
Targeting the Gut in Obesity: Signals from the Inner Surface

 , ,
, ,
Abstract
:
1. Introduction
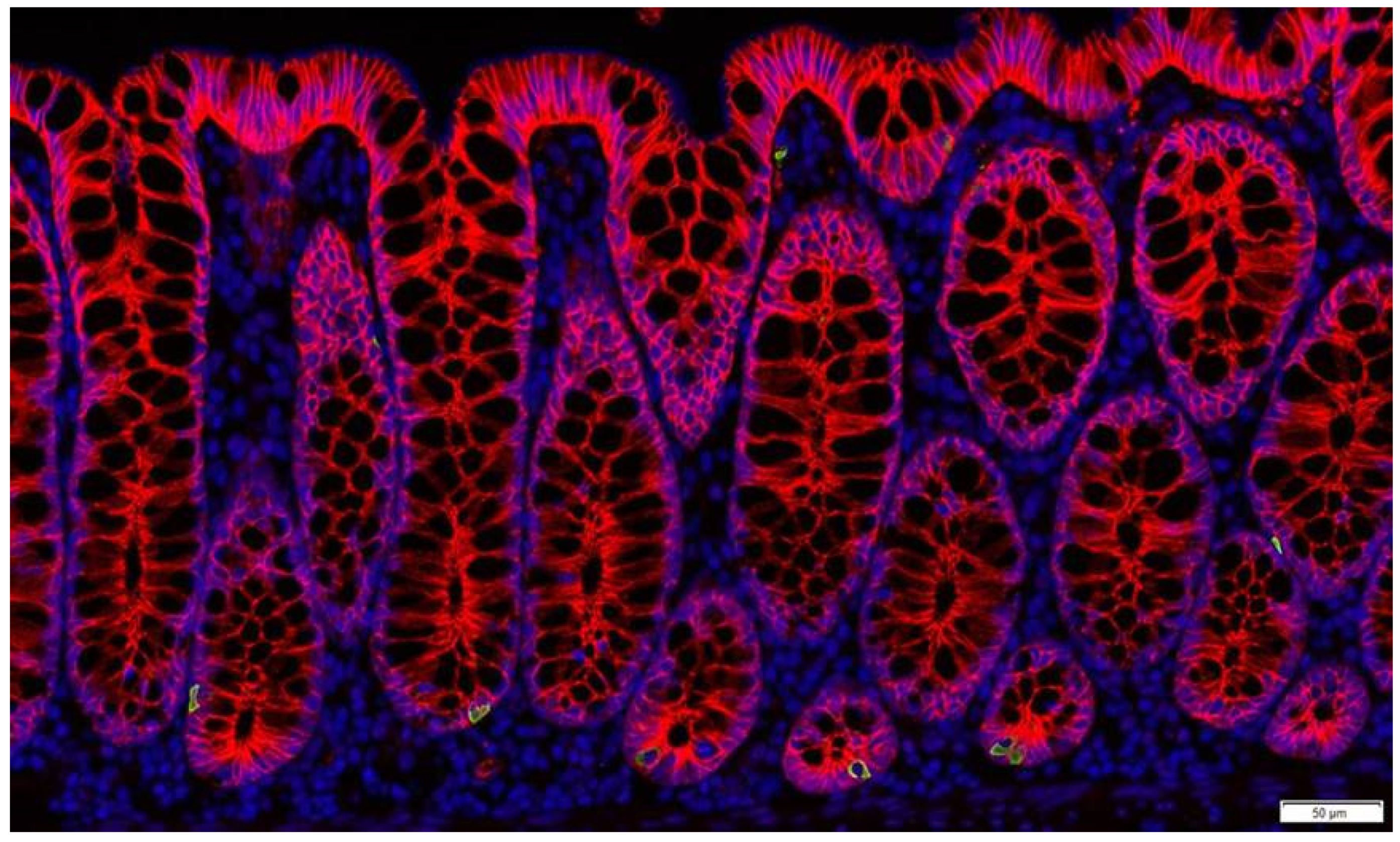
2. Intestinal Epithelium and Signalling Properties of Nutrients
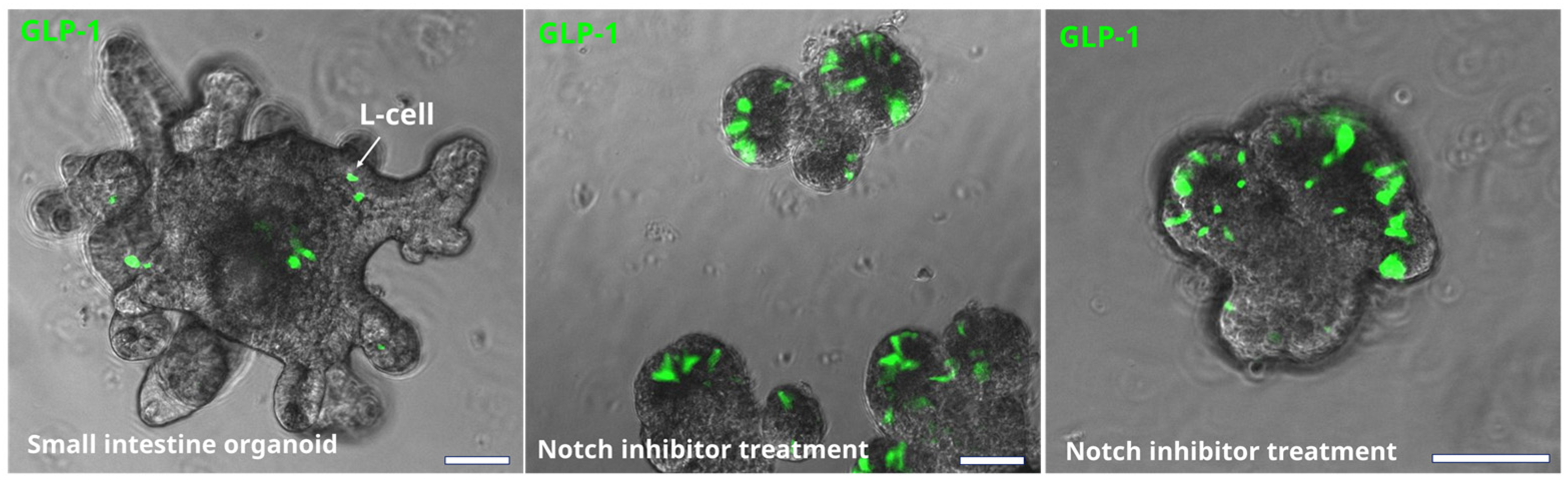
3. Enteroendocrine Cells
4. Microbial Metabolites
5. Enterokines and Gut-Liver Axis
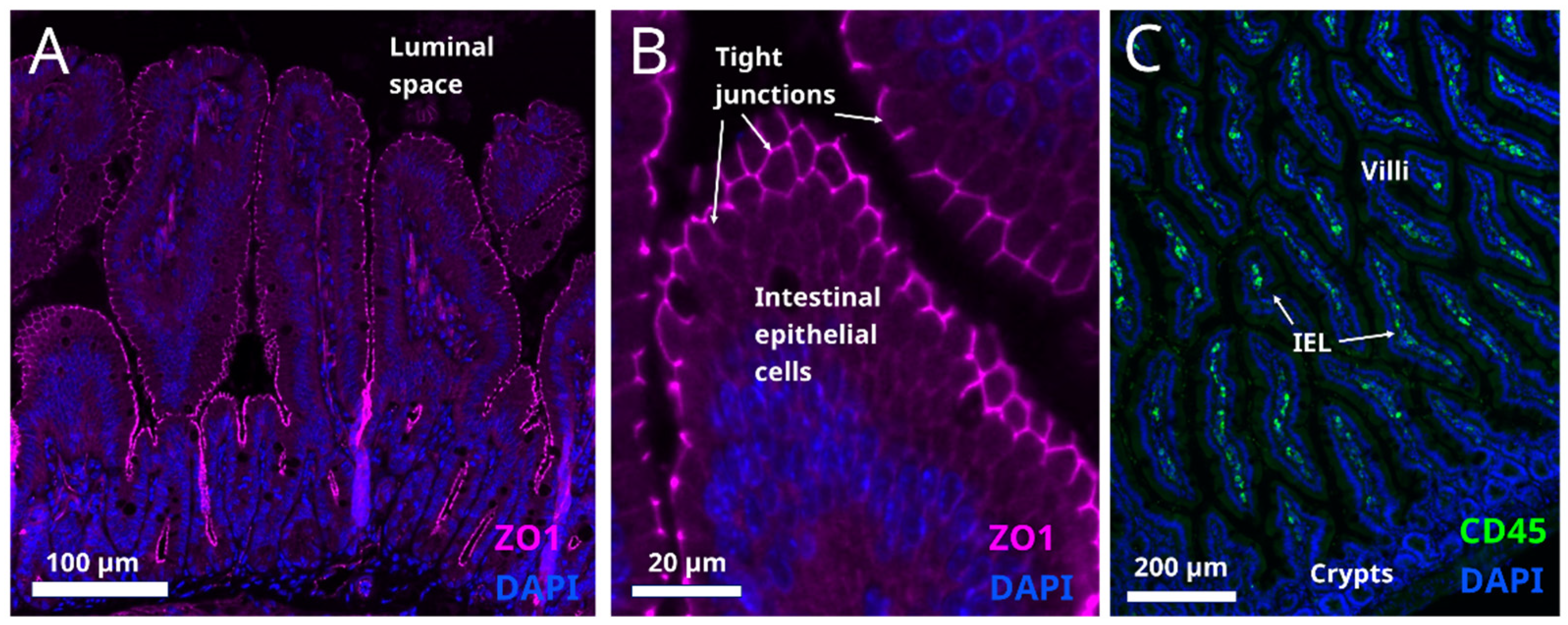
6. Intestinal Barrier Function
7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Der Schoor, S.R.D.; Reeds, P.J.; Stoll, B.; Henry, J.F.; Rosenberger, J.R.; Burrin, D.G.; Van Goudoever, J.B. The High Metabolic Cost of a Functional Gut. Gastroenterology 2002, 123, 1931–1940. [Google Scholar] [CrossRef]
- Clevers, H. The Intestinal Crypt, a Prototype Stem Cell Compartment. Cell 2013, 154, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Gehart, H.; Clevers, H. Tales from the Crypt: New Insights into Intestinal Stem Cells. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 19–34. [Google Scholar] [CrossRef]
- Mah, A.T.; Van Landeghem, L.; Gavin, H.E.; Magness, S.T.; Lund, P.K. Impact of Diet-Induced Obesity on Intestinal Stem Cells: Hyperproliferation but Impaired Intrinsic Function That Requires Insulin/IGF1. Endocrinology 2014, 155, 3302–3314. [Google Scholar] [CrossRef] [Green Version]
- Aliluev, A.; Tritschler, S.; Sterr, M.; Oppenländer, L.; Hinterdobler, J.; Greisle, T.; Irmler, M.; Beckers, J.; Sun, N.; Walch, A.; et al. Diet-Induced Alteration of Intestinal Stem Cell Function Underlies Obesity and Prediabetes in Mice. Nat. Metab. 2021, 3, 1202–1216. [Google Scholar] [CrossRef]
- Pourvali, K.; Monji, H. Obesity and Intestinal Stem Cell Susceptibility to Carcinogenesis. Nutr. Metab. 2021, 18, 37. [Google Scholar] [CrossRef]
- Tysoe, O. PPAR Mediates Intestinal Stem Cell Tumorigenesis. Nat. Rev. Endocrinol. 2021, 17, 514. [Google Scholar] [CrossRef]
- Dailey, M.J. Nutrient-Induced Intestinal Adaption and Its Effect in Obesity. Physiol. Behav. 2014, 136, 74–78. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Bancroft, L.; Nicholas, C.; Lozonschi, I.; Augenlicht, L.H. Targeted Inactivation of P27kip1 Is Sufficient for Large and Small Intestinal Tumorigenesis in the Mouse, Which Can Be Augmented by a Western-Style High-Risk Diet. Cancer Res. 2003, 63, 4990–4996. [Google Scholar]
- Gerbe, F.; Jay, P. Intestinal Tuft Cells: Epithelial Sentinels Linking Luminal Cues to the Immune System. Mucosal Immunol. 2016, 9, 1353–1359. [Google Scholar] [CrossRef]
- Cruciani-Guglielmacci, C.; Fioramonti, X. Editorial: Brain Nutrient Sensing in the Control of Energy Balance: New Insights and Perspectives. Front. Physiol. 2019, 10, 51. [Google Scholar] [CrossRef] [Green Version]
- Keast, R.; Costanzo, A.; Hartley, I. Macronutrient Sensing in the Oral Cavity and Gastrointestinal Tract: Alimentary Tastes. Nutrients 2021, 13, 667. [Google Scholar] [CrossRef]
- Anderson, J.W.; Konz, E.C.; Frederich, R.C.; Wood, C.L. Long-term weight-loss maintenance: A meta-analysis of US studies. Am. J. Clin. Nutr. 2001, 74, 579–584. [Google Scholar] [CrossRef]
- Avenell, A.; Broom, J.; Brown, T.J.; Poobalan, A.; Aucott, L.; Stearns, S.C.; Smith, W.C.; Jung, R.T.; Campbell, M.K.; Grant, M. Systematic review of the long-term effects and economic consequences of treatments for obesity and implications for health improvement. Health Technol. Assess 2004, 8, 1–182. [Google Scholar] [CrossRef]
- Ayyad, C.; Andersen, T. Long-term efficacy of dietary treatment of obesity: A systematic review of studies published between 1931 and 1999. Obes. Rev. 2000, 1, 113–119. [Google Scholar] [CrossRef]
- Look AHEAD Research, Group; Wing, R.R.; Bolin, P.; Brancati, F.L.; Bray, G.A.; Clark, J.M.; Coday, M.; Crow, R.S.; Curtis, J.M.; Egan, C.M.; et al. Cardiovascular Effects of Intensive Lifestyle Intervention in Type 2 Diabetes. N. Engl. J. Med. 2013, 369, 145–154. [Google Scholar]
- Knowler, W.C.; Barrett-Connor, E.; Fowler, S.E.; Hamman, R.F.; Lachin, J.M.; Walker, E.A.; Nathan, D.M.; Diabetes Prevention Program Research Group. Reduction in the Incidence of Type 2 Diabetes with Lifestyle Intervention or Metformin. N. Engl. J. Med. 2002, 346, 393–403. [Google Scholar]
- Dragano, N.R.V.; Solon, C.; Ramalho, A.F.; de Moura, R.F.; Razolli, D.S.; Christiansen, E.; Azevedo, C.; Ulven, T.; Velloso, L.A. Polyunsaturated Fatty Acid Receptors, GPR40 and GPR120, Are Expressed in the Hypothalamus and Control Energy Homeostasis and Inflammation. J. Neuroinflamm. 2017, 14, 91. [Google Scholar] [CrossRef] [Green Version]
- Auguste, S.; Fisette, A.; Fernandes, M.F.; Hryhorczuk, C.; Poitout, V.; Alquier, T.; Fulton, S. Central Agonism of GPR120 Acutely Inhibits Food Intake and Food Reward and Chronically Suppresses Anxiety-Like Behavior in Mice. Int. J. Neuropsychopharmacol. 2016, 19, pyw014. [Google Scholar] [CrossRef]
