
Metabolic Reprogramming of Mammary Epithelial Cells during TGF-β-Induced Epithelial-to-Mesenchymal Transition

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
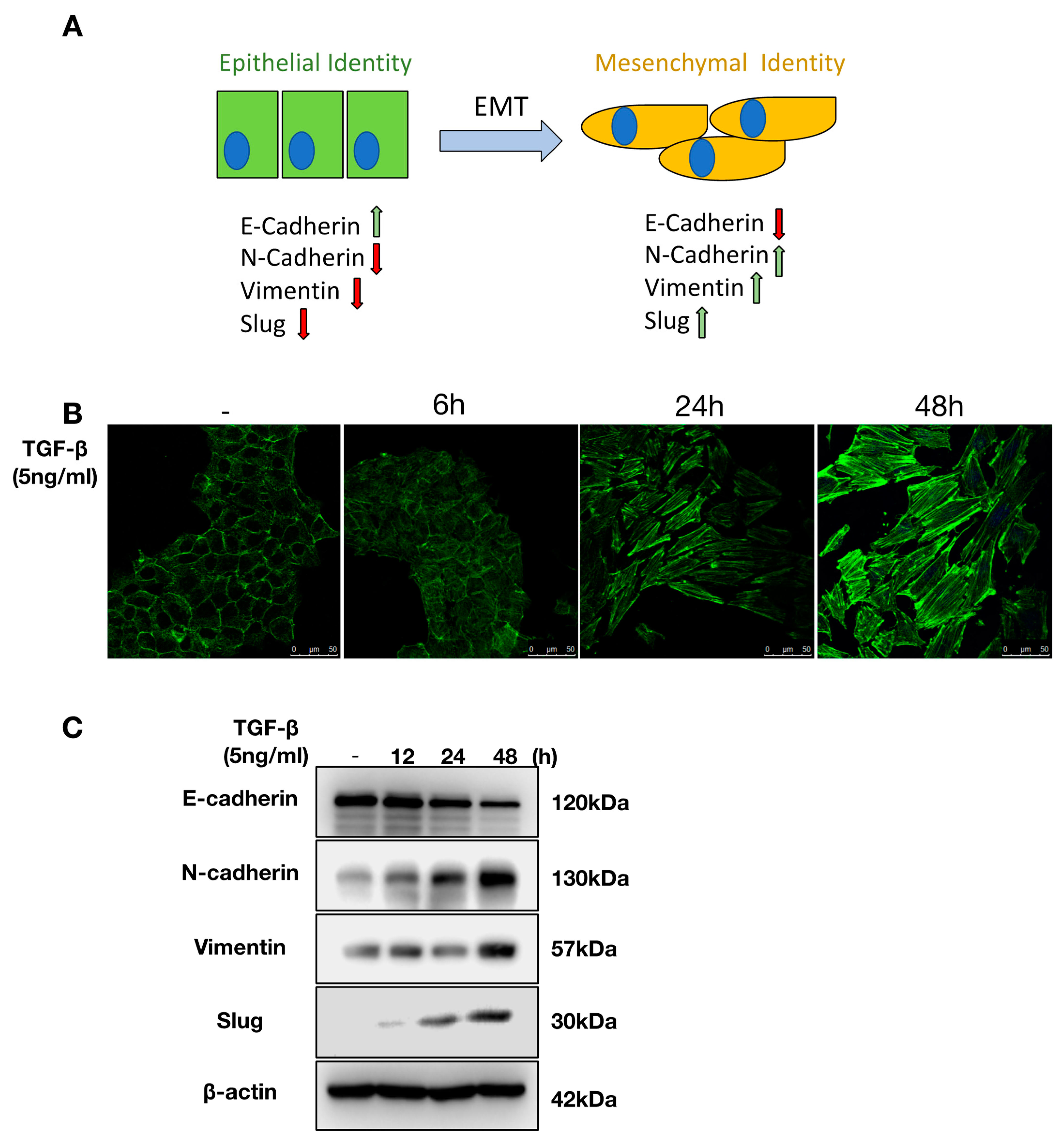
2.1. TGF-β Induces EMT in NMuMG Cells
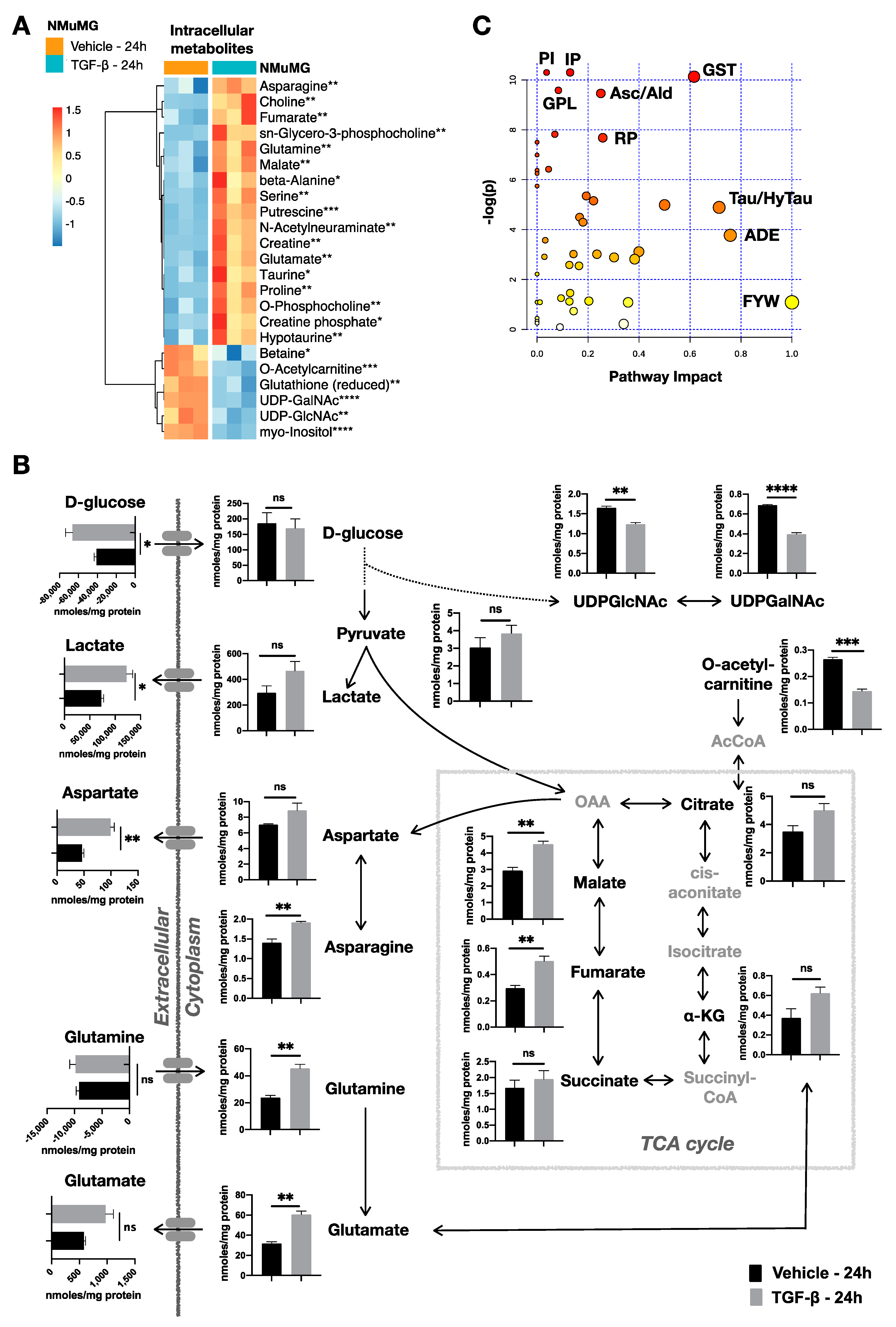
2.2. TGF-β Triggers Metabolic Rewiring in NMuMG Cells
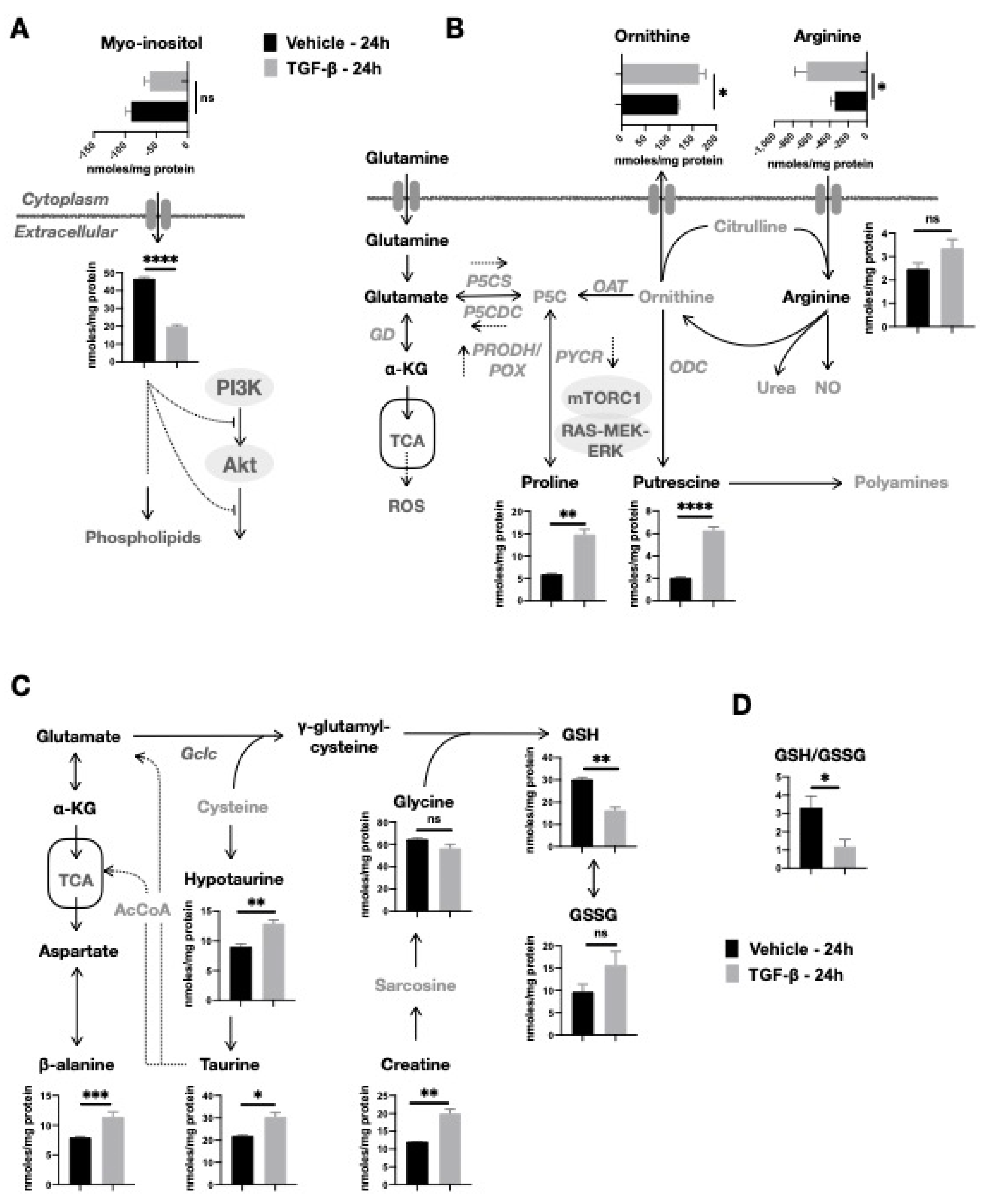
2.3. TGF-β-Induced Depletion of Myo-Inositol and Increase in Putrescine and Proline Levels in NMuMG Cells
2.4. TGF-β Stimulates an Oxidative Cellular Redox State
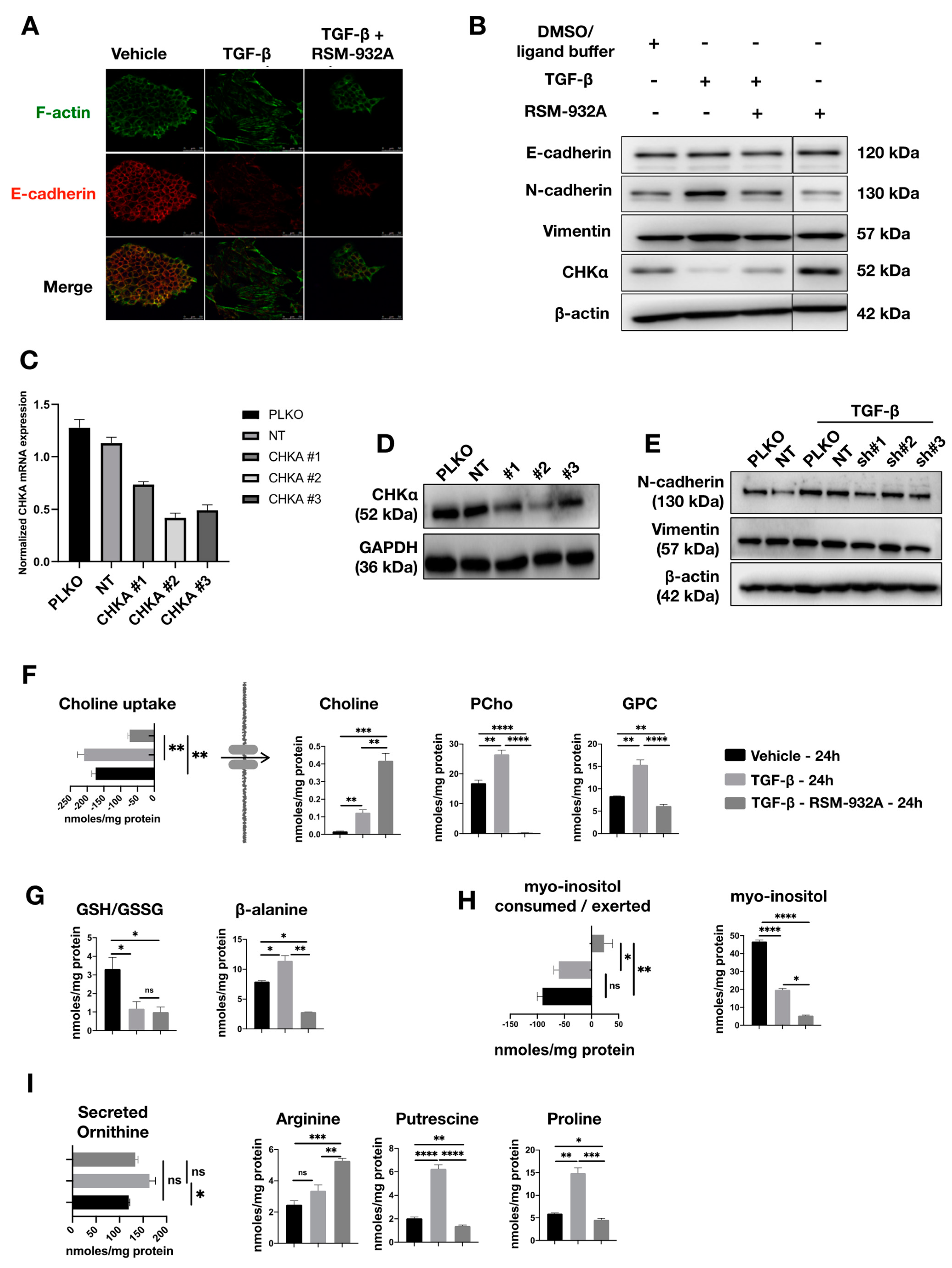
2.5. TGF-β Affects Choline Metabolism
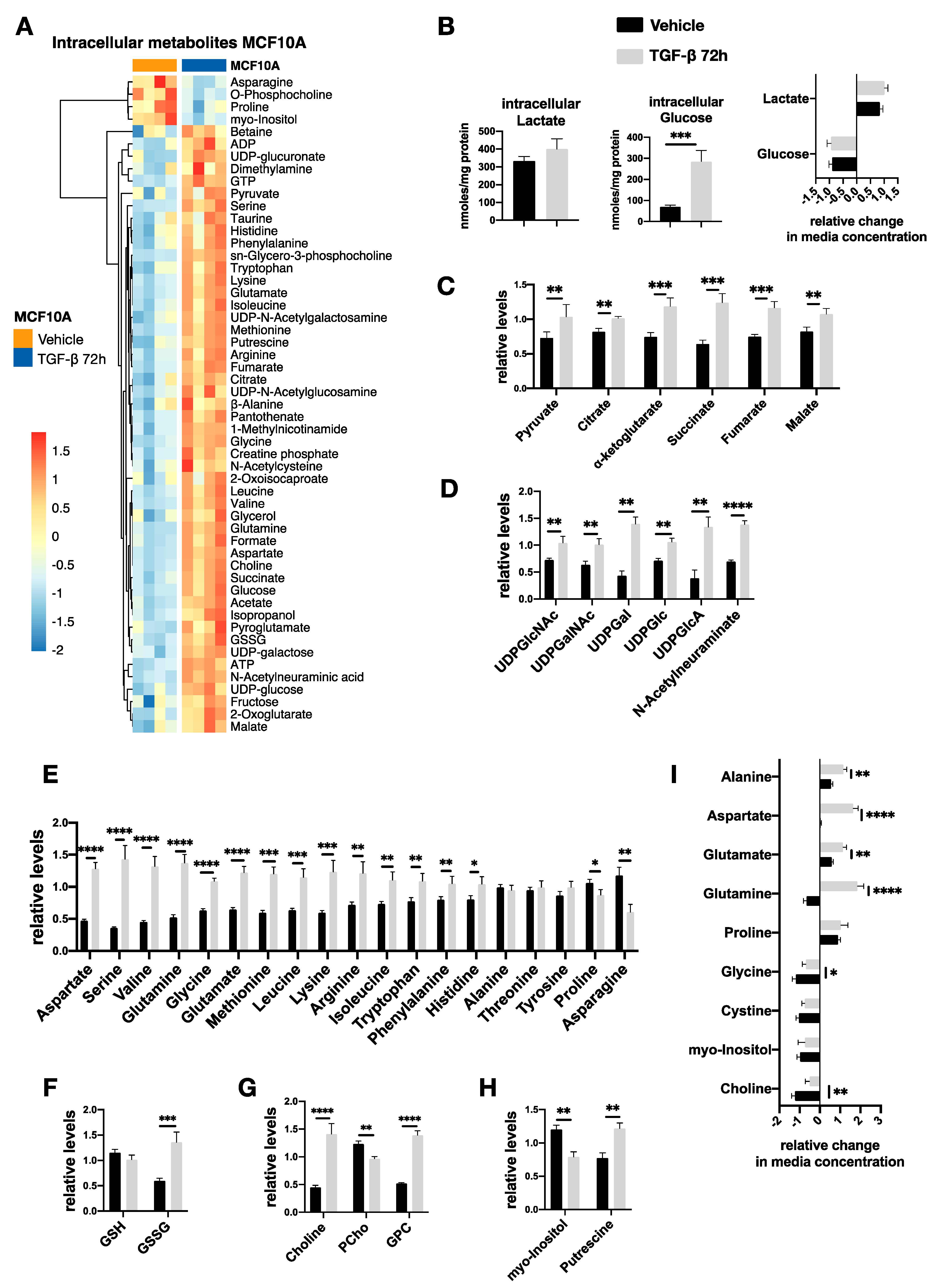
2.6. TGF-β-Induced Metabolic Reprogramming of Premalignant MCF10A-Ras Cells
2.7. TGF-β-Induced Phosphocholine Production may Play a Role in EMT
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Sampling NMuMG and MCF10A-Ras Cells for NMR Analysis
4.3. NMR-Based Metabolomics
4.4. Lentiviral Transduction
4.5. RNA Isolation and Quantitative Real-Time PCR (qRT-PCR)
4.6. Western Blot Analysis
4.7. Immunofluorescence Staining
4.8. Cell Migration and Proliferation Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haensel, D.; Dai, X. Epithelial-to-mesenchymal transition in cutaneous wound healing: Where we are and where we are heading. Dev. Dyn. 2017, 247, 473–480. