | 0.628889 | 283 | −4 | 171 |
| 197 | 0.708 | 334 | 18 | 120 | 3413 | 0.628831 | 554 | 427 | −100 |
| 1168 | 0.703 | 298 | −30 | 156 | 2108 | 0.628062 | 282 | −5 | 172 |
| 2556 | 0.703 | 298 | −30 | 156 | 1332 | 0.628009 | 287 | 3 | 167 |
| 1001 | 0.702 | 297 | −31 | 157 | 3039 | 0.627907 | 324 | 62 | 130 |
| 165 | 0.701 | 303 | −22 | 151 | 3420 | 0.627273 | 276 | −14 | 178 |
| 2221 | 0.701 | 328 | 14 | 126 | 3085 | 0.626506 | 260 | −39 | 194 |
| 2579 | 0.700 | 301 | −24 | 153 | 3115 | 0.626223 | 320 | 57 | 134 |
| 4579 | 0.699 | 588 | 387 | −134 | 1154 | 0.625541 | 289 | 8 | 165 |
| 1195 | 0.698 | 296 | −30 | 158 | 1153 | 0.625541 | 289 | 8 | 165 |
| 900 | 0.698 | 296 | −30 | 158 | 1169 | 0.625282 | 277 | −11 | 177 |
| 2197 | 0.697 | 355 | 55 | 99 | 161 | 0.625282 | 277 | −11 | 177 |
| 212 | 0.697 | 306 | −15 | 148 | 1140 | 0.625282 | 277 | −11 | 177 |
| 211 | 0.697 | 306 | −15 | 148 | 2588 | 0.625282 | 277 | −11 | 177 |
| 2578 | 0.694 | 300 | −22 | 154 | 4598 | 0.62256 | 287 | 7 | 167 |
| 205 | 0.691 | 318 | 6 | 136 | 848 | 0.622517 | 282 | −1 | 172 |
| 2555 | 0.688 | 296 | −24 | 158 | 1155 | 0.62203 | 288 | 9 | 166 |
| 3079 | 0.687 | 398 | 125 | 56 | 3412 | 0.620843 | 280 | −3 | 174 |
| 4573 | 0.687 | 417 | 153 | 37 | 2137 | 0.620536 | 278 | −6 | 176 |
| 2557 | 0.686 | 308 | −5 | 146 | 2407 | 0.620451 | 358 | 123 | 96 |
| 4572 | 0.686 | 410 | 144 | 44 | 1147 | 0.62 | 279 | −4 | 175 |
| 3421 | 1 | 311 | 0 | 143 | 637 | 0.619048 | 325 | 71 | 129 |
| 2202 | 1 | 282 | −42 | 172 | 2201 | 0.618893 | 380 | 160 | 74 |
| 213 | 1 | 311 | 1 | 143 | 2199 | 0.618487 | 368 | 141 | 86 |
| 2067 | 1 | 449 | 206 | 5 | 189 | 0.617849 | 270 | −17 | 184 |
| 126 | 1 | 294 | −21 | 160 | 1156 | 0.61753 | 310 | 48 | 144 |
| 2070 | 1 | 470 | 239 | −16 | 2203 | 0.617021 | 261 | −31 | 193 |
| 1330 | 1 | 293 | −21 | 161 | 2685 | 0.616725 | 354 | 120 | 100 |
| 168 | 1 | 301 | −9 | 153 | 1992 | 0.616279 | 265 | −24 | 189 |
| 4575 | 1 | 417 | 163 | 37 | 3469 | 0.615 | 246 | −54 | 208 |
| 1132 | 1 | 387 | 119 | 67 | 4879 | 0.614232 | 328 | 80 | 126 |
| 3419 | 0.673289 | 305 | −1 | 149 | 2 | 0.614191 | 277 | −3 | 177 |
| 1133 | 0.673043 | 387 | 121 | 67 | 3924 | 0.613333 | 276 | −4 | 178 |
| 190 | 0.671772 | 307 | 3 | 147 | 713 | 0.612691 | 280 | 3 | 174 |
| 1331 | 0.670507 | 291 | −20 | 163 | 2206 | 0.610132 | 277 | 0 | 177 |
| 181 | 0.67033 | 305 | 1 | 149 | 1952 | 0.609977 | 269 | −13 | 185 |
| 1000 | 0.670306 | 307 | 4 | 147 | 990 | 0.609865 | 272 | −8 | 182 |
