
The Interaction between the Gut Microbiome and Bile Acids in Cardiometabolic Diseases


Abstract
:1. Introduction
2. The Gut Microbiome
3. An Overview of the Gut Microbiome in CMD Perspective
4. Gut Microbiome-Derived Metabolites
5. Bile Acid Metabolism
6. FXR and TGR5 Pathway
7. Cardio-Metabolic Disease, Gut Microbiome, and Bile Acid Interplay
8. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
Glossary
| ASCVD | Atherosclerotic cardiovascular disease |
| BA | Bile Acids |
| CMD | Cardio-metabolic disease |
| CVD | Cardiovascular disease |
| FXR | Farnesoid-X receptor |
| GIM | Gastro-intestinal microbiome |
| Holobiont | Superorganism of host and commensal microorganisms |
| LXR | Liver X receptor |
| Prebiotics | Food supplements or components that stimulate the health of the gut ecosystem |
| Probiotics | Living microorganisms believed to be beneficial to human health when ingested |
| SCFA | Short-chain fatty acid |
| Synbiotics | Food supplements containing both pre- and probiotics |
| TGR5 | Takeda G protein-coupled receptor 5 |
| TMAO | Trimethylamine N-oxide |
References
- WHO. Cardiovascular Diseases (CVDs) (Fact Sheet). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 30 September 2021).
- WHO. Obesity and Overweight (Fact Sheet). Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 30 September 2021).
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, R.E.; Peterson, D.A.; Gordon, J.I. Ecological and Evolutionary Forces Shaping Microbial Diversity in the Human Intestine. Cell 2006, 124, 837–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holscher, H.D. Dietary Fiber and Prebiotics and the Gastrointestinal Microbiota. Gut Microbes 2017, 8, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Maruvada, P.; Leone, V.; Kaplan, L.M.; Chang, E.B. The Human Microbiome and Obesity: Moving beyond Associations. Cell Host Microbe 2017, 22, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Alcock, J.; Maley, C.C.; Aktipis, C.A. Is Eating Behavior Manipulated by the Gastrointestinal Microbiota? Evolutionary Pressures and Potential Mechanisms. BioEssays 2014, 36, 940–949. [Google Scholar] [CrossRef]
- Koh, A.; Molinaro, A.; Ståhlman, M.; Khan, M.T.; Schmidt, C.; Mannerås-Holm, L.; Wu, H.; Carreras, A.; Jeong, H.; Olofsson, L.E.; et al. Microbially Produced Imidazole Propionate Impairs Insulin Signaling through MTORC1. Cell 2018, 175, 947–961.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Khurana, S.; Raufman, J.-P.; Pallone, T.L. Bile Acids Regulate Cardiovascular Function. Clin. Transl. Sci. 2011, 4, 210–218. [Google Scholar] [CrossRef]
- Hasan, N.; Yang, H. Factors Affecting the Composition of the Gut Microbiota, and Its Modulation. PeerJ 2019, 7, e7502. [Google Scholar] [CrossRef] [Green Version]
- The Human Microbiome Project Consortium. Structure, Function and Diversity of the Healthy Human Microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deschasaux, M.; Bouter, K.E.; Prodan, A.; Levin, E.; Groen, A.K.; Herrema, H.; Tremaroli, V.; Bakker, G.J.; Attaye, I.; Pinto-Sietsma, S.-J.; et al. Depicting the Composition of Gut Microbiota in a Population with Varied Ethnic Origins but Shared Geography. Nat. Med. 2018, 24, 1526–1531. [Google Scholar] [CrossRef]
- Dwiyanto, J.; Hussain, M.H.; Reidpath, D.; Ong, K.S.; Qasim, A.; Lee, S.W.H.; Lee, S.M.; Foo, S.C.; Chong, C.W.; Rahman, S. Ethnicity Influences the Gut Microbiota of Individuals Sharing a Geographical Location: A Cross-Sectional Study from a Middle-Income Country. Sci. Rep. 2021, 11, 2618. [Google Scholar] [CrossRef]
