
A Novel Ketone-Supplemented Diet Improves Recognition Memory and Hippocampal Mitochondrial Efficiency in Healthy Adult Mice

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Diet
2.2. Two-Object Novel Object Recognition
2.3. Brain Slice Preparation
2.4. Mitochondrial Respirometry
2.5. ATP Quantification
2.6. Western Blot
2.7. Statistics
2.8. Sex as a Biological Variable
3. Results
3.1. Ketogenic Diet Elevates Blood Beta-Hydroxybutyrate
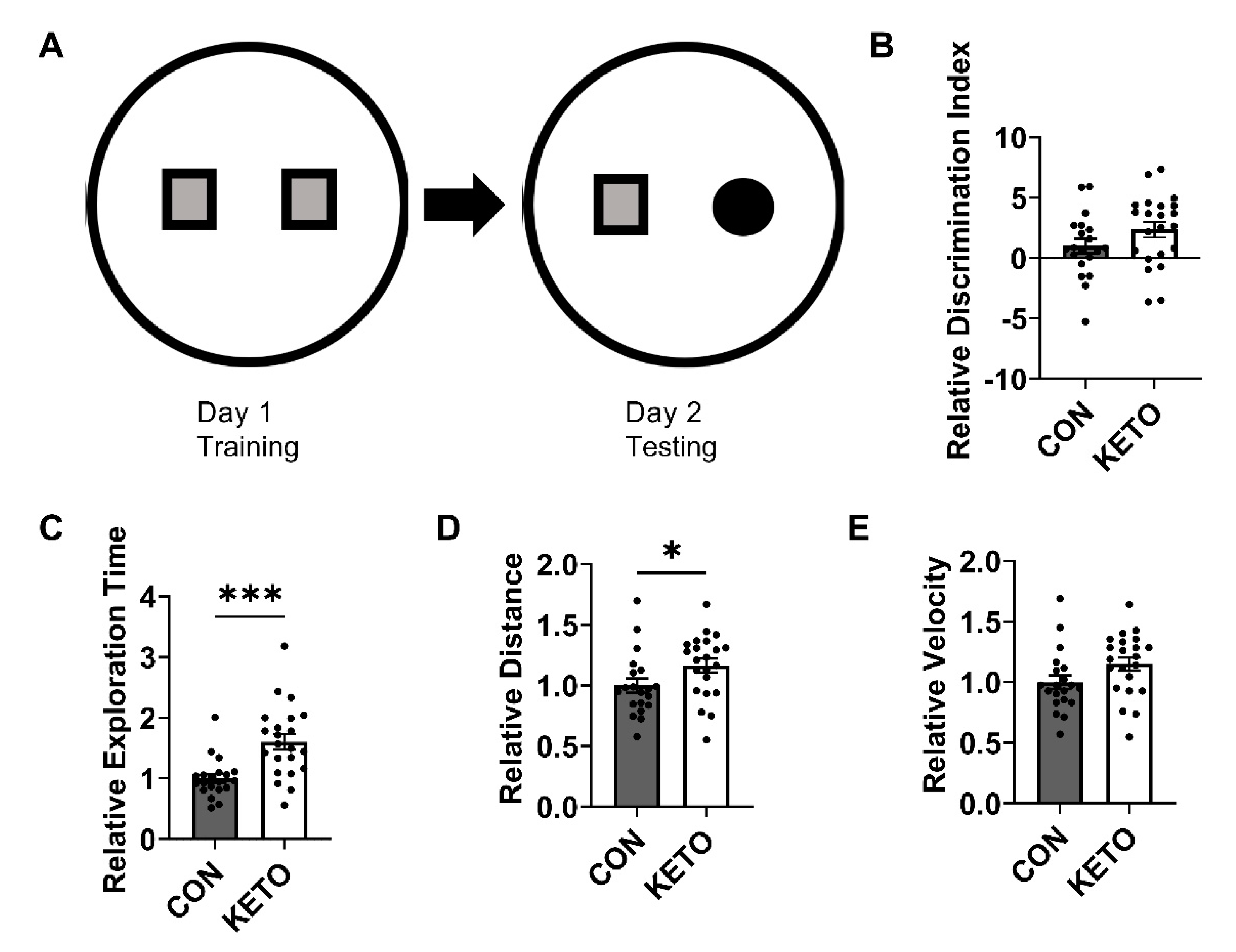
3.2. Ketogenic Diet Improves Recognition Memory and Locomotion
3.3. Ketogenic Diet Enhances Hippocampal Mitochondrial Efficiency
3.4. Ketogenic Diet Induces Sex-Specific Hippocampal Mitochondrial Complex V Expression
3.5. Ketogenic Diet Does Not Alter the Expression of Hippocampal Mitochondrial Dynamics Proteins
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kann, O.; Kovács, R. Mitochondria and neuronal activity. Am. J. Physiol.-Cell Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Arumugam, T.V. Hallmarks of brain aging: Adaptive and pathological modification by metabolic states. Cell Metab. 2018, 27, 1176–1199. [Google Scholar] [CrossRef]
- Gonzalez-Lima, F.; Barksdale, B.R.; Rojas, J.C. Mitochondrial respiration as a target for neuroprotection and cognitive enhancement. Biochem. Pharmacol. 2014, 88, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, E.; Musich, P.R.; Lin, F. Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci. Ther. 2019, 25, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, M.M.; Garbarino, V.R.; Pollet, E.; Palavicini, J.P.; Kellogg, D.L., Jr.; Kraig, E.; Orr, M.E. Biological aging processes underlying cognitive decline and neurodegenerative disease. J. Clin. Investig. 2022, 132, e158453. [Google Scholar] [CrossRef] [PubMed]
- Lantz, B.M.; Foerster, J.M.; Link, D.P.; Holcroft, J.W. Regional distribution of cardiac output: Normal values in man determined by video dilution technique. Am. J. Roentgenol. 1981, 137, 903–907. [Google Scholar] [CrossRef]
- Williams, L.; Leggett, R. Reference values for resting blood flow to organs of man. Clin. Phys. Physiol. Meas. 1989, 10, 187. [Google Scholar] [CrossRef]
- Clarke, D.D.; Sokoloff, L. Circulation and energy metabolism of the brain. In Basic Neurochemistry: Molecular, Cellular, and Medical Aspects, 6th ed.; Lippincott Raven: Philadelphia, PA, USA, 1999; pp. 637–669. [Google Scholar]
- Bliss, T.V.; Lømo, T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 1973, 232, 331–356. [Google Scholar] [CrossRef]
- Divakaruni, S.S.; van Dyke, A.M.; Chandra, R.; LeGates, T.A.; Contreras, M.; Dharmasri, P.A.; Higgs, H.N.; Lobo, M.K.; Thompson, S.M.; Blanpied, T.A. Long-term potentiation requires a rapid burst of dendritic mitochondrial fission during induction. Neuron 2018, 100, 860–875.e7. [Google Scholar] [CrossRef]
- Olesen, M.A.; Torres, A.K.; Jara, C.; Murphy, M.P.; Tapia-Rojas, C. Premature synaptic mitochondrial dysfunction in the hippocampus during aging contributes to memory loss. Redox Biol. 2020, 34, 101558. [Google Scholar] [CrossRef]