| 163 | 0.668161 | 298 | −8 | 156 | 3392 | 0.609865 | 272 | −8 | 182 |
| 2958 | 0.667814 | 388 | 127 | 66 | 166 | 0.609589 | 267 | −16 | 187 |
| 3923 | 0.667431 | 291 | −18 | 163 | 3411 | 0.607692 | 553 | 456 | −99 |
| 926 | 1 | 304 | 3 | 150 | 2189 | 0.606762 | 341 | 108 | 113 |
| 3473 | 1 | 304 | 3 | 150 | 3445 | 0.605905 | 472 | 325 | −18 |
| 2198 | 1 | 375 | 110 | 79 | 4580 | 0.60414 | 467 | 319 | −13 |
| 2065 | 1 | 455 | 231 | −1 | 1861 | 0.60414 | 467 | 319 | −13 |
| 204 | 1 | 290 | −17 | 164 | 1859 | 0.60414 | 467 | 319 | −13 |
| 3395 | 1 | 315 | 21 | 139 | 3410 | 0.603712 | 553 | 462 | −99 |
| 215 | 1 | 291 | −13 | 163 | 4577 | 0.603359 | 467 | 320 | −13 |
| 4571 | 1 | 405 | 160 | 49 | 1642 | 0.602794 | 302 | 47 | 152 |
| 169 | 0.658314 | 289 | −15 | 165 | 1643 | 0.602794 | 302 | 47 | 152 |
| 3114 | 0.657505 | 311 | 19 | 143 | 1644 | 0.602794 | 302 | 47 | 152 |
| 3394 | 0.655602 | 316 | 28 | 138 | 2107 | 0.602687 | 314 | 67 | 140 |
| 198 | 0.655012 | 281 | −25 | 173 | 671 | 0.602637 | 320 | 77 | 134 |
| 1212 | 0.655012 | 281 | −25 | 173 | 4578 | 0.602581 | 467 | 321 | −13 |
| 3124 | 0.653277 | 309 | 19 | 145 | 4581 | 0.602581 | 467 | 321 | −13 |
| 3098 | 0.653277 | 309 | 19 | 145 | 1977 | 0.602076 | 348 | 124 | 106 |
| 2227 | 0.652268 | 302 | 9 | 152 | 3442 | 0.60177 | 272 | −2 | 182 |
| 2471 | 0.65 | 299 | 6 | 155 | 2194 | 0.601643 | 293 | 33 | 161 |
| 3444 | 0.649886 | 284 | −17 | 170 | 2467 | 0.600887 | 542 | 448 | −88 |
| 3629 | 0.649874 | 258 | −57 | 196 | 2636 | 0.600751 | 480 | 345 | −26 |
| 199 | 0.648402 | 284 | −16 | 170 | 2635 | 0.600751 | 480 | 345 | −26 |
| 1137 | 1 | 419 | 193 | 35 | 4750 | 0.600742 | 486 | 355 | −32 |
| 1136 | 1 | 419 | 193 | 35 | 2496 | 0.600671 | 537 | 440 | −83 |
| 1138 | 1 | 419 | 193 | 35 | 4870 | 0.600372 | 323 | 84 | 131 |
| 1139 | 1 | 419 | 193 | 35 | 692 | 0.599589 | 292 | 33 | 162 |
| 1464 | 0.647208 | 255 | −60 | 199 | 2195 | 0.599589 | 292 | 33 | 162 |
| 4574 | 0.646965 | 405 | 172 | 49 | 4549 | 0.599567 | 554 | 470 | −100 |
| 1768 | 1 | 357 | 98 | 97 | 4551 | 0.599341 | 546 | 457 | −92 |
| 3754 | 1 | 486 | 298 | −32 | 1143 | 0.597802 | 272 | 1 | 182 |
| 2222 | 1 | 285 | −13 | 169 | 4128 | 0.597802 | 272 | 1 | 182 |
| 3396 | 1 | 313 | 31 | 141 | 1352 | 0.597802 | 272 | 1 | 182 |
| 155 | 0.645161 | 300 | 11 | 154 | 4601 | 0.597802 | 272 | 1 | 182 |
| 167 | 0.644295 | 288 | −7 | 166 | 2118 | 0.597802 | 272 | 1 | 182 |
| 2888 | 1 | 284 | −13 | 170 | 2667 | 0.596899 | 308 | 62 | 146 |
| 4266 | 0.643991 | 284 | −13 | 170 | 4486 | 0.596491 | 272 | 2 | 182 |