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-Mesenchymal Transition: At the Crossroads of Development and Tumor Metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, C.J.; Massague, J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Zhang, Y.E. Mechanistic insight into contextual TGF-beta signaling. Curr. Opin. Cell Biol. 2018, 51, 1–7. [Google Scholar] [CrossRef]
- Kahata, K.; Maturi, V.; Moustakas, A. TGF-beta Family Signaling in Ductal Differentiation and Branching Morphogenesis. Cold Spring Harb. Perspect. Biol. 2018, 10, a031997. [Google Scholar] [CrossRef] [Green Version]
- Colak, S.; Ten Dijke, P. Targeting TGF-beta Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Schuster, N.; Krieglstein, K. Mechanisms of TGF-beta-mediated apoptosis. Cell Tissue Res. 2002, 307, 1–14. [Google Scholar] [CrossRef]
- Ten Dijke, P.; Goumans, M.J.; Itoh, F.; Itoh, S. Regulation of cell proliferation by Smad proteins. J. Cell Physiol. 2002, 191, 1–16. [Google Scholar] [CrossRef]
- Drabsch, Y.; ten Dijke, P. TGF-beta signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev. 2012, 31, 553–568. [Google Scholar] [CrossRef]
- Inman, G.J. Switching TGFbeta from a tumor suppressor to a tumor promoter. Curr. Opin. Genet Dev. 2011, 21, 93–99. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Morandi, A.; Taddei, M.L.; Chiarugi, P.; Giannoni, E. Targeting the Metabolic Reprogramming That Controls Epithelial-to-Mesenchymal Transition in Aggressive Tumors. Front. Oncol. 