- Hirasawa, A.; Tsumaya, K.; Awaji, T.; Katsuma, S.; Adachi, T.; Yamada, M.; Sugimoto, Y.; Miyazaki, S.; Tsujimoto, G. Free Fatty Acids Regulate Gut Incretin Glucagon-like Peptide-1 Secretion through GPR120. Nat. Med. 2005, 11, 90–94. [Google Scholar] [CrossRef]
- Christensen, L.W.; Kuhre, R.E.; Janus, C.; Svendsen, B.; Holst, J.J. Vascular, but Not Luminal, Activation of FFAR1 (GPR40) Stimulates GLP-1 Secretion from Isolated Perfused Rat Small Intestine. Physiol. Rep. 2015, 3, e12551. [Google Scholar] [CrossRef] [Green Version]
- Sankoda, A.; Harada, N.; Kato, T.; Ikeguchi, E.; Iwasaki, K.; Yamane, S.; Murata, Y.; Hirasawa, A.; Inagaki, N. Free Fatty Acid Receptors, G Protein-Coupled Receptor 120 and G Protein-Coupled Receptor 40, Are Essential for Oil-Induced Gastric Inhibitory Polypeptide Secretion. J. Diabetes Investig. 2019, 10, 1430–1437. [Google Scholar] [CrossRef]
- Pais, R.; Gribble, F.M.; Reimann, F. Signalling Pathways Involved in the Detection of Peptones by Murine Small Intestinal Enteroendocrine L-Cells. Peptides 2016, 77, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Alleleyn, A.M.E.; van Avesaat, M.; Troost, F.J.; Masclee, A.A.M. Gastrointestinal Nutrient Infusion Site and Eating Behavior: Evidence for A Proximal to Distal Gradient within the Small Intestine? Nutrients 2016, 8, 117. [Google Scholar] [CrossRef] [Green Version]
- Rolls, B.J. Carbohydrates, Fats, and Satiety. Am. J. Clin. Nutr. 1995, 61 (Suppl. 4), 960S–967S. [Google Scholar] [CrossRef]
- Chapman, I.M.; Goble, E.A.; Wittert, G.A.; Horowitz, M. Effects of Small-Intestinal Fat and Carbohydrate Infusions on Appetite and Food Intake in Obese and Nonobese Men. Am. J. Clin. Nutr. 1999, 69, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, N.; McKnight, A.D.; Carty, J.R.E.; Arnold, M.; Betley, J.N.; Alhadeff, A.L. Hypothalamic Detection of Macronutrients via Multiple Gut-Brain Pathways. Cell Metab. 2021, 33, 676–687.e5. [Google Scholar] [CrossRef]
- Minokoshi, Y.; Nakajima, K.-I.; Okamoto, S. Homeostatic versus Hedonic Control of Carbohydrate Selection. J. Physiol. 2020, 598, 3831–3844. [Google Scholar] [CrossRef]
- Paradis, S.; Philippe, E.; Cabanac, M. Does intestinal absorption participate in the ponderostat? Physiol. Behav. 2007, 16, 664–668. [Google Scholar] [CrossRef]
- Ramachandran, D.; Clara, R.; Fedele, S.; Michel, L.; Burkard, J.; Kaufman, S.; Diaz, A.A.; Weissfeld, N.; De Bock, K.; Prip-Buus, C.; et al. Enhancing Enterocyte Fatty Acid Oxidation in Mice Affects Glycemic Control Depending on Dietary Fat. Sci. Rep. 2018, 8, 10818. [Google Scholar] [CrossRef]
- Smith, S.R.; Stenlof, K.S.; Greenway, F.L.; McHutchison, J.; Schwartz, S.M.; Dev, V.B.; Berk, E.S.; Kapikian, R. Orlistat 60 Mg Reduces Visceral Adipose Tissue: A 24-Week Randomized, Placebo-Controlled, Multicenter Trial. Obesity 2011, 19, 1796–1803. [Google Scholar] [CrossRef]
- Ehrenkranz, J.R.L.; Lewis, N.G.; Kahn, C.R.; Roth, J. Phlorizin: A Review. Diabetes Metab. Res. Rev. 2005, 21, 31–38. [Google Scholar] [CrossRef]
- Zhang, X.-Z.; Guan, J.; Cai, S.-L.; Du, Q.; Guo, M.-L. Polymeric In Situ Hydrogel Implant of Epigallocatechin Gallate (EGCG) for Prolonged and Improved Antihyperlipidemic and Anti-Obesity Activity: Preparation and Characterization. J. Biomater. Tissue Eng. 2015, 5, 813–817. [Google Scholar] [CrossRef]
- Howarth, N.C.; Saltzman, E.; Roberts, S.B. Dietary fiber and weight regulation. Nutr. Rev. 2001, 59, 129–139. [Google Scholar] [CrossRef]
- Furness, J.B.; Rivera, L.R.; Cho, H.-J.; Bravo, D.M.; Callaghan, B. The Gut as a Sensory Organ. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 729–740. [Google Scholar] [CrossRef]
- Lund, M.L.; Egerod, K.L.; Engelstoft, M.S.; Dmytriyeva, O.; Theodorsson, E.; Patel, B.A.; Schwartz, T.W. Enterochromaffin 5-HT Cells-A Major Target for GLP-1 and Gut Microbial Metabolites. Mol. Metab. 2018, 11, 70–83. [Google Scholar] [CrossRef]
- Kato, S. Role of serotonin 5-HT3 receptors in intestinal inflammation. Biol. Pharm. Bull. 2013, 36, 1406–1409. [Google Scholar] [CrossRef] [Green Version]
- Oh, C.M.; Park, S.; Kim, H. Serotonin as a New Therapeutic Target for Diabetes Mellitus and Obesity. Diabetes Metab. J. 2016, 40, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Binetti, J.; Bertran, L.; Riesco, D.; Aguilar, C.; Martínez, S.; Sabench, F.; Porras, J.A.; Camaron, J.; Castillo, D.D.; Richart, C.; et al. Deregulated Serotonin Pathway in Women with Morbid Obesity and NAFLD. Life 2020, 10, 245. [Google Scholar] [CrossRef]
- Carey, A.L.; Kingwell, B.A. Reducing peripheral serotonin turns up the heat in brown fat. Nat. Med. 2015, 21, 114–116. [Google Scholar] [CrossRef]
- Gribble, F.M.; Reimann, F. Enteroendocrine Cells: Chemosensors in the Intestinal Epithelium. Annu. Rev. Physiol. 2016, 78, 277–299. [Google Scholar] [CrossRef]
- Greiner, T.U.; Bäckhed, F. Microbial Regulation of GLP-1 and L-Cell Biology. Mol. Metab. 2016, 5, 753–758. [Google Scholar] [CrossRef]
- Lassmann, V.; Vague, P.; Vialettes, B.; Simon, M.C. Low Plasma Levels of Pancreatic Polypeptide in Obesity. Diabetes 1980, 29, 428–430. [Google Scholar] [CrossRef]