- LaManna, J.C.; Salem, N.; Puchowicz, M.; Erokwu, B.; Koppaka, S.; Flask, C.; Lee, Z. Ketones suppress brain glucose consumption. Adv. Exp. Med. Biol. 2009, 645, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kuang, Y.; Xu, K.; Harris, D.; Lee, Z.; LaManna, J.; Puchowicz, M.A. Ketosis proportionately spares glucose utilization in brain. J. Cereb. Blood Flow Metab. 2013, 33, 1307–1311. [Google Scholar] [CrossRef]
- Courchesne-Loyer, A.; Croteau, E.; Castellano, C.-A.; St-Pierre, V.; Hennebelle, M.; Cunnane, S.C. Inverse relationship between brain glucose and ketone metabolism in adults during short-term moderate dietary ketosis: A dual tracer quantitative positron emission tomography study. J. Cereb. Blood Flow Metab. 2017, 37, 2485–2493. [Google Scholar] [CrossRef] [PubMed]
- Owen, O.E. Ketone bodies as a fuel for the brain during starvation. Biochem. Mol. Biol. Educ. 2005, 33, 246–251. [Google Scholar] [CrossRef]
- Wilder, R.M. The effects of ketonemia on the course of epilepsy. Mayo Clin. Proc. 1921, 2, 307–308. [Google Scholar]
- Liu, H.; Yang, Y.; Wang, Y.; Tang, H.; Zhang, F.; Zhang, Y.; Zhao, Y. Ketogenic diet for treatment of intractable epilepsy in adults: A meta-analysis of observational studies. Epilepsia Open 2018, 3, 9–17. [Google Scholar] [CrossRef]
- Kraeuter, A.-K.; Phillips, R.; Sarnyai, Z. Ketogenic therapy in neurodegenerative and psychiatric disorders: From mice to men. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 101, 109913. [Google Scholar] [CrossRef]
- Taylor, M.K.; Sullivan, D.K.; Mahnken, J.D.; Burns, J.M.; Swerdlow, R.H. Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimers Dement. Transl. Res. Clin. Interv. 2018, 4, 28–36. [Google Scholar] [CrossRef]
- Brandt, J.; Buchholz, A.; Henry-Barron, B.; Vizthum, D.; Avramopoulos, D.; Cervenka, M.C. Preliminary Report on the feasibility and efficacy of the modified atkins diet for treatment of mild cognitive impairment and early Alzheimer’s disease. J. Alzheimers Dis. 2019, 68, 969–981. [Google Scholar] [CrossRef]
- Lilamand, M.; Porte, B.; Cognat, E.; Hugon, J.; Mouton-Liger, F.; Paquet, C. Are ketogenic diets promising for Alzheimer’s disease? A translational review. Alzheimers Res. Ther. 2020, 12, 1–10. [Google Scholar] [CrossRef]
- VanItallie, T.B.; Nonas, C.; Di Rocco, A.; Boyar, K.; Hyams, K.; Heymsfield, S.B. Treatment of Parkinson disease with diet-induced hyperketonemia: A feasibility study. Neurology 2005, 64, 728–730. [Google Scholar] [CrossRef] [PubMed]
- Włodarek, D. Role of ketogenic diets in neurodegenerative diseases (Alzheimer’s disease and Parkinson’s disease). Nutrients 2019, 11, 169. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Mukherjee, P. Targeting energy metabolism in brain cancer: Review and hypothesis. Nutr. Metab. 2005, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Wang, H.; Liu, J.; Jiye, A.; Zhou, F.; Wang, G. Multi-dimensional roles of ketone bodies in cancer biology: Opportunities for cancer therapy. Pharmacol. Res. 2019, 150, 104500. [Google Scholar] [CrossRef]