2017, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Sciacovelli, M.; Frezza, C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2017, 284, 3132–3144. [Google Scholar] [CrossRef]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquière, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Hamabe, A.; Konno, M.; Tanuma, N.; Shima, H.; Tsunekuni, K.; Kawamoto, K.; Nishida, N.; Koseki, J.; Mimori, K.; Gotoh, N.; et al. Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2014, 111, 15526–15531. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Quek, L.-E.; Sultani, G.; Turner, N. Epithelial-mesenchymal transition induction is associated with augmented glucose uptake and lactate production in pancreatic ductal adenocarcinoma. Cancer Metab. 2016, 4, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-García, A.; Samsó, P.; Fontova, P.; Simon, H.; Manzano, A.; Castaño, E.; Rosa, J.L.; Martinez-Outshoorn, U.; Ventura, F.; Navarro-Sabate, A.; et al. TGF-β1 targets Smad, p38 MAPK, and PI3K/Akt signaling pathways to induce PFKFB3 gene expression and glycolysis in glioblastoma cells. FEBS J. 2017, 284, 3437–3454. [Google Scholar] [CrossRef] [Green Version]
- Colvin, H.; Nishida, N.; Konno, M.; Haraguchi, N.; Takahashi, H.; Nishimura, J.; Hata, T.; Kawamoto, K.; Asai, A.; Tsunekuni, K.; et al. Oncometabolite D-2-Hydroxyglurate Directly Induces Epithelial-Mesenchymal Transition and is Associated with Distant Metastasis in Colorectal Cancer. Sci. Rep. 2016, 6, 36289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Frezza, C. Fumarate drives EMT in renal cancer. Cell Death Differ. 