- Cawthon, C.R.; de La Serre, C.B. The Critical Role of CCK in the Regulation of Food Intake and Diet-Induced Obesity. Peptides 2021, 138, 170492. [Google Scholar] [CrossRef]
- Svane, M.S.; Jørgensen, N.B.; Bojsen-Møller, K.N.; Dirksen, C.; Nielsen, S.; Kristiansen, V.B.; Toräng, S.; Wewer Albrechtsen, N.J.; Rehfeld, J.F.; Hartmann, B.; et al. Peptide YY and Glucagon-like Peptide-1 Contribute to Decreased Food Intake after Roux-En-Y Gastric Bypass Surgery. Int. J. Obes. 2016, 40, 1699–1706. [Google Scholar] [CrossRef]
- Jacobsen, S.H.; Olesen, S.C.; Dirksen, C.; Jørgensen, N.B.; Bojsen-Møller, K.N.; Kielgast, U.; Worm, D.; Almdal, T.; Naver, L.S.; Hvolris, L.E.; et al. Changes in Gastrointestinal Hormone Responses, Insulin Sensitivity, and Beta-Cell Function within 2 Weeks after Gastric Bypass in Non-Diabetic Subjects. Obes. Surg. 2012, 22, 1084–1096. [Google Scholar] [CrossRef]
- Reimann, F.; Tolhurst, G.; Gribble, F.M. G-Protein-Coupled Receptors in Intestinal Chemosensation. Cell Metab. 2012, 15, 421–431. [Google Scholar] [CrossRef] [Green Version]
- Nauck, M.A.; Kemmeries, G.; Holst, J.J.; Meier, J.J. Rapid Tachyphylaxis of the Glucagon-like Peptide 1-Induced Deceleration of Gastric Emptying in Humans. Diabetes 2011, 60, 1561–1565. [Google Scholar] [CrossRef] [Green Version]
- Christoffersen, B.Ø.; Skyggebjerg, R.B.; Bugge, A.; Kirk, R.K.; Vestergaard, B.; Uldam, H.K.; Fels, J.J.; Pyke, C.; Sensfuss, U.; Sanfridson, A.; et al. Long-Acting CCK Analogue NN9056 Lowers Food Intake and Body Weight in Obese Göttingen Minipigs. Int. J. Obes. 2020, 44, 447–456. [Google Scholar] [CrossRef] [Green Version]
- Hauge, M.; Vestmar, M.A.; Husted, A.S.; Ekberg, J.P.; Wright, M.J.; Di Salvo, J.; Weinglass, A.B.; Engelstoft, M.S.; Madsen, A.N.; Lückmann, M.; et al. GPR40 (FFAR1)-Combined Gs and Gq Signaling in Vitro Is Associated with Robust Incretin Secretagogue Action Ex Vivo and in Vivo. Mol. Metab. 2015, 4, 3–14. [Google Scholar] [CrossRef]
- Husted, A.S.; Trauelsen, M.; Rudenko, O.; Hjorth, S.A.; Schwartz, T.W. GPCR-Mediated Signaling of Metabolites. Cell Metab. 2017, 25, 777–796. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhao, Y.; Hu, Y.; Peng, J. Targeting the GPR119/Incretin Axis: A Promising New Therapy for Metabolic-Associated Fatty Liver Disease. Cell. Mol. Biol. Lett. 2021, 26, 32. [Google Scholar] [CrossRef]
- Petersen, N.; Reimann, F.; van Es, J.H.; van den Berg, B.M.; Kroone, C.; Pais, R.; Jansen, E.; Clevers, H.; Gribble, F.M.; de Koning, E.J.P. Targeting Development of Incretin-Producing Cells Increases Insulin Secretion. J. Clin. Investig. 2015, 125, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Lund, M.L.; Sorrentino, G.; Egerod, K.L.; Kroone, C.; Mortensen, B.; Knop, F.K.; Reimann, F.; Gribble, F.M.; Drucker, D.J.; de Koning, E.; et al. L-Cell Differentiation Is Induced by Bile Acids Through GPBAR1 and Paracrine GLP-1 and Serotonin Signaling. Diabetes 2020, 9, 614–623. [Google Scholar] [CrossRef]
- Arora, T.; Akrami, R.; Pais, R.; Bergqvist, L.; Johansson, B.R.; Schwartz, T.W.; Reimann, F.; Gribble, F.M.; Bäckhed, F. Microbial Regulation of the L Cell Transcriptome. Sci. Rep. 2018, 8, 1207. [Google Scholar] [CrossRef]
- Kuhre, R.E.; Deacon, C.F.; Holst, J.J.; Petersen, N. What Is an L-Cell and How Do We Study the Secretory Mechanisms of the L-Cell? Front. Endocrinol. 2021, 12, 694284. [Google Scholar] [CrossRef]
- Grosse, J.; Heffron, H.; Burling, K.; Akhter Hossain, M.; Habib, A.M.; Rogers, G.J.; Richards, P.; Larder, R.; Rimmington, D.; Adriaenssens, A.A.; et al. Insulin-like Peptide 5 Is an Orexigenic Gastrointestinal Hormone. Proc. Natl. Acad. Sci. USA 2014, 111, 11133–11138. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; De Vadder, F.; Tremaroli, V.; Wichmann, A.; Mithieux, G.; Bäckhed, F. Insulin-like Peptide 5 Is a Microbially Regulated Peptide That Promotes Hepatic Glucose Production. Mol. Metab. 2016, 5, 263–270. [Google Scholar] [CrossRef]
- Kamal, N.; Chami, T.; Andersen, A.; Rosell, F.A.; Schuster, M.M.; Whitehead, W.E. Delayed Gastrointestinal Transit Times in Anorexia Nervosa and Bulimia Nervosa. Gastroenterology 1991, 101, 1320–1324. [Google Scholar] [CrossRef]
- Germain, N.; Galusca, B.; Le Roux, C.W.; Bossu, C.; Ghatei, M.A.; Lang, F.; Bloom, S.R.; Estour, B. Constitutional Thinness and Lean Anorexia Nervosa Display Opposite Concentrations of Peptide YY, Glucagon-like Peptide 1, Ghrelin, and Leptin. Am. J. Clin. Nutr. 2007, 85, 967–971. [Google Scholar] [CrossRef]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin Is a Growth-Hormone-Releasing Acylated Peptide from Stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef]
- Cummings, D.E.; Purnell, J.Q.; Frayo, R.S.; Schmidova, K.; Wisse, B.E.; Weigle, D.S. A Preprandial Rise in Plasma Ghrelin Levels Suggests a Role in Meal Initiation in Humans. Diabetes 2001, 50, 1714–1719. [Google Scholar] [CrossRef] [Green Version]