- Ricci, A.; Idzikowski, M.A.; Soares, C.N.; Brietzke, E. Exploring the mechanisms of action of the antidepressant effect of the ketogenic diet. Rev. Neurosci. 2020, 31, 637–648. [Google Scholar] [CrossRef]
- Shamshtein, D.; Liwinski, T. Ketogenic therapy for major depressive disorder: A review of neurobiological evidence. Recent Prog. Nutr. 2022, 2, 1. [Google Scholar] [CrossRef]
- Tillery, E.E.; Ellis, K.D.; Threatt, T.B.; Reyes, H.A.; Plummer, C.S.; Barney, L.R. The use of the ketogenic diet in the treatment of psychiatric disorders. Ment. Health Clin. 2021, 11, 211–219. [Google Scholar] [CrossRef]
- Sarnyai, Z.; Kraeuter, A.-K.; Palmer, C.M. Ketogenic diet for schizophrenia: Clinical implication. Curr. Opin. Psychiatry 2019, 32, 394–401. [Google Scholar] [CrossRef]
- Kapogiannis, D.; Avgerinos, K.I. Brain glucose and ketone utilization in brain aging and neurodegenerative diseases. Int. Rev. Neurobiol. 2020, 154, 79–110. [Google Scholar]
- Jensen, N.J.; Wodschow, H.Z.; Nilsson, M.; Rungby, J. Effects of ketone bodies on brain metabolism and function in neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 8767. [Google Scholar] [CrossRef]
- Yang, H.; Shan, W.; Zhu, F.; Wu, J.; Wang, Q. Ketone bodies in neurological diseases: Focus on neuroprotection and underlying mechanisms. Front. Neurol. 2019, 10, 585. [Google Scholar] [CrossRef] [PubMed]
- Desrochers, S.; Dubreuil, P.; Brunet, J.; Jette, M.; David, F.; Landau, B.R.; Brunengraber, H. Metabolism of (R, S)-1, 3-butanediol acetoacetate esters, potential parenteral and enteral nutrients in conscious pigs. Am. J. Physiol.-Endocrinol. Metab. 1995, 268, E660–E667. [Google Scholar] [CrossRef] [PubMed]
- Desrochers, S.; David, F.; Garneau, M.; Jetté, M.; Brunengraber, H. Metabolism of R-and S-1, 3-butanediol in perfused livers from meal-fed and starved rats. Biochem. J. 1992, 285, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Covarrubias, A.J.; Zhao, M.; Yu, X.; Gut, P.; Ng, C.-P.; Huang, Y.; Haldar, S.; Verdin, E. Ketogenic diet reduces midlife mortality and improves memory in aging mice. Cell Metab. 2017, 26, 547–557.e8. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.A.H.; Deemer, S.E.; Bergeron, J.M.; Little, J.T.; Warren, J.L.; Fisher, G.; Smith, D.L., Jr.; Fontaine, K.R.; Dickinson, S.L.; Allison, D.B.; et al. Dietary R, S-1,3-butanediol diacetoacetate reduces body weight and adiposity in obese mice fed a high-fat diet. FASEB J. 2018, 33, 2409–2421. [Google Scholar] [CrossRef]
- Moore, E.S.; Cleland, T.A.; Williams, W.O.; Peterson, C.M.; Singh, B.; Southard, T.L.; Pasch, B.; Labitt, R.N.; Daugherity, E.K. Comparing phlebotomy by tail tip amputation, facial vein puncture, and tail vein incision in C57BL/6 mice by using physiologic and behavioral metrics of pain and distress. J. Am. Assoc. Lab. Anim. Sci. 2017, 56, 307–317. [Google Scholar]
- Newman, J.C.; Verdin, E. β-Hydroxybutyrate: A signaling metabolite. Annu. Rev. Nutr. 2017, 37, 51–76. [Google Scholar] [CrossRef]