| 4759 | 0.643392 | 258 | −53 | 196 | 3118 | 0.596429 | 334 | 106 | 120 |
| 1118 | 0.642132 | 253 | −60 | 201 | 3073 | 0.595133 | 269 | −2 | 185 |
| 3113 | 0.642127 | 314 | 35 | 140 | 227 | 0.594771 | 273 | 5 | 181 |
| 3925 | 0.641553 | 281 | −16 | 173 | 228 | 0.594771 | 273 | 5 | 181 |
| 2220 | 0.640244 | 315 | 38 | 139 | 3071 | 0.594714 | 270 | 0 | 184 |
| 4899 | 1 | 282 | −13 | 172 | 4570 | 0.594714 | 270 | 0 | 184 |
| 279 | 1 | 282 | −13 | 172 | 4897 | 0.594714 | 270 | 0 | 184 |
| 1447 | 1 | 282 | −13 | 172 | 2684 | 0.594454 | 343 | 123 | 111 |
| 1559 | 1 | 282 | −13 | 172 | 4485 | 0.594421 | 277 | 12 | 177 |
| 281 | 0.639456 | 282 | −13 | 172 | 278 | 0.594298 | 542 | 458 | −88 |
| 164 | 0.639269 | 280 | −16 | 174 | 4896 | 0.593407 | 270 | 1 | 184 |
| 4468 | 0.639098 | 340 | 78 | 114 | 1117 | 0.593407 | 270 | 1 | 184 |
| 1134 | 0.638066 | 409 | 187 | 45 | 1152 | 0.593254 | 299 | 50 | 155 |
| 1135 | 0.638066 | 409 | 187 | 45 | 1448 | 0.593148 | 277 | 13 | 177 |
| 2200 | 1 | 376 | 138 | 78 | 4129 | 0.593148 | 277 | 13 | 177 |
| 1949 | 0.634033 | 272 | −25 | 182 | 1329 | 0.593148 | 277 | 13 | 177 |
| 4592 | 0.633047 | 295 | 12 | 159 |
| Compound | ALog p | M. Wt | HBA | HBD | Rotatable Bonds | Rings | Aromatic Rings | MFPSA | Minimum Distance |
|---|---|---|---|---|---|---|---|---|---|
| 126 | 2.73 | 272.30 | 4 | 2 | 2 | 3 | 2 | 0.22 | 0.731 |
| 165 | 2.84 | 270.28 | 4 | 1 | 2 | 3 | 2 | 0.211 | 0.731 |
| 2579 | 2.88 | 268.26 | 4 | 1 | 2 | 3 | 2 | 0.215 | 0.730 |
| 182 | 2.71 | 256.30 | 3 | 1 | 2 | 3 | 2 | 0.151 | 0.718 |
| 189 | 2.41 | 267.28 | 3 | 1 | 1 | 4 | 3 | 0.216 | 0.696 |
| 2203 | 3.24 | 252.27 | 2 | 2 | 0 | 4 | 3 | 0.244 | 0.661 |
| 204 | 4.18 | 270.32 | 3 | 1 | 2 | 3 | 2 | 0.139 | 0.660 |
| 164 | 3.46 | 272.30 | 4 | 2 | 2 | 3 | 2 | 0.22 | 0.659 |
| 3395 | 3.55 | 298.33 | 4 | 2 | 4 | 3 | 2 | 0.229 | 0.656 |
| 181 | 3.18 | 300.35 | 4 | 1 | 3 | 3 | 2 | 0.153 | 0.652 |
| 2202 | 3.48 | 236.27 | 1 | 1 | 0 | 4 | 3 | 0.167 | 0.649 |
| 2137 | 4.14 | 266.33 | 2 | 2 | 2 | 3 | 2 | 0.145 | 0.619 |
| 3396 | 3.64 | 284.35 | 3 | 2 | 4 | 3 | 2 | 0.173 | 0.587 |
| 212 | 3.21 | 265.26 | 3 | 1 | 1 | 4 | 3 | 0.207 | 0.556 |
| 211 | 3.21 | 265.26 | 3 | 1 | 1 | 4 | 3 | 0.207 | 0.556 |
| 213 | 3.43 | 279.29 | 3 | 0 | 2 | 4 | 3 | 0.148 | 0.545 |
| 2197 | 3.31 | 323.39 | 4 | 2 | 6 | 3 | 3 | 0.21 | 0.545 |
| 1875 | 4.69 | 264.32 | 2 | 2 | 1 | 3 | 3 | 0.147 | 0.502 |
| 3421 | 3.01 | 281.31 | 3 | 1 | 2 | 4 | 3 | 0.157 | 0.493 |