- Tschöp, M.; Wawarta, R.; Riepl, R.L.; Friedrich, S.; Bidlingmaier, M.; Landgraf, R.; Folwaczny, C. Post-Prandial Decrease of Circulating Human Ghrelin Levels. J. Endocrinol. Investig. 2001, 24, RC19–RC21. [Google Scholar] [CrossRef]
- Cowley, M.A.; Smith, R.G.; Diano, S.; Tschöp, M.; Pronchuk, N.; Grove, K.L.; Strasburger, C.J.; Bidlingmaier, M.; Esterman, M.; Heiman, M.L.; et al. The Distribution and Mechanism of Action of Ghrelin in the CNS Demonstrates a Novel Hypothalamic Circuit Regulating Energy Homeostasis. Neuron 2003, 37, 649–661. [Google Scholar] [CrossRef] [Green Version]
- Kohno, D.; Gao, H.-Z.; Muroya, S.; Kikuyama, S.; Yada, T. Ghrelin Directly Interacts with Neuropeptide-Y-Containing Neurons in the Rat Arcuate Nucleus: Ca2+ Signaling via Protein Kinase A and N-Type Channel-Dependent Mechanisms and Cross-Talk with Leptin and Orexin. Diabetes 2003, 52, 948–956. [Google Scholar] [CrossRef] [Green Version]
- López, M.; Lage, R.; Saha, A.K.; Pérez-Tilve, D.; Vázquez, M.J.; Varela, L.; Sangiao-Alvarellos, S.; Tovar, S.; Raghay, K.; Rodríguez-Cuenca, S.; et al. Hypothalamic Fatty Acid Metabolism Mediates the Orexigenic Action of Ghrelin. Cell Metab. 2008, 7, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Atasoy, D.; Su, H.H.; Sternson, S.M. Hunger States Switch a Flip-Flop Memory Circuit via a Synaptic AMPK-Dependent Positive Feedback Loop. Cell 2011, 146, 992–1003. [Google Scholar] [CrossRef] [Green Version]
- Torz, L.J.; Osborne-Lawrence, S.; Rodriguez, J.; He, Z.; Cornejo, M.P.; Mustafá, E.R.; Jin, C.; Petersen, N.; Hedegaard, M.A.; Nybo, M.; et al. Metabolic Insights from a GHSR-A203E Mutant Mouse Model. Mol. Metab. 2020, 39, 101004. [Google Scholar] [CrossRef]
- Sun, Y.; Butte, N.F.; Garcia, J.M.; Smith, R.G. Characterization of Adult Ghrelin and Ghrelin Receptor Knockout Mice under Positive and Negative Energy Balance. Endocrinology 2008, 149, 843–850. [Google Scholar] [CrossRef] [Green Version]
- Castañeda, T.R.; Tong, J.; Datta, R.; Culler, M.; Tschöp, M.H. Ghrelin in the Regulation of Body Weight and Metabolism. Front. Neuroendocrinol. 2010, 31, 44–60. [Google Scholar] [CrossRef]
- Grunddal, K.V.; Diep, T.A.; Petersen, N.; Tough, I.R.; Skov, L.J.; Liu, L.; Buijink, J.A.; Mende, F.; Jin, C.; Jepsen, S.L.; et al. Selective Release of Gastrointestinal Hormones Induced by an Orally Active GPR39 Agonist. Mol. Metab. 2021, 49, 101207. [Google Scholar] [CrossRef]
- Lindqvist, A.; Shcherbina, L.; Fischer, A.-H.T.; Wierup, N. Ghrelin Is a Regulator of Glucagon-Like Peptide 1 Secretion and Transcription in Mice. Front. Endocrinol. 2017, 8, 135. [Google Scholar] [CrossRef] [Green Version]
- Gagnon, J.; Baggio, L.L.; Drucker, D.J.; Brubaker, P.L. Ghrelin Is a Novel Regulator of GLP-1 Secretion. Diabetes 2015, 64, 1513–1521. [Google Scholar] [CrossRef] [Green Version]
- Jepsen, S.L.; Vestergaard, E.T.; Larraufie, P.; Gribble, F.M.; Reimann, F.; Jørgensen, J.O.L.; Holst, J.J.; Kuhre, R.E. Ghrelin Does Not Directly Stimulate Secretion of Glucagon-like Peptide-1. J. Clin. Endocrinol. Metab. 2020, 105, dgz046. [Google Scholar] [CrossRef]
- Chanoine, J.-P.; De Waele, K.; Walia, P. Ghrelin and the Growth Hormone Secretagogue Receptor in Growth and Development. Int. J. Obes. 2009, 33 (Suppl. 1), S48–S52. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.-J.; Liang, G.; Li, R.L.; Xie, X.; Sleeman, M.W.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Goldstein, J.L.; Brown, M.S. Ghrelin O-Acyltransferase (GOAT) Is Essential for Growth Hormone-Mediated Survival of Calorie-Restricted Mice. Proc. Natl. Acad. Sci. USA 2010, 107, 7467–7472. [Google Scholar] [CrossRef] [Green Version]
- Abizaid, A.; Hougland, J.L. Ghrelin Signaling: GOAT and GHS-R1a Take a LEAP in Complexity. Trends Endocrinol. Metab. TEM 2020, 31, 107–117. [Google Scholar] [CrossRef]
- Singh, R.K.; Chang, H.-W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of Diet on the Gut Microbiome and Implications for Human Health. J. Transl. Med. 2017, 15, 73. [Google Scholar] [CrossRef] [Green Version]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-Chain Fatty Acids Stimulate Glucagon-like Peptide-1 Secretion via the G-Protein-Coupled Receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Nøhr, M.K.; Pedersen, M.H.; Gille, A.; Egerod, K.L.; Engelstoft, M.S.; Husted, A.S.; Sichlau, R.M.; Grunddal, K.V.; Poulsen, S.S.; Han, S.; et al. GPR41/FFAR3 and GPR43/FFAR2 as Cosensors for Short-Chain Fatty Acids in Enteroendocrine Cells vs. FFAR3 in Enteric Neurons and FFAR2 in Enteric Leukocytes. Endocrinology 2013, 154, 3552–3564. [Google Scholar] [CrossRef]
- Fellows, R.; Denizot, J.; Stellato, C.; Cuomo, A.; Jain, P.; Stoyanova, E.; Balázsi, S.; Hajnády, Z.; Liebert, A.; Kazakevych, J.; et al. Microbiota Derived Short Chain Fatty Acids Promote Histone Crotonylation in the Colon through Histone Deacetylases. Nat. Commun. 2018, 9, 105. [Google Scholar] [CrossRef]