- Lincoln, B.C.; Des Rosiers, C.; Brunengraber, H. Metabolism of S-3-hydroxybutyrate in the perfused rat liver. Arch. Biochem. Biophys. 1987, 259, 149–156. [Google Scholar] [CrossRef]
- Antunes, M.; Biala, G. The novel object recognition memory: Neurobiology, test procedure, and its modifications. Cogn. Process. 2012, 13, 93–110. [Google Scholar] [CrossRef]
- Grayson, B.; Leger, M.; Piercy, C.; Adamson, L.; Harte, M.; Neill, J.C. Assessment of disease-related cognitive impairments using the novel object recognition (NOR) task in rodents. Behav. Brain Res. 2015, 285, 176–193. [Google Scholar] [CrossRef]
- Lueptow, L.M. Novel object recognition test for the investigation of learning and memory in mice. JoVE J. Vis. Exp. 2017, 126, e55718. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.A.; Lartey, F.M.; Schüler, E.; Rafat, M.; King, G.; Kim, A.; Ko, R.; Semaan, S.; Gonzalez, S.; Jenkins, M. Reduced cognitive deficits after FLASH irradiation of whole mouse brain are associated with less hippocampal dendritic spine loss and neuroinflammation. Radiother. Oncol. 2019, 139, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Bennion, D.; Jensen, T.; Walther, C.; Hamblin, J.; Wallmann, A.; Couch, J.; Blickenstaff, J.; Castle, M.; Dean, L.; Beckstead, S. Transient receptor potential vanilloid 1 agonists modulate hippocampal CA1 LTP via the GABAergic system. Neuropharmacology 2011, 61, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Hurst, K.; Badgley, C.; Ellsworth, T.; Bell, S.; Friend, L.; Prince, B.; Welch, J.; Cowan, Z.; Williamson, R.; Lyon, C.; et al. A putative lysophosphatidylinositol receptor GPR55 modulates hippocampal synaptic plasticity. Hippocampus 2017, 27, 985–998. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.M.; Marriott, D.; Trotter, J.; Hammond, T.; Lyman, D.; Call, T.; Walker, B.; Christensen, N.; Haynie, D.; Badura, Z.; et al. Running exercise mitigates the negative consequences of chronic stress on dorsal hippocampal long-term potentiation in male mice. Neurobiol. Learn. Mem. 2018, 149, 28–38. [Google Scholar] [CrossRef]
- Parker, B.A.; Walton, C.M.; Carr, S.T.; Andrus, J.L.; Cheung, E.C.; Duplisea, M.J.; Wilson, E.K.; Draney, C.; Lathen, D.R.; Kenner, K.B. β-Hydroxybutyrate elicits favorable mitochondrial changes in skeletal muscle. Int. J. Mol. Sci. 2018, 19, 2247. [Google Scholar] [CrossRef]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; de la Monte, S.M. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J. Alzheimers Dis. 2005, 8, 247–268. [Google Scholar] [CrossRef]
- Inagaki, T.; Gautreaux, C.; Luine, V. Acute estrogen treatment facilitates recognition memory consolidation and alters monoamine levels in memory-related brain areas. Horm. Behav. 2010, 58, 415–426. [Google Scholar] [CrossRef]
- Barha, C.K.; Galea, L.A. Influence of different estrogens on neuroplasticity and cognition in the hippocampus. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2010, 1800, 1056–1067. [Google Scholar] [CrossRef]
- Frick, K.; Berger-Sweeney, J. Spatial reference memory and neocortical neurochemistry vary with the estrous cycle in C57BL/6 mice. Behav. Neurosci. 2001, 115, 229. [Google Scholar] [CrossRef]