- Snijder, M.B.; Galenkamp, H.; Prins, M.; Derks, E.M.; Peters, R.J.G.; Zwinderman, A.H.; Stronks, K. Cohort Profile: The Healthy Life in an Urban Setting (HELIUS) Study in Amsterdam, The Netherlands. BMJ Open 2017, 7, e017873. [Google Scholar] [CrossRef] [Green Version]
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in Lean and Overweight Healthy Subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef] [PubMed]
- van Son, J.; Serlie, M.J.; Ståhlman, M.; Bäckhed, F.; Nieuwdorp, M.; Aron-Wisnewsky, J. Plasma Imidazole Propionate Is Positively Correlated with Blood Pressure in Overweight and Obese Humans. Nutrients 2021, 13, 2706. [Google Scholar] [CrossRef] [PubMed]
- Kerimi, A.; Kraut, N.U.; da Encarnacao, J.A.; Williamson, G. The Gut Microbiome Drives Inter- and Intra-Individual Differences in Metabolism of Bioactive Small Molecules. Sci. Rep. 2020, 10, 19590. [Google Scholar] [CrossRef] [PubMed]
- Healey, G.R.; Murphy, R.; Brough, L.; Butts, C.A.; Coad, J. Interindividual Variability in Gut Microbiota and Host Response to Dietary Interventions. Nutr. Rev. 2017, 75, 1059–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.-Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA Gene Sequencing for Species and Strain-Level Microbiome Analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef] [Green Version]
- Gensollen, T.; Iyer, S.S.; Kasper, D.L.; Blumberg, R.S. How Colonization by Microbiota in Early Life Shapes the Immune System. Science 2016, 352, 539–544. [Google Scholar] [CrossRef] [Green Version]
- Winkler, E.S.; Shrihari, S.; Hykes, B.L.; Handley, S.A.; Andhey, P.S.; Huang, Y.-J.S.; Swain, A.; Droit, L.; Chebrolu, K.K.; Mack, M.; et al. The Intestinal Microbiome Restricts Alphavirus Infection and Dissemination through a Bile Acid-Type I IFN Signaling Axis. Cell 2020, 182, 901–918.e18. [Google Scholar] [CrossRef]
- Romaní-Pérez, M.; Bullich-Vilarrubias, C.; López-Almela, I.; Liébana-García, R.; Olivares, M.; Sanz, Y. The Microbiota and the Gut–Brain Axis in Controlling Food Intake and Energy Homeostasis. Int. J. Mol. Sci. 2021, 22, 5830. [Google Scholar] [CrossRef]
- Tang, W.H.W.; Hazen, S.L. The Gut Microbiome and Its Role in Cardiovascular Diseases. Circulation 2017, 135, 1008–1010. [Google Scholar] [CrossRef]
- Warmbrunn, M.V.; Herrema, H.; Aron-Wisnewsky, J.; Soeters, M.R.; Van Raalte, D.H.; Nieuwdorp, M. Gut Microbiota: A Promising Target against Cardiometabolic Diseases. Expert Rev. Endocrinol. Metab. 2020, 15, 13–27. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Wang, F.; Yuan, J.; Li, J.; Jiang, D.; Zhang, J.; Li, H.; Wang, R.; Tang, J.; Huang, T.; et al. Effects of Dietary Fat on Gut Microbiota and Faecal Metabolites, and Their Relationship with Cardiometabolic Risk Factors: A 6-Month Randomised Controlled-Feeding Trial. Gut 2019, 68, 1417–1429. [Google Scholar] [CrossRef] [Green Version]
- de Groot, P.; Nikolic, T.; Pellegrini, S.; Sordi, V.; Imangaliyev, S.; Rampanelli, E.; Hanssen, N.; Attaye, I.; Bakker, G.; Duinkerken, G.; et al. Faecal Microbiota Transplantation Halts Progression of Human New-Onset Type 1 Diabetes in a Randomised Controlled Trial. Gut 2021, 70, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.W.M.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of Intestinal Microbiota From Lean Donors Increases Insulin Sensitivity in Individuals With Metabolic Syndrome. Gastroenterology 2012, 143, 913–916.e7. [Google Scholar] [CrossRef]