2016, 24, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, Y.; Wu, G. SDHB deficiency promotes TGFbeta-mediated invasion and metastasis of colorectal cancer through transcriptional repression complex SNAIL1-SMAD3/4. Transl. Oncol. 2016, 9, 512–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostidis, S.; Addie, R.D.; Morreau, H.; Mayboroda, O.; Giera, M. Quantitative NMR analysis of intra- and extracellular metabolism of mammalian cells: A tutorial. Anal. Chim. Acta 2017, 980, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Arlauckas, S.P.; Popov, A.V.; Delikatny, E.J. Choline kinase alpha—Putting the ChoK-hold on tumor metabolism. Prog. Lipid Res. 2016, 63, 28–40. [Google Scholar] [CrossRef] [Green Version]
- Hua, W.; Ten Dijke, P.; Kostidis, S.; Giera, M.; Hornsveld, M. TGFbeta-induced metabolic reprogramming during epithelial-to-mesenchymal transition in cancer. Cell. Mol. Life Sci. 2002, 77, 2103–2123. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; Kumar, J.; Hermanson, K.; Sun, Y.; Qureshi, H.; Perley, D.; Scheidegger, A.; Singh, B.; Dhasarathy, A. The calcium channel proteins ORAI3 and STIM1 mediate TGF-β induced Snai1 expression. Oncotarget 2018, 9, 29468–29483. [Google Scholar] [CrossRef] [Green Version]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Law, B.K.; Aakre, M.E.; Edgerton, M.; Shyr, Y.; Bhowmick, N.A.; Moses, H.L. Transforming