- Larraufie, P.; Martin-Gallausiaux, C.; Lapaque, N.; Dore, J.; Gribble, F.M.; Reimann, F.; Blottiere, H.M. SCFAs Strongly Stimulate PYY Production in Human Enteroendocrine Cells. Sci. Rep. 2018, 8, 74. [Google Scholar] [CrossRef]
- Xu, D.; Yu, B.-P.; Luo, H.-S.; Chen, L.-D. Control of Gallbladder Contractions by Cholecystokinin through Cholecystokinin-A Receptors on Gallbladder Interstitial Cells of Cajal. World J. Gastroenterol. 2008, 14, 2882–2887. [Google Scholar] [CrossRef]
- Russell, D.W. The Enzymes, Regulation, and Genetics of Bile Acid Synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [Green Version]
- Chiang, J.Y.L. Bile Acids: Regulation of Synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef] [Green Version]
- Carey, M.C.; Small, D.M. Micelle Formation by Bile Salts. Physical-Chemical and Thermodynamic Considerations. Arch. Intern. Med. 1972, 130, 506–527. [Google Scholar] [CrossRef]
- Kuhre, R.E.; Wewer Albrechtsen, N.J.; Larsen, O.; Jepsen, S.L.; Balk-Møller, E.; Andersen, D.B.; Deacon, C.F.; Schoonjans, K.; Reimann, F.; Gribble, F.M.; et al. Bile Acids Are Important Direct and Indirect Regulators of the Secretion of Appetite- and Metabolism-Regulating Hormones from the Gut and Pancreas. Mol. Metab. 2018, 11, 84–95. [Google Scholar] [CrossRef]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G Protein-Coupled Receptor Responsive to Bile Acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [Green Version]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a Nuclear Receptor for Bile Acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef]
- Li, F.; Jiang, C.; Krausz, K.W.; Li, Y.; Albert, I.; Hao, H.; Fabre, K.M.; Mitchell, J.B.; Patterson, A.D.; Gonzalez, F.J. Microbiome Remodelling Leads to Inhibition of Intestinal Farnesoid X Receptor Signalling and Decreased Obesity. Nat. Commun. 2013, 4, 2384. [Google Scholar] [CrossRef]
- Devaraj, S.; Hemarajata, P.; Versalovic, J. The Human Gut Microbiome and Body Metabolism: Implications for Obesity and Diabetes. Clin. Chem. 2013, 59, 617–628. [Google Scholar] [CrossRef] [Green Version]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet Rapidly and Reproducibly Alters the Human Gut Microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Chimerel, C.; Emery, E.; Summers, D.K.; Keyser, U.; Gribble, F.M.; Reimann, F. Bacterial Metabolite Indole Modulates Incretin Secretion from Intestinal Enteroendocrine L Cells. Cell Rep. 2014, 9, 1202–1208. [Google Scholar] [CrossRef] [Green Version]
- Koh, A.; Molinaro, A.; Ståhlman, M.; Khan, M.T.; Schmidt, C.; Mannerås-Holm, L.; Wu, H.; Carreras, A.; Jeong, H.; Olofsson, L.E.; et al. Microbially Produced Imidazole Propionate Impairs Insulin Signaling through MTORC1. Cell 2018, 175, 947–961.e17. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An Obesity-Associated Gut Microbiome with Increased Capacity for Energy Harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Zhang, H.; DiBaise, J.K.; Zuccolo, A.; Kudrna, D.; Braidotti, M.; Yu, Y.; Parameswaran, P.; Crowell, M.D.; Wing, R.; Rittmann, B.E.; et al. Human gut microbiota in obesity and after gastric bypass. Proc. Natl. Acad. Sci. USA 2009, 106, 2365–2370. [Google Scholar] [CrossRef] [Green Version]
- Ilhan, Z.E.; DiBaise, J.K.; Dautel, S.E.; Isern, N.G.; Kim, Y.M.; Hoyt, D.W.; Schepmoes, A.A.; Brewer, H.M.; Weitz, K.K.; Metz, T.O.; et al. Temporospatial shifts in the human gut microbiome and metabolome after gastric bypass surgery. NPJ Biofilms Microbiomes 2020, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Basolo, A.; Parrington, S.; Ando, T.; Hollstein, T.; Piaggi, P.; Krakoff, J. Procedures for Measuring Excreted and Ingested Calories to Assess Nutrient Absorption Using Bomb Calorimetry. Obesity 2020, 28, 2315–2322. [Google Scholar] [CrossRef]
- Wortelboer, K.; Nieuwdorp, M.; Herrema, H. Fecal microbiota transplantation beyond Clostridioides difficile infections. EBioMedicine 2019, 44, 716–729. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.H.; Cho, Y.-S. Fecal Microbiota Transplantation: Current Applications, Effectiveness, and Future Perspectives. Clin. Endosc. 2016, 49, 257–265. [Google Scholar] [CrossRef]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.W.M.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of Intestinal Microbiota from Lean Donors Increases Insulin Sensitivity in Individuals with Metabolic Syndrome. Gastroenterology 2012, 143, 913–916.e7. [Google Scholar] [CrossRef]
- Yu, E.W.; Gao, L.; Stastka, P.; Cheney, M.C.; Mahabamunuge, J.; Torres Soto, M.; Ford, C.B.; Bryant, J.A.; Henn, M.R.; Hohmann, E.L. Fecal Microbiota Transplantation for the Improvement of Metabolism in Obesity: The FMT-TRIM Double-Blind Placebo-Controlled Pilot Trial. PLoS Med. 2020, 17, e1003051. [Google Scholar] [CrossRef]