- Sonnenburg, J.L.; Bäckhed, F. Diet–Microbiota Interactions as Moderators of Human Metabolism. Nature 2016, 535, 56–64. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A Core Gut Microbiome in Obese and Lean Twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An Obesity-Associated Gut Microbiome with Increased Capacity for Energy Harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut Microbiota from Twins Discordant for Obesity Modulate Metabolism in Mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.T.; Parajuli, N.; Sung, M.M.; Bairwa, S.C.; Levasseur, J.; Soltys, C.-L.M.; Wishart, D.S.; Madsen, K.; Schertzer, J.D.; Dyck, J.R.B. Fecal Transplant from Resveratrol-Fed Donors Improves Glycaemia and Cardiovascular Features of the Metabolic Syndrome in Mice. Am. J. Physiol.-Endocrinol. Metab. 2018, 315, E511–E519. [Google Scholar] [CrossRef]
- Mazagova, M.; Wang, L.; Anfora, A.T.; Wissmueller, M.; Lesley, S.A.; Miyamoto, Y.; Eckmann, L.; Dhungana, S.; Pathmasiri, W.; Sumner, S.; et al. Commensal Microbiota Is Hepatoprotective and Prevents Liver Fibrosis in Mice. FASEB J. 2015, 29, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Mell, B.; Jala, V.R.; Mathew, A.V.; Byun, J.; Waghulde, H.; Zhang, Y.; Haribabu, B.; Vijay-Kumar, M.; Pennathur, S.; Joe, B. Evidence for a Link between Gut Microbiota and Hypertension in the Dahl Rat. Physiol. Genom. 2015, 47, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aron-Wisnewsky, J.; Clément, K. The Gut Microbiome, Diet, and Links to Cardiometabolic and Chronic Disorders. Nat. Rev. Nephrol. 2016, 12, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Ma, S.; Ning, M.; Yang, W.; Ye, Y.; Zhang, L.; Shen, J.; Leng, Y. TGR5 Agonist Ameliorates Insulin Resistance in Skeletal Muscles and Improves Glucose Homeostasis in Diabetic Mice. Metabolism 2019, 99, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression From NAFLD to NASH. Transplantation 2019, 103, e1–e13. [Google Scholar] [CrossRef]
- Pais, R.; Barritt, A.S.; Calmus, Y.; Scatton, O.; Runge, T.; Lebray, P.; Poynard, T.; Ratziu, V.; Conti, F. NAFLD, and Liver Transplantation: Current Burden and Expected Challenges. J. Hepatol. 2016, 65, 1245–1257. [Google Scholar] [CrossRef] [Green Version]
- Houttu, V.; Boulund, U.; Grefhorst, A.; Soeters, M.R.; Pinto-Sietsma, S.-J.; Nieuwdorp, M.; Holleboom, A.G. The Role of the Gut Microbiome and Exercise in Non-Alcoholic Fatty Liver Disease. Ther. Adv. Gastroenterol. 2020, 13, 175628482094174. [Google Scholar] [CrossRef]
- Houttu, V.; Csader, S.; Nieuwdorp, M.; Holleboom, A.G.; Schwab, U. Dietary Interventions in Patients With Non-Alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Front. Nutr. 2021, 8, 716783. [Google Scholar] [CrossRef]
- Raja, G.; Gupta, H.; Gebru, Y.A.; Youn, G.S.; Choi, Y.R.; Kim, H.S.; Yoon, S.J.; Kim, D.J.; Kim, T.-J.; Suk, K.T. Recent Advances of Microbiome-Associated Metabolomics Profiling in Liver Disease: Principles, Mechanisms, and Applications. Int. J. Mol. Sci. 2021, 22, 1160. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Jeong, J.-J.; Won, S.-M.; Sharma, S.P.; Gebru, Y.A.; Ganesan, R.; Gupta, H.; Suk, K.T.; Kim, D.J. Gut Microbiota-Related Cellular and Molecular Mechanisms in the Progression of Nonalcoholic Fatty Liver Disease. Cells 2021, 10, 2634. [Google Scholar] [CrossRef] [PubMed]
- Won, S.-M.; Park, E.; Jeong, J.-J.; Ganesan, R.; Gupta, H.; Gebru, Y.; Sharma, S.; Kim, D.-J.; Suk, K.-T. The Gut Microbiota-Derived Immune Response in Chronic Liver Disease. Int. J. Mol. Sci. 2021, 22, 8309. [Google Scholar] [CrossRef] [PubMed]