growth factor beta-regulated gene expression in a mouse mammary gland epithelial cell line. Breast Cancer Res. 2003, 5, R187–R198. [Google Scholar] [CrossRef] [Green Version]
- Jansen, J.; Jansen, K.; Neven, E.; Poesen, R.; Othman, A.; van Mil, A.; Sluijter, J.; Toraño, J.S.; Zaal, E.A.; Berkers, C.R.; et al. Remote sensing and signaling in kidney proximal tubules stimulates gut microbiome-derived organic anion secretion. Proc. Natl. Acad. Sci. USA 2019, 116, 16105–16110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Drabsch, Y.; Dekker, T.J.A.; De Vinuesa, A.G.; Li, Y.; Hawinkels, L.J.A.C.; Sheppard, K.-A.; Goumans, M.-J.; Luwor, R.B.; De Vries, C.J.; et al. Nuclear receptor NR4A1 promotes breast cancer invasion and metastasis by activating TGF-β signalling. Nat. Commun. 2014, 5, 3388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhou, F.; de Vinuesa, A.G.; de Kruijf, E.M.; Mesker, W.E.; Hui, L.; Drabsch, Y.; Li, Y.; Bauer, A.; Rousseau, A.; et al. TRAF4 Promotes TGF-β Receptor Signaling and Drives Breast Cancer Metastasis. Mol. Cell 2013, 51, 559–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Chen, K.; Xing, G.; Li, L.; Ma, B.; Hu, Z.; Duan, L.; Liu, X. Phospholipid remodeling is critical for stem cell pluripotency by facilitating mesenchymal-to-epithelial transition. Sci. Adv. 2019, 5, eaax7525. [Google Scholar] [CrossRef] [Green Version]