| 3412 | 3.09 | 228.24 | 3 | 3 | 2 | 2 | 2 | 0.264 | 0.843 |
| 2982 | 3.29 | 270.32 | 3 | 2 | 6 | 2 | 2 | 0.204 | 0.822 |
| 166 | 2.63 | 242.23 | 4 | 1 | 2 | 3 | 2 | 0.245 | 0.816 |
| 3323 | 3.14 | 222.24 | 2 | 0 | 1 | 3 | 2 | 0.123 | 0.815 |
| 197 | 3.29 | 262.31 | 1 | 0 | 2 | 4 | 4 | 0.116 | 0.811 |
| 215 | 2.69 | 223.23 | 3 | 0 | 0 | 4 | 3 | 0.154 | 0.809 |
| 2981 | 4.37 | 252.31 | 2 | 1 | 5 | 2 | 2 | 0.139 | 0.807 |
| 198 | 2.32 | 257.28 | 4 | 2 | 3 | 3 | 2 | 0.196 | 0.788 |
| 3469 | 2.46 | 182.22 | 1 | 1 | 0 | 3 | 3 | 0.154 | 0.785 |
| 3444 | 3.04 | 268.26 | 4 | 1 | 1 | 3 | 2 | 0.241 | 0.781 |
| 2578 | 3.20 | 270.28 | 4 | 2 | 4 | 2 | 2 | 0.239 | 0.774 |
| 1330 | 3.20 | 270.28 | 4 | 2 | 4 | 2 | 2 | 0.239 | 0.774 |
| 1331 | 3.20 | 270.28 | 4 | 2 | 4 | 2 | 2 | 0.239 | 0.774 |
| 2195 | 4.00 | 338.35 | 5 | 3 | 3 | 3 | 2 | 0.257 | 0.772 |
| 3040 | 4.83 | 350.41 | 4 | 2 | 4 | 3 | 2 | 0.185 | 0.768 |
| 4598 | 2.86 | 298.29 | 5 | 1 | 3 | 3 | 2 | 0.22 | 0.761 |
| 3518 | 2.99 | 226.27 | 2 | 1 | 1 | 3 | 2 | 0.133 | 0.757 |
| 168 | 2.98 | 251.24 | 3 | 2 | 0 | 4 | 3 | 0.278 | 0.754 |
| 2677 | 3.30 | 265.31 | 3 | 0 | 2 | 3 | 2 | 0.107 | 0.743 |
| 199 | 2.73 | 271.31 | 4 | 1 | 4 | 3 | 2 | 0.14 | 0.741 |
| 1952 | 3.88 | 230.26 | 3 | 2 | 2 | 2 | 2 | 0.205 | 0.739 |
| TTT | 3.65 | 304.39 | 2 | 2 | 3 | 3 | 3 | 0.171 |
| Compound | ΔG | Compound | ΔG |
|---|---|---|---|
| 126 | −10.70 | 2982 | −11.85 |
| 165 | −9.81 | 166 | −7.96 |
| 2579 | −9.17 | 3323 | −10.11 |
| 182 | −12.74 | 197 | −9.72 |
| 189 | −8.65 | 215 | −13.18 |
| 2203 | −8.14 | 2981 | −13.72 |
| 204 | −12.21 | 198 | −8.01 |
| 164 | −13.22 | 3469 | −5.40 |
| 3395 | −7.62 | 3444 | −8.88 |
| 181 | −11.55 | 2578 | −9.51 |
| 2202 | −9.22 | 1330 | −12.20 |
| 2137 | −9.98 | 1331 | −14.09 |
| 3396 | −14.14 | 2195 | −16.52 |
| 212 | −12.39 | 3040 | −14.25 |
| 211 | −10.35 | 4598 | −10.13 |
| 213 | −12.63 | 3518 | −7.54 |
| 2197 | −13.10 | 168 | −16.41 |
| 1875 | −8.65 | 2677 | −9.54 |
| 3421 | −7.76 | 199 | −10.79 |
| 3412 | −14.00 | 1952 | −12.93 |
| TTT | −9.30 |
| Compound | BBB Level a | HIA b | Aq c | CYP2D6 d | PPB e |
|---|---|---|---|---|---|
| 164 | 2 | 0 | 3 | f | t |
| 181 | 1 | 0 | 2 | t | t |
| 182 | 1 | 0 | 3 | t | t |
| 204 | 1 | 0 | 2 | t | t |
| 212 | 1 | 0 | 2 | f | t |
| 213 | 1 | 0 | 2 | f | t |
| 215 | 1 | 0 | 2 | t | t |
| 1330 | 2 | 0 | 3 | f | t |
| 1331 | 2 | 0 | 3 | f | t |
| 1952 | 1 | 0 | 3 | f | t |
| 2195 | 2 | 0 | 2 | t | t |
| 2197 | 2 | 0 | 2 | t | t |
| 2981 | 1 | 0 | 2 | f | t |
| 2982 | 2 | 0 | 3 | t | t |