- Madatali Abuwani, A.; Priyadarshini Dash, S.; Ganesan, R.; Renu, K.; Vellingiri, B.; Kandasamy, S.; Sundara Rajan, C.R.; Valsala Gopalakrishnan, A. Gut Microbiome and Metabolic Response in Non-Alcoholic Fatty Liver Disease. Clin. Chim. Acta 2021, 523, 304–314. [Google Scholar] [CrossRef]
- Hanssen, N.M.J.; de Vos, W.M.; Nieuwdorp, M. Fecal Microbiota Transplantation in Human Metabolic Diseases: From a Murky Past to a Bright Future? Cell Metab. 2021, 33, 1098–1110. [Google Scholar] [CrossRef]
- Shanahan, F.; Ghosh, T.S.; O’Toole, P.W. The Healthy Microbiome—What Is the Definition of a Healthy Gut Microbiome? Gastroenterology 2021, 160, 483–494. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The Human Microbiome Project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Zhou, J.; Wu, L.; Deng, Y.; Zhi, X.; Jiang, Y.-H.; Tu, Q.; Xie, J.; Van Nostrand, J.D.; He, Z.; Yang, Y. Reproducibility and Quantitation of Amplicon Sequencing-Based Detection. ISME J. 2011, 5, 1303–1313. [Google Scholar] [CrossRef]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the Microbiome: Advantages of Whole Genome Shotgun versus 16S Amplicon Sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef] [Green Version]
- Chatelier, E.L.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of Human Gut Microbiome Correlates with Metabolic Markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Cotillard, A.; Kennedy, S.P.; Kong, L.C.; Prifti, E.; Pons, N.; Le Chatelier, E.; Almeida, M.; Quinquis, B.; Levenez, F.; Galleron, N.; et al. Dietary Intervention Impact on Gut Microbial Gene Richness. Nature 2013, 500, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Amar, J.; Chabo, C.; Waget, A.; Klopp, P.; Vachoux, C.; Bermúdez-Humarán, L.G.; Smirnova, N.; Bergé, M.; Sulpice, T.; Lahtinen, S.; et al. Intestinal Mucosal Adherence and Translocation of Commensal Bacteria at the Early Onset of Type 2 Diabetes: Molecular Mechanisms and Probiotic Treatment. EMBO Mol. Med. 2011, 3, 559–572. [Google Scholar] [CrossRef]
- Fine, R.L.; Manfredo Vieira, S.; Gilmore, M.S.; Kriegel, M.A. Mechanisms and Consequences of Gut Commensal Translocation in Chronic Diseases. Gut Microbes 2020, 11, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.S.; Wang, J.; Yannie, P.J.; Ghosh, S. Intestinal Barrier Dysfunction, LPS Translocation, and Disease Development. J. Endocr. Soc. 2020, 4, bvz039. [Google Scholar] [CrossRef] [Green Version]
- Scheithauer, T.P.M.; Herrema, H.; Yu, H.; Bakker, G.J.; Winkelmeijer, M.; Soukhatcheva, G.; Dai, D.; Ma, C.; Havik, S.R.; Balvers, M.; et al. Gut-Derived Bacterial Flagellin Induces Beta-Cell Inflammation and Dysfunction. Mol. Biol. 2021; preprint. [Google Scholar] [CrossRef]
- Visconti, A.; Le Roy, C.I.; Rosa, F.; Rossi, N.; Martin, T.C.; Mohney, R.P.; Li, W.; de Rinaldis, E.; Bell, J.T.; Venter, J.C.; et al. Interplay between the Human Gut Microbiome and Host Metabolism. Nat. Commun. 2019, 10, 4505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macfarlane, G.T.; Gibson, G.R.; Beatty, E.; Cummings, J.H. Estimation of Short-Chain Fatty Acid Production from Protein by Human Intestinal Bacteria Based on Branched-Chain Fatty Acid Measurements. FEMS Microbiol. Ecol. 1992, 10, 81–88. [Google Scholar] [CrossRef]
- Bridgeman, S.C.; Northrop, W.; Melton, P.E.; Ellison, G.C.; Newsholme, P.; Mamotte, C.D.S. Butyrate Generated by Gut Microbiota and Its Therapeutic Role in Metabolic Syndrome. Pharmacol. Res. 2020, 160, 105174. [Google Scholar] [CrossRef]
- Blaak, E.E.; Canfora, E.E.; Theis, S.; Frost, G.; Groen, A.K.; Mithieux, G.; Nauta, A.; Scott, K.; Stahl, B.; van Harsselaar, J.; et al. Short Chain Fatty Acids in Human Gut and Metabolic Health. Benef. Microbes 2020, 11, 411–455. [Google Scholar] [CrossRef]