- Jumpertz, R.; Le, D.S.; Turnbaugh, P.J.; Trinidad, C.; Bogardus, C.; Gordon, J.I.; Krakoff, J. Energy-Balance Studies Reveal Associations between Gut Microbes, Caloric Load, and Nutrient Absorption in Humans. Am. J. Clin. Nutr. 2011, 94, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, N.; Tan, H.-Y.; Li, S.; Zhang, C.; Feng, Y. Function of Akkermansia Muciniphila in Obesity: Interactions With Lipid Metabolism, Immune Response and Gut Systems. Front. Microbiol. 2020, 11, 219. [Google Scholar] [CrossRef] [Green Version]
- Depommier, C.; Everard, A.; Druart, C.; Plovier, H.; Van Hul, M.; Vieira-Silva, S.; Falony, G.; Raes, J.; Maiter, D.; Delzenne, N.M.; et al. Supplementation with Akkermansia Muciniphila in Overweight and Obese Human Volunteers: A Proof-of-Concept Exploratory Study. Nat. Med. 2019, 25, 1096–1103. [Google Scholar] [CrossRef]
- Dosoky, N.S.; Chen, Z.; Guo, Y.; McMillan, C.; Flynn, C.R.; Davies, S.S. Two-Week Administration of Engineered Escherichia Coli Establishes Persistent Resistance to Diet-Induced Obesity Even without Antibiotic Pre-Treatment. Appl. Microbiol. Biotechnol. 2019, 103, 6711–6723. [Google Scholar] [CrossRef]
- Borgeraas, H.; Johnson, L.K.; Skattebu, J.; Hertel, J.K.; Hjelmesaeth, J. Effects of probiotics on body weight, body mass index, fat mass and fat percentage in subjects with overweight or obesity: A systematic review and meta-analysis of randomized controlled trials. Obes. Rev. 2018, 19, 219–232. [Google Scholar] [CrossRef] [Green Version]
- Koutnikova, H.; Genser, B.; Monteiro-Sepulveda, M.; Faurie, J.M.; Rizkalla, S.; Schrezenmeir, J.; Clément, K. Impact of bacterial probiotics on obesity, diabetes and non-alcoholic fatty liver disease related variables: A systematic review and meta-analysis of randomised controlled trials. BMJ Open 2019, 9, e017995. [Google Scholar] [CrossRef]
- Ilmonen, J.; Isolauri, E.; Poussa, T.; Laitinen, K. Impact of dietary counselling and probiotic intervention on maternal anthropometric measurements during and after pregnancy: A randomized placebo-controlled trial. Clin. Nutr. 2011, 30, 156–164. [Google Scholar] [CrossRef]
- Fon Tacer, K.; Bookout, A.L.; Ding, X.; Kurosu, H.; John, G.B.; Wang, L.; Goetz, R.; Mohammadi, M.; Kuro-o, M.; Mangelsdorf, D.J.; et al. Research Resource: Comprehensive Expression Atlas of the Fibroblast Growth Factor System in Adult Mouse. Mol. Endocrinol. 2010, 24, 2050–2064. [Google Scholar] [CrossRef] [Green Version]
- Marcelin, G.; Jo, Y.-H.; Li, X.; Schwartz, G.J.; Zhang, Y.; Dun, N.J.; Lyu, R.-M.; Blouet, C.; Chang, J.K.; Chua, S. Central Action of FGF19 Reduces Hypothalamic AGRP/NPY Neuron Activity and Improves Glucose Metabolism. Mol. Metab. 2014, 3, 19–28. [Google Scholar] [CrossRef]
- Kurosu, H.; Choi, M.; Ogawa, Y.; Dickson, A.S.; Goetz, R.; Eliseenkova, A.V.; Mohammadi, M.; Rosenblatt, K.P.; Kliewer, S.A.; Kuro-O, M. Tissue-Specific Expression of BetaKlotho and Fibroblast Growth Factor (FGF) Receptor Isoforms Determines Metabolic Activity of FGF19 and FGF21. J. Biol. Chem. 2007, 282, 26687–26695. [Google Scholar] [CrossRef] [Green Version]
- Holt, J.A.; Luo, G.; Billin, A.N.; Bisi, J.; McNeill, Y.Y.; Kozarsky, K.F.; Donahee, M.; Wang, D.Y.; Mansfield, T.A.; Kliewer, S.A.; et al. Definition of a Novel Growth Factor-Dependent Signal Cascade for the Suppression of Bile Acid Biosynthesis. Genes Dev. 2003, 17, 1581–1591. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast Growth Factor 15 Functions as an Enterohepatic Signal to Regulate Bile Acid Homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Potthoff, M.J.; Boney-Montoya, J.; Choi, M.; He, T.; Sunny, N.E.; Satapati, S.; Suino-Powell, K.; Xu, H.E.; Gerard, R.D.; Finck, B.N.; et al. FGF15/19 Regulates Hepatic Glucose Metabolism by Inhibiting the CREB-PGC-1α Pathway. Cell Metab. 2011, 13, 729–738. [Google Scholar] [CrossRef] [Green Version]
- Song, K.-H.; Li, T.; Owsley, E.; Strom, S.; Chiang, J.Y.L. Bile Acids Activate Fibroblast Growth Factor 19 Signaling in Human Hepatocytes to Inhibit Cholesterol 7alpha-Hydroxylase Gene Expression. Hepatology 2009, 49, 297–305. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-C.; Seok, S.; Zhang, Y.; Ma, J.; Kong, B.; Guo, G.; Kemper, B.; Kemper, J.K. Intestinal FGF15/19 Physiologically Repress Hepatic Lipogenesis in the Late Fed-State by Activating SHP and DNMT3A. Nat. Commun. 2020, 11, 5969. [Google Scholar] [CrossRef]
- Kir, S.; Beddow, S.A.; Samuel, V.T.; Miller, P.; Previs, S.F.; Suino-Powell, K.; Xu, H.E.; Shulman, G.I.; Kliewer, S.A.; Mangelsdorf, D.J. FGF19 as a Postprandial, Insulin-Independent Activator of Hepatic Protein and Glycogen Synthesis. Science 2011, 331, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; John, L.M.; Adams, S.H.; Yu, X.X.; Tomlinson, E.; Renz, M.; Williams, P.M.; Soriano, R.; Corpuz, R.; Moffat, B.; et al. Fibroblast Growth Factor 19 Increases Metabolic Rate and Reverses Dietary and Leptin-Deficient Diabetes. Endocrinology 2004, 145, 2594–2603. [Google Scholar] [CrossRef] [Green Version]
