
Modelling hCDKL5 Heterologous Expression in Bacteria

 ,
,  ,
,  , , ,
, , ,  ,
,  and
and 
Abstract
1. Introduction
- (i)
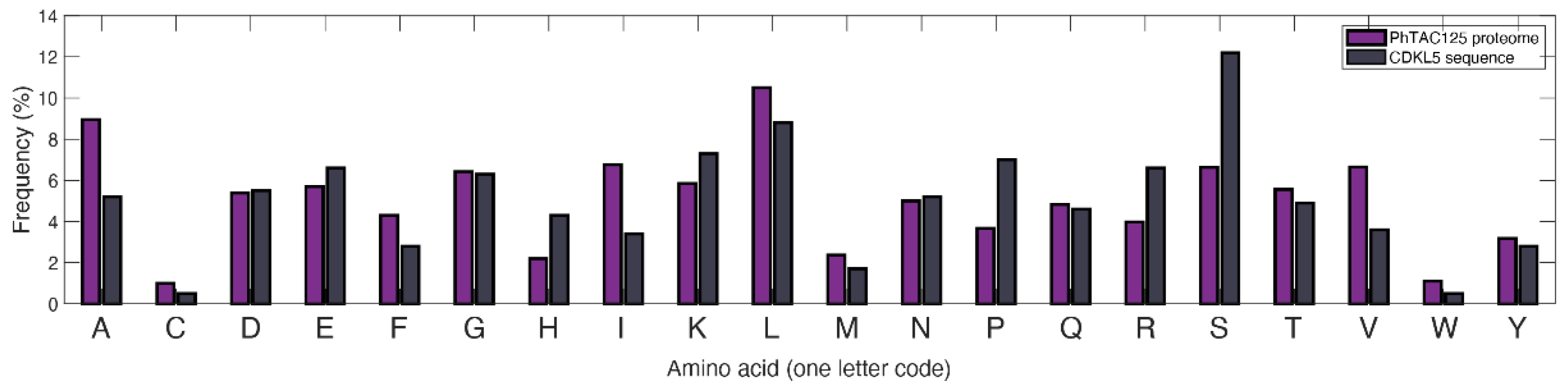
- Almost two-thirds of its sequence is predicted to be intrinsically disordered, and the lack of a precise 3D structure makes this region more susceptible to proteolytic attack by host-encoded proteases.
- (ii)
- The cytoplasmic accumulation of the enzyme in eukaryotic cells is associated to considerable toxicity, and the only permissive production strategy is its extracellular secretion, often accompanied with unwanted glycosylation [26]. PhTAC125 is the only prokaryotic cell factory in which full-length hCDKL5 production has been demonstrated, and the implementation of its efficient production process is the obligatory step towards any possible application (Calvanese et al., 2021, in press).
2. Results and Discussion
2.1. An Updated Metabolic Reconstruction of PhTAC125
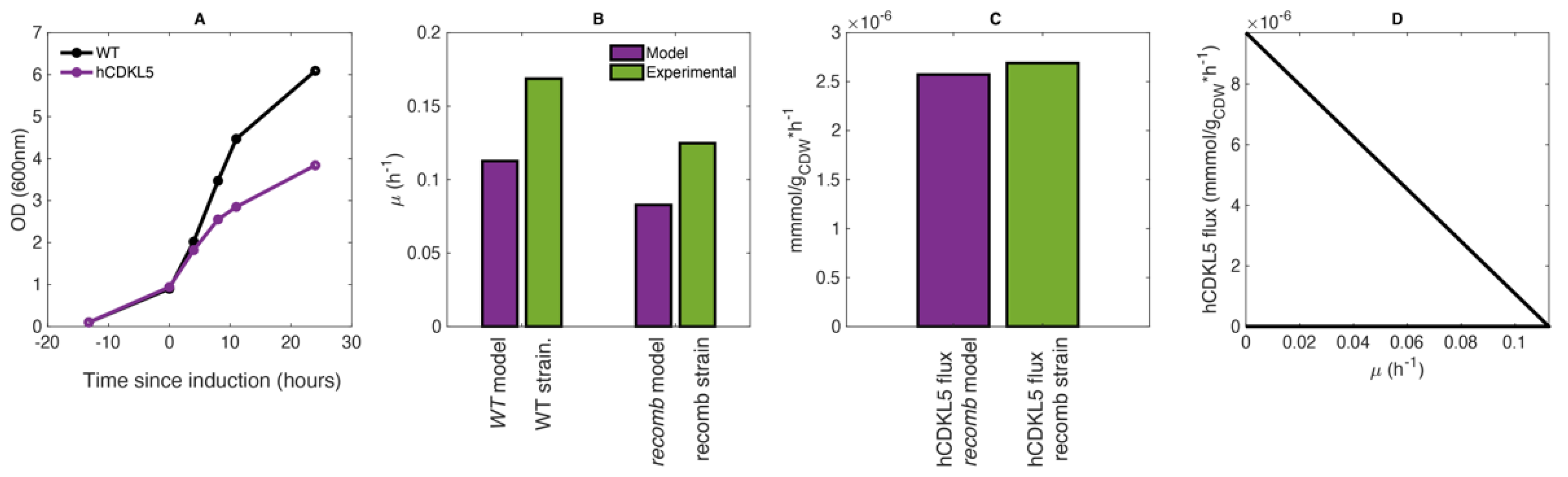
2.2. CDKL5 Production in Controlled Growth Conditions
2.3. Estimation of Average hCDKL5 Production Flux and Nutrients Uptake Rates
2.4. Recombinant Model Construction to Account for hCDKL5 Production
- A wt model by constraining the iMF721_v2_CDKL5 reconstruction with glutamate/gluconate uptake rates to the values experimentally determined and setting the biomass assembly reaction as the BOF of the model
- A recomb model by constraining the iMF721_v2_CDKL5 reconstruction with glutamate/gluconate uptake and growth rates to the values experimentally determined and setting the hCDKL5 production reaction as the BOF of the model