- Spencer, J.L.; Waters, E.M.; Milner, T.A.; McEwen, B.S. Estrous cycle regulates activation of hippocampal Akt, LIM kinase, and neurotrophin receptors in C57BL/6 mice. Neuroscience 2008, 155, 1106–1119. [Google Scholar] [CrossRef] [PubMed]
- Razmara, A.; Duckles, S.P.; Krause, D.N.; Procaccio, V. Estrogen suppresses brain mitochondrial oxidative stress in female and male rats. Brain Res. 2007, 1176, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Champlin, A.K.; Dorr, D.L.; Gates, A.H. Determining the stage of the estrous cycle in the mouse by the appearance of the vagina. Biol. Reprod. 1973, 8, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Kashiwaya, Y.; Bergman, C.; Lee, J.-H.; Wan, R.; King, M.T.; Mughal, M.R.; Okun, E.; Clarke, K.; Mattson, M.P.; Veech, R.L. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1530–1539. [Google Scholar] [CrossRef]
- Ciarlone, S.L.; Grieco, J.C.; D’Agostino, D.P.; Weeber, E.J. Ketone ester supplementation attenuates seizure activity, and improves behavior and hippocampal synaptic plasticity in an Angelman syndrome mouse model. Neurobiol. Dis. 2016, 96, 38–46. [Google Scholar] [CrossRef]
- Pawlosky, R.J.; Kashiwaya, Y.; King, M.T.; Veech, R.L. A dietary ketone ester normalizes abnormal behavior in a mouse model of Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 1044. [Google Scholar] [CrossRef]
- Hammond, K.A.; Diamond, J. Maximal sustained energy budgets in humans and animals. Nature 1997, 386, 457–462. [Google Scholar] [CrossRef]
- Cervos-Navarro, J.; Diemer, N. Selective vulnerability in brain hypoxia. Crit. Rev. Neurobiol. 1991, 6, 149–182. [Google Scholar]
- Schmidt-Kastner, R. Genomic approach to selective vulnerability of the hippocampus in brain ischemia–hypoxia. Neuroscience 2015, 309, 259–279. [Google Scholar] [CrossRef]
- Prins, M.L.; Fujima, L.S.; Hovda, D.A. Age-dependent reduction of cortical contusion volume by ketones after traumatic brain injury. J. Neurosci. Res. 2005, 82, 413–420. [Google Scholar] [CrossRef]
- Hu, Z.-G.; Wang, H.-D.; Qiao, L.; Yan, W.; Tan, Q.-F.; Yin, H.-X. The protective effect of the ketogenic diet on traumatic brain injury-induced cell death in juvenile rats. Brain Inj. 2009, 23, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.G.; Wang, H.D.; Jin, W.; Yin, H.X. Ketogenic diet reduces cytochrome c release and cellular apoptosis following traumatic brain injury in juvenile rats. Ann. Clin. Lab. Sci. 2009, 39, 76–83. [Google Scholar] [PubMed]
- Cocco, T.; Pacelli, C.; Sgobbo, P.; Villani, G. Control of OXPHOS efficiency by complex I in brain mitochondria. Neurobiol. Aging 2009, 30, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Nanayakkara, G.; Shao, Y.; Cueto, R.; Wang, L.; Yang, W.Y.; Tian, Y.; Wang, H.; Yang, X. Mitochondrial proton leak plays a critical role in pathogenesis of cardiovascular diseases. In Mitochondrial Dynamics in Cardiovascular Medicine; Springer: Cham, Switzerland, 2017; pp. 359–370. [Google Scholar]