| 3040 | 1 | 0 | 2 | f | t |
| 3396 | 1 | 0 | 3 | f | t |
| 3412 | 2 | 0 | 3 | f | t |
| Remdesivir | 4 | 3 | 3 | f | f |
| Compound | FDA Rat Carcinogenicity | TD50 (Rat) a | MTD b | Rat Oral LD50 b | LOAEL b | Ocular Irritancy | Skin Irritancy |
|---|---|---|---|---|---|---|---|
| Hippacine (164) | Not carcinogenic | 63.019 | 0.285 | 0.441 | 0.052 | Mild | None |
| Naamine D (2197) | Not carcinogenic | 4.022 | 0.086 | 2.440 | 0.015 | Moderate | None |
| (±)-Enterofuran (3412) | Carcinogenic | 87.484 | 0.690 | 2.483 | 0.089 | Severe | None |
| Daphnelone (2982) | Carcinogenic | 184.723 | 0.829 | 0.646 | 0.173 | Severe | None |
| 4,2′-dihydroxy-4′-methoxychalcone (1330) | Not carcinogenic | 259.532 | 0.320 | 1.010 | 0.060 | None | None |
| 2′,5′-dihydroxy-4-methoxychalcone (1331) | Not carcinogenic | 259.532 | 0.320 | 1.010 | 0.060 | Mild | None |
| Wighteone (2195) | Not carcinogenic | 42.573 | 0.525 | 0.962 | 0.053 | Severe | None |
| Remdesivir | Not carcinogenic | 9.246 | 0.235 | 0.309 | 0.004 | Mild | Mild |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elkaeed, E.B.; Khalifa, M.M.; Alsfouk, B.A.; Alsfouk, A.A.; El-Attar, A.-A.M.M.; Eissa, I.H.; Metwaly, A.M. The Discovery of Potential SARS-CoV-2 Natural Inhibitors among 4924 African Metabolites Targeting the Papain-like Protease: A Multi-Phase In Silico Approach. Metabolites 2022, 12, 1122. https://doi.org/10.3390/metabo12111122
Elkaeed EB, Khalifa MM, Alsfouk BA, Alsfouk AA, El-Attar A-AMM, Eissa IH, Metwaly AM. The Discovery of Potential SARS-CoV-2 Natural Inhibitors among 4924 African Metabolites Targeting the Papain-like Protease: A Multi-Phase In Silico Approach. Metabolites. 2022; 12(11):1122. https://doi.org/10.3390/metabo12111122
Chicago/Turabian StyleElkaeed, Eslam B., Mohamed M. Khalifa, Bshra A. Alsfouk, Aisha A. Alsfouk, Abdul-Aziz M. M. El-Attar, Ibrahim H. Eissa, and Ahmed M. Metwaly. 2022. "The Discovery of Potential SARS-CoV-2 Natural Inhibitors among 4924 African Metabolites Targeting the Papain-like Protease: A Multi-Phase In Silico Approach" Metabolites 12, no. 11: 1122. https://doi.org/10.3390/metabo12111122
APA StyleElkaeed, E. B., Khalifa, M. M., Alsfouk, B. A., Alsfouk, A. A., El-Attar, A.-A. M. M., Eissa, I. H., & Metwaly, A. M. (2022). The Discovery of Potential SARS-CoV-2 Natural Inhibitors among 4924 African Metabolites Targeting the Papain-like Protease: A Multi-Phase In Silico Approach. Metabolites, 12(11), 1122. https://doi.org/10.3390/metabo12111122