- Bouter, K.E.; Bakker, G.J.; Levin, E.; Hartstra, A.V.; Kootte, R.S.; Udayappan, S.D.; Katiraei, S.; Bahler, L.; Gilijamse, P.W.; Tremaroli, V.; et al. Differential Metabolic Effects of Oral Butyrate Treatment in Lean versus Metabolic Syndrome Subjects. Clin. Transl. Gastroenterol. 2018, 9, e155. [Google Scholar] [CrossRef]
- Chambers, E.S.; Viardot, A.; Psichas, A.; Morrison, D.J.; Murphy, K.G.; Zac-Varghese, S.E.K.; MacDougall, K.; Preston, T.; Tedford, C.; Finlayson, G.S.; et al. Effects of Targeted Delivery of Propionate to the Human Colon on Appetite Regulation, Body Weight Maintenance and Adiposity in Overweight Adults. Gut 2015, 64, 1744–1754. [Google Scholar] [CrossRef] [Green Version]
- Crimarco, A.; Springfield, S.; Petlura, C.; Streaty, T.; Cunanan, K.; Lee, J.; Fielding-Singh, P.; Carter, M.M.; Topf, M.A.; Wastyk, H.C.; et al. A Randomized Crossover Trial on the Effect of Plant-Based Compared with Animal-Based Meat on Trimethylamine-N-Oxide and Cardiovascular Disease Risk Factors in Generally Healthy Adults: Study With Appetizing Plantfood—Meat Eating Alternative Trial (SWAP-MEAT). Am. J. Clin. Nutr. 2020, 112, 1188–1199. [Google Scholar] [CrossRef]
- Zhao, S.; Jang, C.; Liu, J.; Uehara, K.; Gilbert, M.; Izzo, L.; Zeng, X.; Trefely, S.; Fernandez, S.; Carrer, A.; et al. Dietary Fructose Feeds Hepatic Lipogenesis via Microbiota-Derived Acetate. Nature 2020, 579, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human Nutrition, the Gut Microbiome and the Immune System. Nature 2011, 474, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinero, N.; Ruiz, L.; Sánchez, B.; Margolles, A.; Delgado, S. Intestinal Bacteria Interplay With Bile and Cholesterol Metabolism: Implications on Host Physiology. Front. Physiol. 2019, 10, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begley, M.; Gahan, C.G.M.; Hill, C. The Interaction between Bacteria and Bile. FEMS Microbiol. Rev. 2005, 29, 625–651. [Google Scholar] [CrossRef] [Green Version]
- Islam, K.B.M.S.; Fukiya, S.; Hagio, M.; Fujii, N.; Ishizuka, S.; Ooka, T.; Ogura, Y.; Hayashi, T.; Yokota, A. Bile Acid Is a Host Factor That Regulates the Composition of the Cecal Microbiota in Rats. Gastroenterology 2011, 141, 1773–1781. [Google Scholar] [CrossRef]
- Li, T.; Chiang, J.Y.L. Bile Acids as Metabolic Regulators. Curr. Opin. Gastroenterol. 2015, 31, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Dawson, P.A. Animal Models to Study Bile Acid Metabolism. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2019, 1865, 895–911. [Google Scholar] [CrossRef]
- Duboc, H.; Taché, Y.; Hofmann, A.F. The Bile Acid TGR5 Membrane Receptor: From Basic Research to Clinical Application. Dig. Liver Dis. 2014, 46, 302–312. [Google Scholar] [CrossRef] [Green Version]
- Chiang, J.Y. Recent Advances in Understanding Bile Acid Homeostasis. F1000Res 2017, 6, 2029. [Google Scholar] [CrossRef]
- Choudhuri, S.; Klaassen, C.D. Molecular Regulation of Bile Acid Homeostasis. Drug Metab. Dispos 2021, 50, DMD-MR-2021-000643. [Google Scholar] [CrossRef]
- Fiorucci, S.; Distrutti, E.; Carino, A.; Zampella, A.; Biagioli, M. Bile Acids and Their Receptors in Metabolic Disorders. Prog. Lipid Res. 2021, 82, 101094. [Google Scholar] [CrossRef]