2.5. The PhTAC125 Recomb Model Accurately Simulates hCDKL5 Production
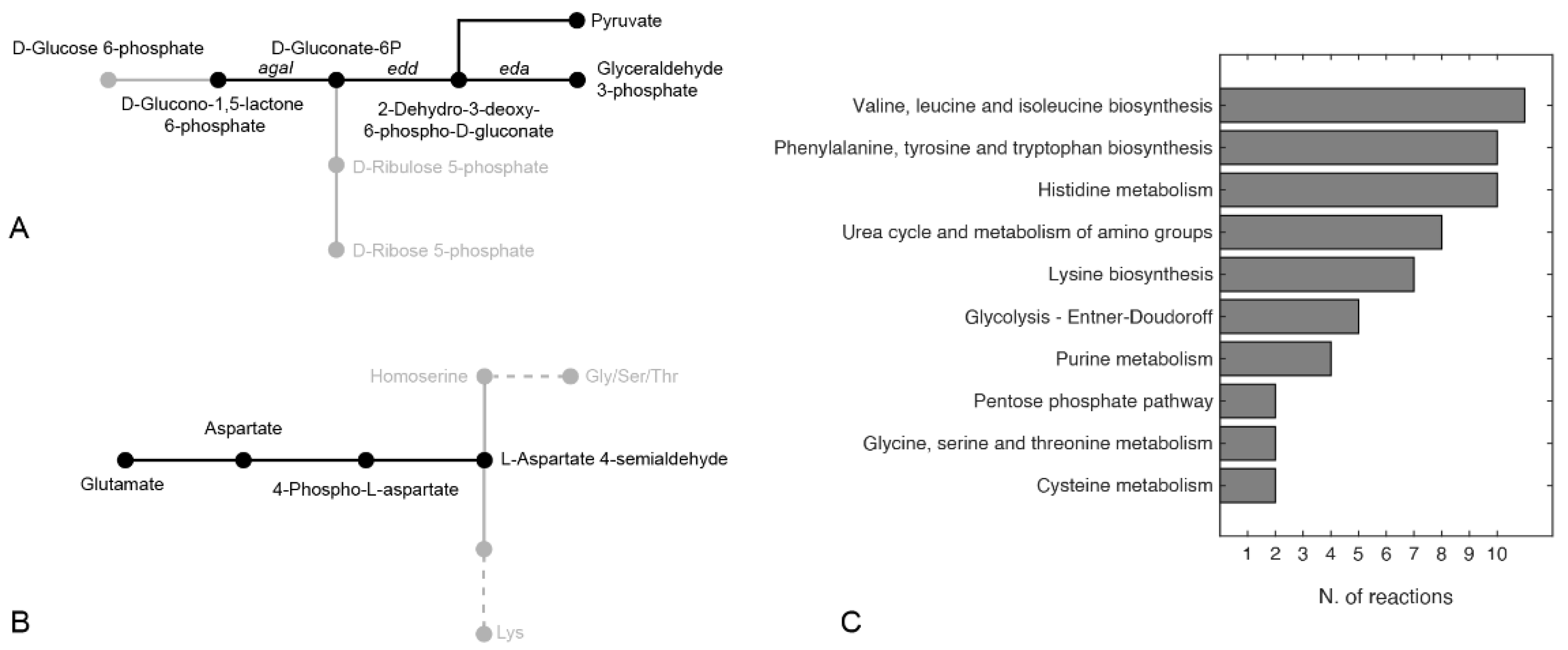
2.6. PhTAC125 Metabolic Rewiring Following hCDKL5 Induction
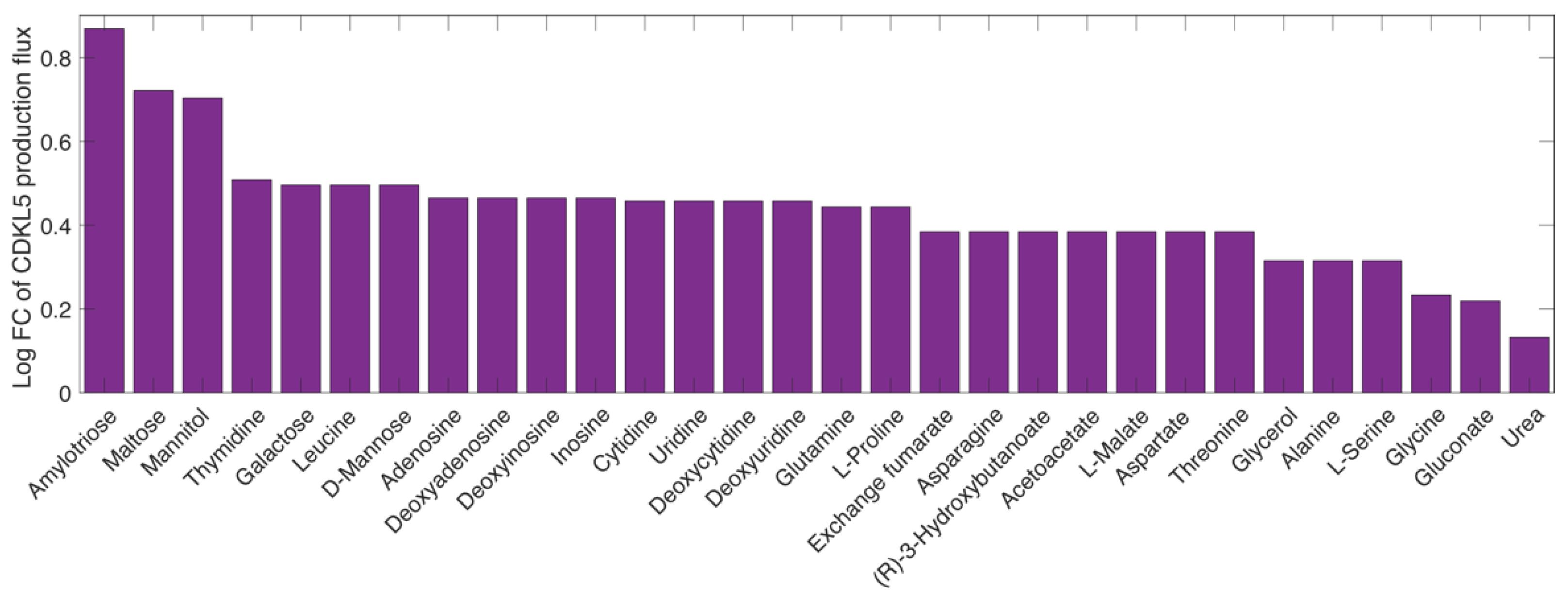
2.7. Finding the Optimal Growth Medium
2.8. Finding Hypothetical Targets for hCDKL5 Overproduction
3. Materials and Methods
3.1. Bacterial Strains and Conjugation Experiments
3.2. hCDKL5 Production
3.3. Glutamate and Gluconate Consumption Experiment
3.4. Metabolomic Data
3.5. PhTAC125 Genome-Scale Metabolic Reconstruction and Constraint-Based Simulations
3.6. Identification of Core Reactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Goeddel, D.V.; Kleid, D.G.; Bolivar, F.; Heyneker, H.L.; Yansura, D.G.; Crea, R.; Hirose, T.; Kraszewski, A.; Itakura, K.; Riggs, A.D. Expression in Escherichia Coli of Chemically Synthesized Genes for Human Insulin. Proc. Natl. Acad. Sci. USA 1979, 76, 106–110. [Google Scholar] [CrossRef]
- Burdette, L.A.; Leach, S.A.; Wong, H.T.; Tullman-Ercek, D. Developing Gram-Negative Bacteria for the Secretion of Heterologous Proteins. Microb. Cell Factories 2018, 17, 196. [Google Scholar] [CrossRef]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant Protein Expression in Escherichia Coli: Advances and Challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef] [PubMed]
- Glick, B.R. Metabolic Load and Heterologous Gene Expression. Biotechnol. Adv. 1995, 13, 247–261. [Google Scholar] [CrossRef]