- Ramírez de Molina, A.; Gutiérrez, R.; Ramos, M.A.; Silva, J.M.; Silva, J.; Bonilla, F.; Sánchez, J.J.; Lacal, J.C. Increased choline kinase activity in human breast carcinomas: Clinical evidence for a potential novel antitumor strategy. Oncogene 2002, 21, 4317–4322. [Google Scholar] [CrossRef] [Green Version]
- Ramírez de Molina, A.; Rodríguez-González, A.; Gutiérrez, R.; Martínez-Piñeiro, L.; Sánchez, J.; Bonilla, F.; Rosell, R.; Lacal, J. Overexpression of choline kinase is a frequent feature in human tumor-derived cell lines and in lung, prostate, and colorectal human cancers. Biochem. Biophys. Res. Commun. 2002, 296, 580–583. [Google Scholar] [CrossRef]
- Mariotto, E.; Viola, G.; Ronca, R.; Persano, L.; Aveic, S.; Bhujwalla, Z.M.; Mori, N.; Accordi, B.; Serafin, V.; López-Cara, L.C.; et al. Choline Kinase Alpha Inhibition by EB-3D Triggers Cellular Senescence, Reduces Tumor Growth and Metastatic Dissemination in Breast Cancer. Cancers 2018, 10, 391. [Google Scholar] [CrossRef] [Green Version]
- Koch, K.; Hartmann, R.; Schröter, F.; Suwala, A.K.; Maciaczyk, D.; Krüger, A.C.; Willbold, D.; Kahlert, U.; Maciaczyk, J. Reciprocal regulation of the cholinic phenotype and epithelial-mesenchymal transition in glioblastoma cells. Oncotarget 2016, 7, 73414–73431. [Google Scholar] [CrossRef] [Green Version]