- Wang, H.; He, Q.; Wang, G.; Xu, X.; Hao, H. FXR Modulators for Enterohepatic and Metabolic Diseases. Expert Opin. Ther. Pat. 2018, 28, 765–782. [Google Scholar] [CrossRef]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.-U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Orešič, M.; Bäckhed, F. Gut Microbiota Regulates Bile Acid Metabolism by Reducing the Levels of Tauro-Beta-Muricholic Acid, a Naturally Occurring FXR Antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Houten, S.M.; Mataki, C.; Christoffolete, M.A.; Kim, B.W.; Sato, H.; Messaddeq, N.; Harney, J.W.; Ezaki, O.; Kodama, T.; et al. Bile Acids Induce Energy Expenditure by Promoting Intracellular Thyroid Hormone Activation. Nature 2006, 439, 484–489. [Google Scholar] [CrossRef]
- Chaudhari, S.N.; Harris, D.A.; Aliakbarian, H.; Luo, J.N.; Henke, M.T.; Subramaniam, R.; Vernon, A.H.; Tavakkoli, A.; Sheu, E.G.; Devlin, A.S. Bariatric Surgery Reveals a Gut-Restricted TGR5 Agonist with Anti-Diabetic Effects. Nat. Chem. Biol. 2021, 17, 20–29. [Google Scholar] [CrossRef]
- Ryan, K.K.; Tremaroli, V.; Clemmensen, C.; Kovatcheva-Datchary, P.; Myronovych, A.; Karns, R.; Wilson-Pérez, H.E.; Sandoval, D.A.; Kohli, R.; Bäckhed, F.; et al. FXR Is a Molecular Target for the Effects of Vertical Sleeve Gastrectomy. Nature 2014, 509, 183–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan, S.Y.; Jiao, J.; Qi, J.; Wang, Y.; Wei, P.; McCormick, J.B.; Fisher-Hoch, S.P.; Beretta, L. Bile Acid Changes Associated With Liver Fibrosis and Steatosis in the Mexican-American Population of South Texas. Hepatol. Commun. 2020, 4, 555–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prinz, P.; Hofmann, T.; Ahnis, A.; Elbelt, U.; Goebel-Stengel, M.; Klapp, B.F.; Rose, M.; Stengel, A. Plasma Bile Acids Show a Positive Correlation with Body Mass Index and Are Negatively Associated with Cognitive Restraint of Eating in Obese Patients. Front. Neurosci. 2015, 9, 199. [Google Scholar] [CrossRef] [Green Version]
- Wewalka, M.; Patti, M.-E.; Barbato, C.; Houten, S.M.; Goldfine, A.B. Fasting Serum Taurine-Conjugated Bile Acids Are Elevated in Type 2 Diabetes and Do Not Change With Intensification of Insulin. J. Clin. Endocrinol. Metab. 2014, 99, 1442–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillard, J.; Clerbaux, L.-A.; Nachit, M.; Sempoux, C.; Staels, B.; Bindels, L.B.; Tailleux, A.; Leclercq, I.A. Bile Acids Contribute to the Development of Non-Alcoholic Steatohepatitis in Mice. JHEP Rep. 2022, 4, 100387. [Google Scholar] [CrossRef] [PubMed]
- Mouzaki, M.; Wang, A.Y.; Bandsma, R.; Comelli, E.M.; Arendt, B.M.; Zhang, L.; Fung, S.; Fischer, S.E.; McGilvray, I.G.; Allard, J.P. Bile Acids and Dysbiosis in Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0151829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christakos, S.; Dhawan, P.; Verstuyf, A.; Verlinden, L.; Carmeliet, G. Vitamin D: Metabolism, Molecular Mechanism of Action, and Pleiotropic Effects. Physiol. Rev. 2016, 96, 365–408. [Google Scholar] [CrossRef]
- Jia, W.; Xie, G.; Jia, W. Bile Acid–Microbiota Crosstalk in Gastrointestinal Inflammation and Carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, J.Y.L.; Ferrell, J.M. Bile Acids as Metabolic Regulators and Nutrient Sensors. Annu. Rev. Nutr. 2019, 39, 175–200. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F. Detoxification of Lithocholic Acid, A Toxic Bile Acid: Relevance to Drug Hepatotoxicity. Drug Metab. Rev. 2004, 36, 703–722. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X Nuclear Receptor Ligand Obeticholic Acid for Non-Cirrhotic, Non-Alcoholic Steatohepatitis (FLINT): A Multicentre, Randomised, Placebo-Controlled Trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the Gut Microbiota in Disease. Microb. Ecol. Health Dis. 2015, 26. [Google Scholar] [CrossRef]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Mannerås-Holm, L.; Ståhlman, M.; Olsson, L.M.; Serino, M.; Planas-Fèlix, M.; et al. Metformin Alters the Gut Microbiome of Individuals with Treatment-Naive Type 2 Diabetes, Contributing to the Therapeutic Effects of the Drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef]