- Morton, G.J.; Matsen, M.E.; Bracy, D.P.; Meek, T.H.; Nguyen, H.T.; Stefanovski, D.; Bergman, R.N.; Wasserman, D.H.; Schwartz, M.W. FGF19 Action in the Brain Induces Insulin-Independent Glucose Lowering. J. Clin. Investig. 2013, 123, 4799–4808. [Google Scholar] [CrossRef] [Green Version]
- Ryan, K.K.; Kohli, R.; Gutierrez-Aguilar, R.; Gaitonde, S.G.; Woods, S.C.; Seeley, R.J. Fibroblast Growth Factor-19 Action in the Brain Reduces Food Intake and Body Weight and Improves Glucose Tolerance in Male Rats. Endocrinology 2013, 154, 9–15. [Google Scholar] [CrossRef]
- Lan, T.; Morgan, D.A.; Rahmouni, K.; Sonoda, J.; Fu, X.; Burgess, S.C.; Holland, W.L.; Kliewer, S.A.; Mangelsdorf, D.J. FGF19, FGF21, and an FGFR1/β-Klotho-Activating Antibody Act on the Nervous System to Regulate Body Weight and Glycemia. Cell Metab. 2017, 26, 709–718.e3. [Google Scholar] [CrossRef]
- Mráz, M.; Lacinová, Z.; Kaválková, P.; Haluzíková, D.; Trachta, P.; Drápalová, J.; Hanušová, V.; Haluzík, M. Serum Concentrations of Fibroblast Growth Factor 19 in Patients with Obesity and Type 2 Diabetes Mellitus: The Influence of Acute Hyperinsulinemia, Very-Low Calorie Diet and PPAR-α Agonist Treatment. Physiol. Res. 2011, 60, 627–636. [Google Scholar] [CrossRef]
- Cariello, M.; Piglionica, M.; Gadaleta, R.M.; Moschetta, A. The Enterokine Fibroblast Growth Factor 15/19 in Bile Acid Metabolism. Handb. Exp. Pharmacol. 2019, 256, 73–93. [Google Scholar] [CrossRef]
- Uriarte, I.; Fernandez-Barrena, M.G.; Monte, M.J.; Latasa, M.U.; Chang, H.C.Y.; Carotti, S.; Vespasiani-Gentilucci, U.; Morini, S.; Vicente, E.; Concepcion, A.R.; et al. Identification of Fibroblast Growth Factor 15 as a Novel Mediator of Liver Regeneration and Its Application in the Prevention of Post-Resection Liver Failure in Mice. Gut 2013, 62, 899–910. [Google Scholar] [CrossRef]
- Henriksson, E.; Andersen, B. FGF19 and FGF21 for the Treatment of NASH-Two Sides of the Same Coin? Differential and Overlapping Effects of FGF19 and FGF21 from Mice to Human. Front. Endocrinol. 2020, 11, 601349. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; Moschetta, A. Metabolic Messengers: Fibroblast Growth Factor 15/19. Nat. Metab. 2019, 1, 588–594. [Google Scholar] [CrossRef]
- Ge, X.; Yang, H.; Bednarek, M.A.; Galon-Tilleman, H.; Chen, P.; Chen, M.; Lichtman, J.S.; Wang, Y.; Dalmas, O.; Yin, Y.; et al. LEAP2 Is an Endogenous Antagonist of the Ghrelin Receptor. Cell Metab. 2018, 27, 461–469.e6. [Google Scholar] [CrossRef] [Green Version]
- Al-Massadi, O.; Müller, T.; Tschöp, M.; Diéguez, C.; Nogueiras, R. Ghrelin and LEAP-2: Rivals in Energy Metabolism. Trends Pharmacol. Sci. 2018, 39, 685–694. [Google Scholar] [CrossRef]
- Mani, B.K.; Puzziferri, N.; He, Z.; Rodriguez, J.A.; Osborne-Lawrence, S.; Metzger, N.P.; Chhina, N.; Gaylinn, B.; Thorner, M.O.; Thomas, E.L.; et al. LEAP2 Changes with Body Mass and Food Intake in Humans and Mice. J. Clin. Investig. 2019, 129, 3909–3923. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, Stress, and Diabetes. J. Clin. Investig. 2005, 115, 1111–1119. [Google Scholar] [CrossRef] [Green Version]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A High-Fat Diet Is Associated with Endotoxemia That Originates from the Gut. Gastroenterology 2012, 142, 1100–1101.e2. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Osto, M.; Geurts, L.; Everard, A. Involvement of Gut Microbiota in the Development of Low-Grade Inflammation and Type 2 Diabetes Associated with Obesity. Gut Microbes 2012, 3, 279–288. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Kahles, F.; Rattik, S.; Nairz, M.; McAlpine, C.S.; Anzai, A.; Selgrade, D.; Fenn, A.M.; Chan, C.T.; Mindur, J.E.; et al. Gut Intraepithelial T Cells Calibrate Metabolism and Accelerate Cardiovascular Disease. Nature 2019, 566, 115–119. [Google Scholar] [CrossRef]
- Pickens, C.A.; Sordillo, L.M.; Zhang, C.; Fenton, J.I. Obesity Is Positively Associated with Arachidonic Acid-Derived 5- and 11-Hydroxyeicosatetraenoic Acid (HETE). Metabolism 2017, 70, 177–191. [Google Scholar] [CrossRef]
- Innes, J.K.; Calder, P.C. Omega-6 Fatty Acids and Inflammation. Prostaglandins Leukot. Essent. Fat. Acids 2018, 132, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Thaler, J.P.; Yi, C.-X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity Is Associated with Hypothalamic Injury in Rodents and Humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKbeta/NF-KappaB and ER Stress Link Overnutrition to Energy Imbalance and Obesity. Cell 2008, 135, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Scherer, T.; Lindtner, C.; Zielinski, E.; O’Hare, J.; Filatova, N.; Buettner, C. Short Term Voluntary Overfeeding Disrupts Brain Insulin Control of Adipose Tissue Lipolysis. J. Biol. Chem. 2012, 287, 33061–33069. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Zhang, H.; Yin, Y.; Li, J.; Tang, Y.; Purkayastha, S.; Li, L.; Cai, D. Obesity- and Aging-Induced Excess of Central Transforming Growth Factor-β Potentiates Diabetic Development via an RNA Stress Response. Nat. Med. 2014, 20, 1001–1008. [Google Scholar] [CrossRef] [Green Version]