- Serviddio, G.; Bellanti, F.; Romano, A.D.; Tamborra, R.; Rollo, T.; Altomare, E.; Vendemiale, G. Bioenergetics in aging: Mitochondrial proton leak in aging rat liver, kidney and heart. Redox Rep. 2007, 12, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Krauss, S.; Zhang, C.-Y.; Lowell, B.B. The mitochondrial uncoupling-protein homologues. Nat. Rev. Mol. Cell Biol. 2005, 6, 248–261. [Google Scholar] [CrossRef]
- Lowell, B.B.; Spiegelman, B.M. Towards a molecular understanding of adaptive thermogenesis. Nature 2000, 404, 652–660. [Google Scholar] [CrossRef]
- Brand, M.D.; Steverding, D.; Kadenbach, B.; Stevenson, P.M.; Hafner, R.P. The mechanism of the increase in mitochondrial proton permeability induced by thyroid hormones. Eur. J. Biochem. 1992, 206, 775–781. [Google Scholar] [CrossRef]
- Salin, K.; Auer, S.K.; Rey, B.; Selman, C.; Metcalfe, N.B. Variation in the link between oxygen consumption and ATP production, and its relevance for animal performance. Proc. R. Soc. B Biol. Sci. 2015, 282, 20151028. [Google Scholar] [CrossRef]
- Serrano, F.; Klann, E. Reactive oxygen species and synaptic plasticity in the aging hippocampus. Ageing Res. Rev. 2004, 3, 431–443. [Google Scholar] [CrossRef]
- Brawek, B.; Löffler, M.; Wagner, K.; Huppertz, H.-J.; Wendling, A.-S.; Weyerbrock, A.; Jackisch, R.; Feuerstein, T.J. Reactive oxygen species (ROS) in the human neocortex: Role of aging and cognition. Brain Res. Bull. 2010, 81, 484–490. [Google Scholar] [CrossRef]
- Rego, A.C.; Oliveira, C.R. Mitochondrial dysfunction and reactive oxygen species in excitotoxicity and apoptosis: Implications for the pathogenesis of neurodegenerative diseases. Neurochem. Res. 2003, 28, 1563–1574. [Google Scholar] [CrossRef] [PubMed]
- Kausar, S.; Wang, F.; Cui, H. The role of mitochondria in reactive oxygen species generation and its implications for neurodegenerative diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.; Bonucci, A.; Maggi, E.; Corsi, M.; Businaro, R. Anti-oxidant and anti-inflammatory activity of ketogenic diet: New perspectives for neuroprotection in Alzheimer’s disease. Antioxidants 2018, 7, 63. [Google Scholar] [CrossRef]
- Stafford, P.; Abdelwahab, M.G.; Kim, D.Y.; Preul, M.C.; Rho, J.M.; Scheck, A.C. The ketogenic diet reverses gene expression patterns and reduces reactive oxygen species levels when used as an adjuvant therapy for glioma. Nutr. Metab. 2010, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Greco, T.; Glenn, T.C.; Hovda, D.A.; Prins, M.L. Ketogenic diet decreases oxidative stress and improves mitochondrial respiratory complex activity. J. Cereb. Blood Flow Metab. 2016, 36, 1603–1613. [Google Scholar] [CrossRef]
- Rolls, E. The mechanisms for pattern completion and pattern separation in the hippocampus. Front. Syst. Neurosci. 2013, 7, 74. [Google Scholar] [CrossRef]
- Ranganath, C.; Yonelinas, A.P.; Cohen, M.X.; Dy, C.J.; Tom, S.M.; D’Esposito, M. Dissociable correlates of recollection and familiarity within the medial temporal lobes. Neuropsychologia 2004, 42, 2–13. [Google Scholar] [CrossRef]