- Falcon, S.C.; Hudson, C.S.; Huang, Y.; Mortimore, M.; Golec, J.M.; Charlton, P.A.; Weber, P.; Sundaram, H. A non-catalytic role of choline kinase alpha is important in promoting cancer cell survival. Oncogenesis 2013, 2, e38. [Google Scholar] [CrossRef] [Green Version]
- Asim, M.; Massie, C.E.; Orafidiya, F.; Pértega-Gomes, N.; Warren, A.Y.; Esmaeili, M.; Selth, L.A.; Zecchini, H.I.; Luko, K.; Qureshi, A.; et al. Choline Kinase Alpha as an Androgen Receptor Chaperone and Prostate Cancer Therapeutic Target. J. Natl. Cancer Inst. 2015, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, J.; Morgani, S.M.; David, C.J.; Wang, Q.; Er, E.E.; Huang, Y.-H.; Basnet, H.; Zou, Y.; Shu, W.; Soni, R.K.; et al. TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature 2020, 577, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Jr, R.A.C.; Stewart, T.M.; Pegg, A.E. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef]
- Zabala-Letona, A.; Arruabarrena-Aristorena, A.; Martín-Martín, N.; Fernandez-Ruiz, S.; Sutherland, J.D.; Clasquin, M.; Tomas-Cortazar, J.; Jimenez, J.; Torres, I.; Quang, P.; et al. mTORC1-dependent AMD1 regulation sustains polyamine metabolism in prostate cancer. Nature 2017, 547, 109–113. [Google Scholar] [CrossRef]
- Schwörer, S.; Berisa, M.; Violante, S.; Qin, W.; Zhu, J.; Hendrickson, R.C.; Cross, J.R.; Thompson, C.B. Proline biosynthesis is a vent for TGFβ-induced mitochondrial redox stress. EMBO J. 2020, 39, e103334. [Google Scholar] [CrossRef]