- Li, Z.; Rinas, U. Recombinant Protein Production Associated Growth Inhibition Results Mainly from Transcription and Not from Translation. Microb. Cell Factories 2020, 19, 83. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.; Hwa, T. Bacterial Growth Laws and Their Applications. Curr. Opin. Biotechnol. 2011, 22, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, F.; Rinas, U. Stress Induced by Recombinant Protein Production in Escherichia coli. In Physiological Stress Responses in Bioprocesses; Advances in Biochemical Engineering/Biotechnology; Springer: Berlin/Heidelberg, Germany, 2004; Volume 89, pp. 73–92. ISBN 978-3-540-20311-7. [Google Scholar]
- Grabherr, R.; Nilsson, E.; Striedner, G.; Bayer, K. Stabilizing Plasmid Copy Number to Improve Recombinant Protein Production. Biotechnol. Bioeng. 2002, 77, 142–147. [Google Scholar] [CrossRef]
- Sharma, A.K.; Shukla, E.; Janoti, D.S.; Mukherjee, K.J.; Shiloach, J. A Novel Knock out Strategy to Enhance Recombinant Protein Expression in Escherichia Coli. Microb. Cell Factories 2020, 19, 148. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.; Cardoso, M.; Moreira, I.S.; Gonçalves, J.; Correia, J.D.G.; Verde, S.C.; Melo, R. Integrated in Silico and Experimental Approach towards the Design of a Novel Recombinant Protein Containing an Anti-HER2 ScFv. IJMS 2021, 22, 3547. [Google Scholar] [CrossRef]
- Fang, X.; Lloyd, C.J.; Palsson, B.O. Reconstructing Organisms in Silico: Genome-Scale Models and Their Emerging Applications. Nat. Rev. Microbiol. 2020, 18, 731–743. [Google Scholar] [CrossRef]