- Eichenbaum, H.; Yonelinas, A.P.; Ranganath, C. The medial temporal lobe and recognition memory. Annu. Rev. Neurosci. 2007, 30, 123–152. [Google Scholar] [CrossRef]
- Jeneson, A.; Kirwan, C.B.; Hopkins, R.O.; Wixted, J.T.; Squire, L.R. Recognition memory and the hippocampus: A test of the hippocampal contribution to recollection and familiarity. Learn. Mem. 2010, 17, 63–70. [Google Scholar] [CrossRef]
- Wixted, J.T.; Squire, L.R. The role of the human hippocampus in familiarity-based and recollection-based recognition memory. Behav. Brain Res. 2010, 215, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Merkow, M.B.; Burke, J.F.; Kahana, M.J. The human hippocampus contributes to both the recollection and familiarity components of recognition memory. Proc. Natl. Acad. Sci. USA 2015, 112, 14378–14383. [Google Scholar] [CrossRef] [PubMed]
- Schoemaker, D.; Gauthier, S.; Pruessner, J.C. Recollection and familiarity in aging individuals with mild cognitive impairment and Alzheimer’s disease: A literature review. Neuropsychol. Rev. 2014, 24, 313–331. [Google Scholar] [CrossRef] [PubMed]
- Koen, J.D.; Yonelinas, A.P. The effects of healthy aging, amnestic mild cognitive impairment, and Alzheimer’s disease on recollection and familiarity: A meta-analytic review. Neuropsychol. Rev. 2014, 24, 332–354. [Google Scholar] [CrossRef]
- Murray, A.J.; Knight, N.S.; Cole, M.A.; Cochlin, L.E.; Carter, E.; Tchabanenko, K.; Pichulik, T.; Gulston, M.K.; Atherton, H.J.; Schroeder, M.A.; et al. Novel ketone diet enhances physical and cognitive performance. FASEB J. 2016, 30, 4021–4032. [Google Scholar] [CrossRef]
- Koppel, S.J.; Pei, D.; Wilkins, H.M.; Weidling, I.W.; Wang, X.; Menta, B.W.; Perez-Ortiz, J.; Kalani, A.; Manley, S.; Novikova, L. A ketogenic diet differentially affects neuron and astrocyte transcription. J. Neurochem. 2021, 157, 1930–1945. [Google Scholar] [CrossRef]
- Chih, C.-P.; Lipton, P.; Roberts, E.L. Do active cerebral neurons really use lactate rather than glucose? Trends Neurosci. 2001, 24, 573–578. [Google Scholar] [CrossRef]
- Von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef]
- Herculano-Houzel, S. The glia/neuron ratio: How it varies uniformly across brain structures and species and what that means for brain physiology and evolution. Glia 2014, 62, 1377–1391. [Google Scholar] [CrossRef]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef]
- Dye, L.; Boyle, N.B.; Champ, C.; Lawton, C. The relationship between obesity and cognitive health and decline. Proc. Nutr. Soc. 2017, 76, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Morys, F.; Dadar, M.; Dagher, A. Association between midlife obesity and its metabolic consequences, cerebrovascular disease, and cognitive decline. J. Clin. Endocrinol. Metab. 2021, 106, e4260–e4274. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Ngandu, T.; Fratiglioni, L.; Viitanen, M.; Kåreholt, I.; Winblad, B.; Helkala, E.-L.; Tuomilehto, J.; Soininen, H.; Nissinen, A. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch. Neurol. 2005, 62, 1556–1560. [Google Scholar] [CrossRef] [PubMed]