- Jais, A.; Brüning, J.C. Hypothalamic Inflammation in Obesity and Metabolic Disease. J. Clin. Investig. 2017, 127, 24–32. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Yang, G.; Zhang, Q.; Meng, L.; Xin, Y.; Jiang, X. The Role of Short-Chain Fatty Acids in Intestinal Barrier Function, Inflammation, Oxidative Stress, and Colonic Carcinogenesis. Pharmacol. Res. 2021, 165, 105420. [Google Scholar] [CrossRef]
- Orgeron, M.L.; Stone, K.P.; Wanders, D.; Cortez, C.C.; Van, N.T.; Gettys, T.W. The impact of dietary methionine restriction on biomarkers of metabolic health. Prog. Mol. Biol. Transl. Sci. 2014, 121, 351–376. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, R.; Azevedo, I. Chronic Inflammation in Obesity and the Metabolic Syndrome. Mediat. Inflamm. 2010, 2010, 289645. [Google Scholar] [CrossRef]
- Mujawdiya, P.K.; Sharma, P.; Sharad, S.; Kapur, S. Reversal of Increase in Intestinal Permeability by Mangifera Indica Seed Kernel Extract in High-Fat Diet-Induced Obese Mice. Pharmaceuticals 2020, 13, 190. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, Y.; Zhang, M.; Pang, X.; Xu, J.; Kang, C.; Li, M.; Zhang, C.; Zhang, Z.; Zhang, Y.; et al. Structural Changes of Gut Microbiota during Berberine-Mediated Prevention of Obesity and Insulin Resistance in High-Fat Diet-Fed Rats. PLoS ONE 2012, 7, e42529. [Google Scholar] [CrossRef] [Green Version]
- Scheithauer, T.P.M.; Rampanelli, E.; Nieuwdorp, M.; Vallance, B.A.; Verchere, C.B.; van Raalte, D.H.; Herrema, H. Gut Microbiota as a Trigger for Metabolic Inflammation in Obesity and Type 2 Diabetes. Front. Immunol. 2020, 11, 571731. [Google Scholar] [CrossRef]
- Petersen, N.; Frimurer, T.M.; Terndrup Pedersen, M.; Egerod, K.L.; Wewer Albrechtsen, N.J.; Holst, J.J.; Grapin-Botton, A.; Jensen, K.B.; Schwartz, T.W. Inhibiting RHOA Signaling in Mice Increases Glucose Tolerance and Numbers of Enteroendocrine and Other Secretory Cells in the Intestine. Gastroenterology 2018, 155, 1164–1176.e2. [Google Scholar] [CrossRef]
- Cao, Z.; Lin, S.; Liu, J. Bacteria-Based Microdevices for the Oral Delivery of Macromolecules. Pharmaceutics 2021, 13, 1610. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, W.; Peng, H.; Li, Y.; Leng, T.; Xie, C.; Zhang, L. Oral Gene Therapy of HFD-Obesity via Nonpathogenic Yeast Microcapsules Mediated shRNA Delivery. Pharmaceutics 2021, 13, 1536. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Pathway | Available Compounds | Biological Effect | Side Effects | References |
|---|---|---|---|---|
| SGLT1 inhibitor | Phloridzin, Sotagliflozin | Reduced glucose absorption | Osmotic diarrhoea | [26] |
| Lipase inhibitor | Orlistat | Reduced lipid absorption | Oily stool, diarrhoea | [25] |
| GPR119 agonists | AR231453 APD597 AS1669058 | Increased production of satiety hormones | not determined | [41] |
| Goat Inhibitor | GLWL-01 | Reduced ghrelin signaling | Mild nausea | [67] |
| TGR5 agonists | NT-777 | Increased production of satiety hormones | Gall bladder enlargement | [77,78,79] |
| FXR agonist | Fexaramine | Increased FGF15/19 production, reduced inflammation, beneficial microbiota changes | Itchy skin (pruritis) | [80] |
| Probiotic dietary supplements | Akkermansia muciniphila, Lactobacillus, Streptococcus, Bifidobacterium | Increased gut hormone production, reduced inflammation | Gastrointestinal discomfort | [86] |
| FGF19 | NGM282, Aldafermin (tested for NASH and type 2 diabetes treatment) | Suppression of hepatic lipogenesis, increase in adipose thermogenesis in the liver | Gastrointestinal discomfort | [127] |
| LEAP2 | LEAP2 peptide | Anti-ghrelin action | Reduced growth hormone secretion? | Cl.Trials.gov Identifier: NCT04621409 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petersen, N.; Greiner, T.U.; Torz, L.; Bookout, A.; Gerstenberg, M.K.; Castorena, C.M.; Kuhre, R.E. Targeting the Gut in Obesity: Signals from the Inner Surface. Metabolites 2022, 12, 39. https://doi.org/10.3390/metabo12010039
Petersen N, Greiner TU, Torz L, Bookout A, Gerstenberg MK, Castorena CM, Kuhre RE. Targeting the Gut in Obesity: Signals from the Inner Surface. Metabolites. 2022; 12(1):39. https://doi.org/10.3390/metabo12010039
Chicago/Turabian StylePetersen, Natalia, Thomas U. Greiner, Lola Torz, Angie Bookout, Marina Kjærgaard Gerstenberg, Carlos M. Castorena, and Rune Ehrenreich Kuhre. 2022. "Targeting the Gut in Obesity: Signals from the Inner Surface" Metabolites 12, no. 1: 39. https://doi.org/10.3390/metabo12010039
APA StylePetersen, N., Greiner, T. U., Torz, L., Bookout, A., Gerstenberg, M. K., Castorena, C. M., & Kuhre, R. E. (2022). Targeting the Gut in Obesity: Signals from the Inner Surface. Metabolites, 12(1), 39. https://doi.org/10.3390/metabo12010039