- Chen, M.; Yi, L.; Zhang, Y.; Zhou, X.; Ran, L.; Yang, J.; Zhu, J.; Zhang, Q.; Mi, M. Resveratrol Attenuates Trimethylamine- N -Oxide (TMAO)-Induced Atherosclerosis by Regulating TMAO Synthesis and Bile Acid Metabolism via Remodeling of the Gut Microbiota. mBio 2016, 7, e2210–e2215. [Google Scholar] [CrossRef] [Green Version]
- Attaye, I.; Pinto-Sietsma, S.-J.; Herrema, H.; Nieuwdorp, M. A Crucial Role for Diet in the Relationship Between Gut Microbiota and Cardiometabolic Disease. Annu. Rev. Med. 2020, 71, 149–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.M.; Hazen, S.L. The Gut Microbial Endocrine Organ: Bacterially Derived Signals Driving Cardiometabolic Diseases. Annu. Rev. Med. 2015, 66, 343–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal Microbiota Metabolism of L-Carnitine, a Nutrient in Red Meat, Promotes Atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Yan, Q.; Gu, Y.; Li, X.; Yang, W.; Jia, L.; Chen, C.; Han, X.; Huang, Y.; Zhao, L.; Li, P.; et al. Alterations of the Gut Microbiome in Hypertension. Front. Cell. Infect. Microbiol. 2017, 7, 381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durgan, D.J.; Ganesh, B.P.; Cope, J.L.; Ajami, N.J.; Phillips, S.C.; Petrosino, J.F.; Hollister, E.B.; Bryan, R.M. Role of the Gut Microbiome in Obstructive Sleep Apnea–Induced Hypertension. Hypertension 2016, 67, 469–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B.; et al. Gut Microbiota Dysbiosis Contributes to the Development of Hypertension. Microbiome 2017, 5, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakai, M.; Ribeiro, R.V.; Stevens, B.R.; Gill, P.; Muralitharan, R.R.; Yiallourou, S.; Muir, J.; Carrington, M.; Head, G.A.; Kaye, D.M.; et al. Essential Hypertension Is Associated With Changes in Gut Microbial Metabolic Pathways: A Multisite Analysis of Ambulatory Blood Pressure. Hypertension 2021, 78, 804–815. [Google Scholar] [CrossRef]
- Verhaar, B.J.H.; Collard, D.; Prodan, A.; Levels, J.H.M.; Zwinderman, A.H.; Bäckhed, F.; Vogt, L.; Peters, M.J.L.; Muller, M.; Nieuwdorp, M.; et al. Associations between Gut Microbiota, Faecal Short-Chain Fatty Acids, and Blood Pressure across Ethnic Groups: The HELIUS Study. Eur. Heart J. 2020, 41, 4259–4267. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Jin, J.; Su, X.; Yin, X.; Gao, J.; Wang, X.; Zhang, S.; Bu, P.; Wang, M.; Zhang, Y.; et al. Intestinal Flora Modulates Blood Pressure by Regulating the Synthesis of Intestinal-Derived Corticosterone in High Salt-Induced Hypertension. Circ. Res. 2020, 126, 839–853. [Google Scholar] [CrossRef]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut Dysbiosis Is Linked to Hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef] [Green Version]
- Vallianou, N.G.; Geladari, E.; Kounatidis, D. Microbiome and Hypertension: Where Are We Now? J. Cardiovasc. Med. 2020, 21, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Chávez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. Bile Acid Control of Metabolism and Inflammation in Obesity, Type 2 Diabetes, Dyslipidemia, and Nonalcoholic Fatty Liver Disease. Gastroenterology 2017, 152, 1679–1694.e3. [Google Scholar] [CrossRef] [PubMed]