- Whitmer, R.A.; Gunderson, E.P.; Quesenberry, C.P.; Zhou, J.; Yaffe, K. Body mass index in midlife and risk of Alzheimer disease and vascular dementia. Curr. Alzheimer Res. 2007, 4, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Profenno, L.A.; Porsteinsson, A.P.; Faraone, S.V. Meta-analysis of Alzheimer’s disease risk with obesity, diabetes, and related disorders. Biol. Psychiatry 2010, 67, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, D.; Rothenberg, E.; Blennow, K.; Steen, B.; Skoog, I. An 18-year follow-up of overweight and risk of Alzheimer disease. Arch. Intern. Med. 2003, 163, 1524–1528. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef]
- Henkel, N.D.; Wu, X.; O’Donovan, S.M.; Devine, E.A.; Jiron, J.M.; Rowland, L.M.; Sarnyai, Z.; Ramsey, A.J.; Wen, Z.; Hahn, M.K. Schizophrenia: A disorder of broken brain bioenergetics. Mol. Psychiatry 2022, 27, 2393–2404. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CON | KETO | |
|---|---|---|
| Component | kcal% | kcal% |
| Fat | 13.4 | 90 |
| Protein | 28.7 | 4.7 |
| Carbohydrate | 57.9 | 2 |
| Ketone Ester | 0 | 3.3 |
| Target | Dilution | Host | Company | ID |
|---|---|---|---|---|
| DRP1 | 1:1000 | Rabbit | Novus Biologicals | NB110-55288 |
| pDRP1 | 1:1000 | Rabbit | Cell Signaling | 3455 |
| OPA1 | 1:1000 | Rabbit | Novus Biologicals | NB110-55290 |
| OXPHOS | 1:5000 | Mouse | Thermo | 45-8099 |
| β-actin | 1:1000 | Rabbit | Cell Signaling | 13E5 |
| β-actin | 1:1000 | Mouse | Cell Signaling | 8H10D10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saito, E.R.; Warren, C.E.; Hanegan, C.M.; Larsen, J.G.; du Randt, J.D.; Cannon, M.; Saito, J.Y.; Campbell, R.J.; Kemberling, C.M.; Miller, G.S.; et al. A Novel Ketone-Supplemented Diet Improves Recognition Memory and Hippocampal Mitochondrial Efficiency in Healthy Adult Mice. Metabolites 2022, 12, 1019. https://doi.org/10.3390/metabo12111019
Saito ER, Warren CE, Hanegan CM, Larsen JG, du Randt JD, Cannon M, Saito JY, Campbell RJ, Kemberling CM, Miller GS, et al. A Novel Ketone-Supplemented Diet Improves Recognition Memory and Hippocampal Mitochondrial Efficiency in Healthy Adult Mice. Metabolites. 2022; 12(11):1019. https://doi.org/10.3390/metabo12111019
Chicago/Turabian StyleSaito, Erin R., Cali E. Warren, Cameron M. Hanegan, John G. Larsen, Johannes D. du Randt, Mio Cannon, Jeremy Y. Saito, Rachel J. Campbell, Colin M. Kemberling, Gavin S. Miller, and et al. 2022. "A Novel Ketone-Supplemented Diet Improves Recognition Memory and Hippocampal Mitochondrial Efficiency in Healthy Adult Mice" Metabolites 12, no. 11: 1019. https://doi.org/10.3390/metabo12111019
APA StyleSaito, E. R., Warren, C. E., Hanegan, C. M., Larsen, J. G., du Randt, J. D., Cannon, M., Saito, J. Y., Campbell, R. J., Kemberling, C. M., Miller, G. S., Edwards, J. G., & Bikman, B. T. (2022). A Novel Ketone-Supplemented Diet Improves Recognition Memory and Hippocampal Mitochondrial Efficiency in Healthy Adult Mice. Metabolites, 12(11), 1019. https://doi.org/10.3390/metabo12111019