- Elia, I.; Broekaert, D.; Christen, S.; Boon, R.; Radaelli, E.; Orth, M.; Verfaillie, C.; Grünewald, T.G.P.; Fendt, S.-M. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat. Commun. 2017, 8, 15267. [Google Scholar] [CrossRef] [PubMed]
- Phang, J.M. Proline Metabolism in Cell Regulation and Cancer Biology: Recent Advances and Hypotheses. Antioxid. Redox Signal. 2019, 30, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hua, W.; Kostidis, S.; Mayboroda, O.; Giera, M.; Hornsveld, M.; ten Dijke, P. Metabolic Reprogramming of Mammary Epithelial Cells during TGF-β-Induced Epithelial-to-Mesenchymal Transition. Metabolites 2021, 11, 626. https://doi.org/10.3390/metabo11090626
Hua W, Kostidis S, Mayboroda O, Giera M, Hornsveld M, ten Dijke P. Metabolic Reprogramming of Mammary Epithelial Cells during TGF-β-Induced Epithelial-to-Mesenchymal Transition. Metabolites. 2021; 11(9):626. https://doi.org/10.3390/metabo11090626
Chicago/Turabian StyleHua, Wan, Sarantos Kostidis, Oleg Mayboroda, Martin Giera, Marten Hornsveld, and Peter ten Dijke. 2021. "Metabolic Reprogramming of Mammary Epithelial Cells during TGF-β-Induced Epithelial-to-Mesenchymal Transition" Metabolites 11, no. 9: 626. https://doi.org/10.3390/metabo11090626
APA StyleHua, W., Kostidis, S., Mayboroda, O., Giera, M., Hornsveld, M., & ten Dijke, P. (2021). Metabolic Reprogramming of Mammary Epithelial Cells during TGF-β-Induced Epithelial-to-Mesenchymal Transition. Metabolites, 11(9), 626. https://doi.org/10.3390/metabo11090626