- Ma, K. Farnesoid X Receptor Is Essential for Normal Glucose Homeostasis. J. Clin. Investig. 2006, 116, 1102–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witjes, J.J.; Smits, L.P.; Pekmez, C.T.; Prodan, A.; Meijnikman, A.S.; Troelstra, M.A.; Bouter, K.E.C.; Herrema, H.; Levin, E.; Holleboom, A.G.; et al. Donor Fecal Microbiota Transplantation Alters Gut Microbiota and Metabolites in Obese Individuals With Steatohepatitis. Hepatol. Commun. 2020, 4, 1578–1590. [Google Scholar] [CrossRef] [PubMed]
- Koopen, A.; Witjes, J.; Wortelboer, K.; Majait, S.; Prodan, A.; Levin, E.; Herrema, H.; Winkelmeijer, M.; Aalvink, S.; Bergman, J.J.G.H.M.; et al. Duodenal Anaerobutyricum Soehngenii Infusion Stimulates GLP-1 Production, Ameliorates Glycaemic Control and Beneficially Shapes the Duodenal Transcriptome in Metabolic Syndrome Subjects: A Randomised Double-Blind Placebo-Controlled Cross-over Study. Gut 2021, 70, gutjnl-2020-323297. [Google Scholar] [CrossRef] [PubMed]
- Bogitsh, B.; Carter, C.; Oeltmann, T. Human Parasitology, 4th ed.; Academic Press: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Donachie, S.P.; Fraser, C.J.; Hill, E.C.; Butler, M.A. The Problem with ‘Microbiome’. Diversity 2021, 13, 138. [Google Scholar] [CrossRef]
- Xu, W.; Chen, T.; Pei, Y.; Guo, H.; Li, Z.; Yang, Y.; Zhang, F.; Yu, J.; Li, X.; Yang, Y.; et al. Characterization of Shallow Whole-Metagenome Shotgun Sequencing as a High-Accuracy and Low-Cost Method by Complicated Mock Microbiomes. Front. Microbiol. 2021, 12, 678319. [Google Scholar] [CrossRef]
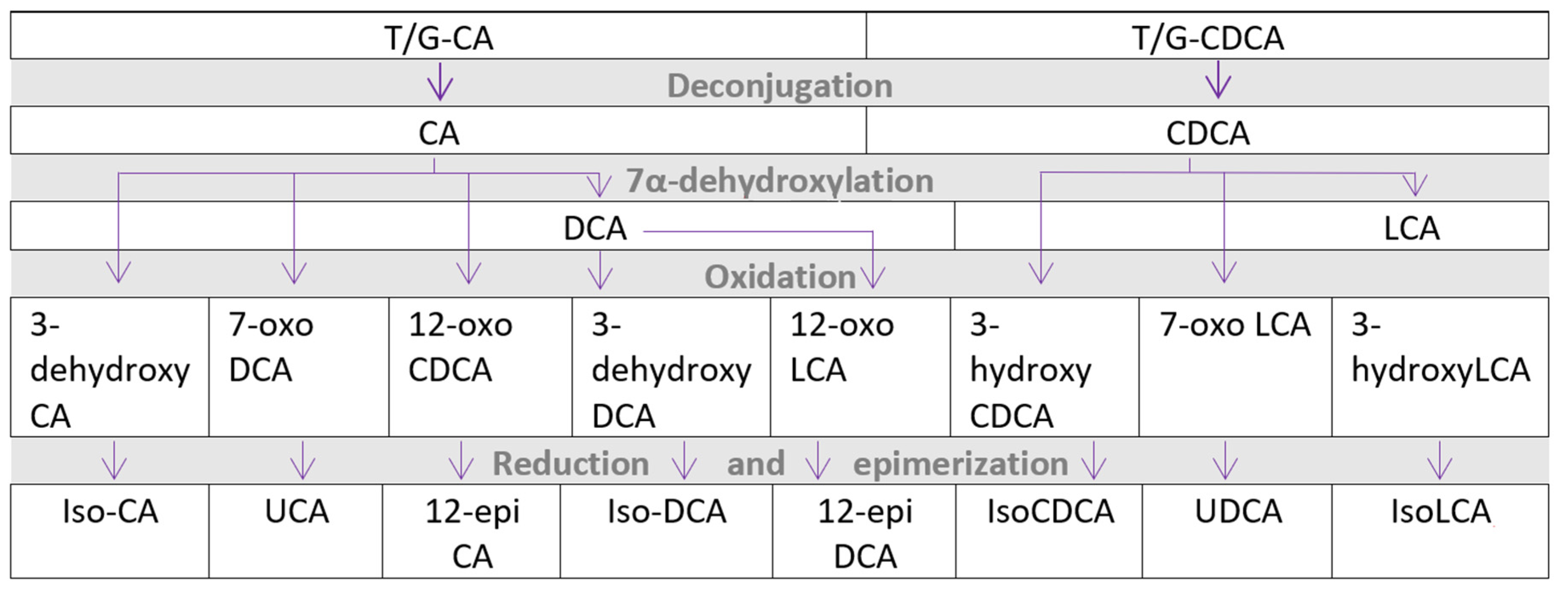
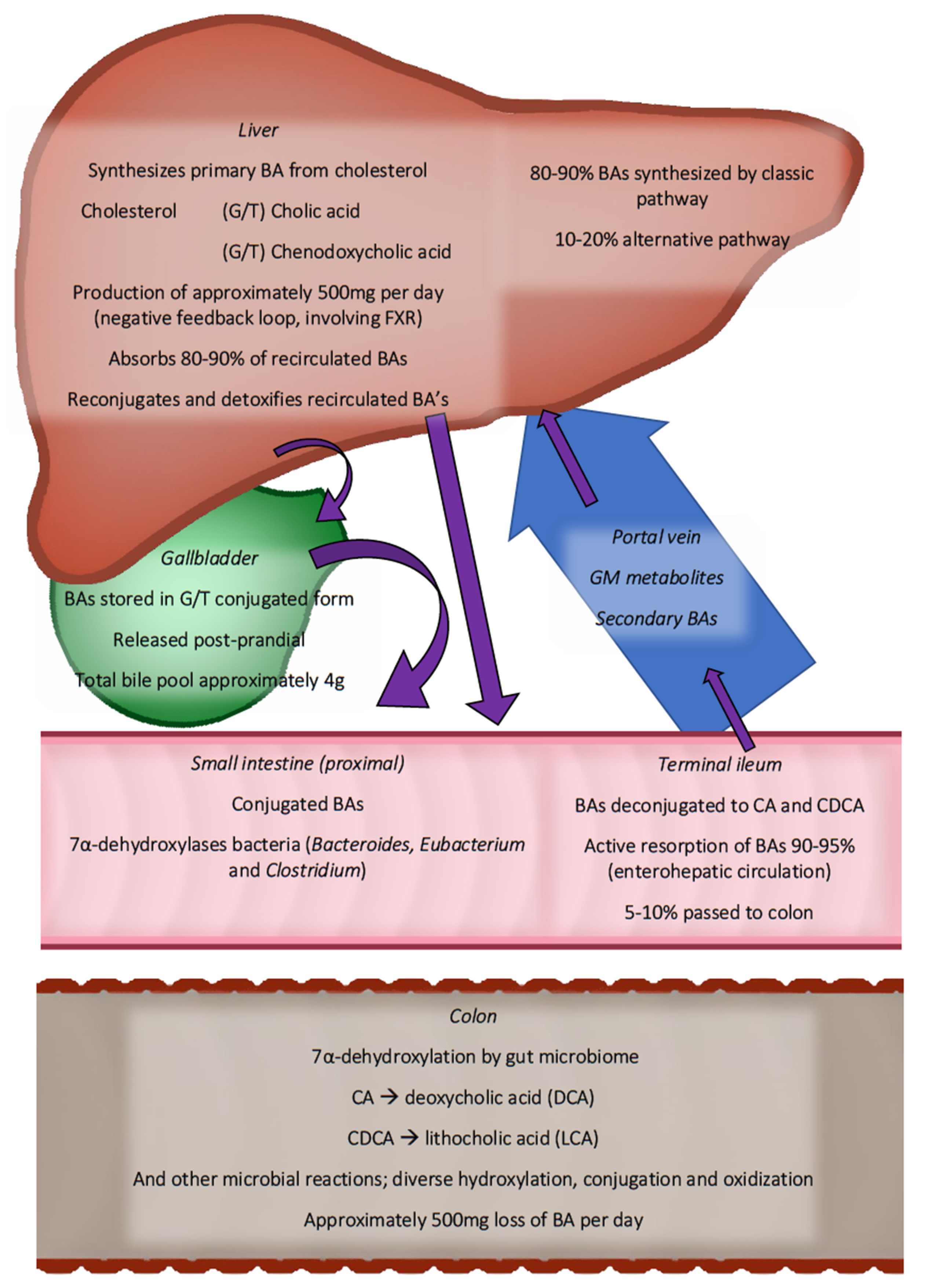

{kind=link}
{kind=link}
| Bile Acids | Source | Associated Disease | References |
|---|---|---|---|
| Total bile pool | After vertical sleeve gastrectomy in mice, bile acid pool is increased. This caused more weight loss and improved glucose tolerance through a farnesoid-X-receptor-mediated pathway. | [79] Mice | |
| Liver fibrosis: Higher concentration | [80] Human | ||
| Primary bile acids | Synthesised in liver | Obesity: Increased concentration, intestinal FGF levels decreased. | [81] Human |
| Type 2 diabetes: Taurine-conjugated BAs are increased. | [82] Human | ||
| NASH: Less concentrated, lowering TGR5 activation. | [83] Mice | ||
| Chendeoxycholic acid | Primary | NAFLD: increased concentration. | [84] Human |
| Secondary bile acid pool | Microbial metabolites, synthesised by (among others) Bacteroides, Bifidobacterium, Clostridium, Eubacterium, Lactobacillus, Listeria, and Ruminococcus | Agonists of FXR and other nuclear receptors. Increased expression of TGR5. Increased insulin sensitivity. | [85,86,87] Both human and mice studies |
| Lithocholic acid | Secondary BA | Cytotoxic in unsulphated form. Liver fibrosis: high concentrations. | [80,88] Human |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Callender, C.; Attaye, I.; Nieuwdorp, M. The Interaction between the Gut Microbiome and Bile Acids in Cardiometabolic Diseases. Metabolites 2022, 12, 65. https://doi.org/10.3390/metabo12010065
Callender C, Attaye I, Nieuwdorp M. The Interaction between the Gut Microbiome and Bile Acids in Cardiometabolic Diseases. Metabolites. 2022; 12(1):65. https://doi.org/10.3390/metabo12010065
Chicago/Turabian StyleCallender, Cengiz, Ilias Attaye, and Max Nieuwdorp. 2022. "The Interaction between the Gut Microbiome and Bile Acids in Cardiometabolic Diseases" Metabolites 12, no. 1: 65. https://doi.org/10.3390/metabo12010065
APA StyleCallender, C., Attaye, I., & Nieuwdorp, M. (2022). The Interaction between the Gut Microbiome and Bile Acids in Cardiometabolic Diseases. Metabolites, 12(1), 65. https://doi.org/10.3390/metabo12010065