- Zielinski, D.C.; Patel, A.; Palsson, B.O. The Expanding Computational Toolbox for Engineering Microbial Phenotypes at the Genome Scale. Microorganisms 2020, 8, 2050. [Google Scholar] [CrossRef]
- Gu, C.; Kim, G.B.; Kim, W.J.; Kim, H.U.; Lee, S.Y. Current Status and Applications of Genome-Scale Metabolic Models. Genome Biol. 2019, 20, 121. [Google Scholar] [CrossRef] [PubMed]
- Presta, L.; Bosi, E.; Mansouri, L.; Dijkshoorn, L.; Fani, R.; Fondi, M. Constraint-Based Modeling Identifies New Putative Targets to Fight Colistin-Resistant A. Baumannii Infections. Sci. Rep. 2017, 7, 3706. [Google Scholar] [CrossRef] [PubMed]
- Fondi, M.; Bosi, E.; Presta, L.; Natoli, D.; Fani, R. Modelling Microbial Metabolic Rewiring during Growth in a Complex Medium. BMC Genom. 2016, 17, 970. [Google Scholar] [CrossRef]
- Sohn, S.B.; Graf, A.B.; Kim, T.Y.; Gasser, B.; Maurer, M.; Ferrer, P.; Mattanovich, D.; Lee, S.Y. Genome-Scale Metabolic Model of Methylotrophic Yeast Pichia Pastoris and Its Use for in Silico Analysis of Heterologous Protein Production. Biotechnol. J. 2010, 5, 705–715. [Google Scholar] [CrossRef]
- Nocon, J.; Steiger, M.G.; Pfeffer, M.; Sohn, S.B.; Kim, T.Y.; Maurer, M.; Rußmayer, H.; Pflügl, S.; Ask, M.; Haberhauer-Troyer, C.; et al. Model Based Engineering of Pichia Pastoris Central Metabolism Enhances Recombinant Protein Production. Metab. Eng. 2014, 24, 129–138. [Google Scholar] [CrossRef]
- Lule, I.; D’Huys, P.-J.; Van Mellaert, L.; Anné, J.; Bernaerts, K.; Van Impe, J. Metabolic Impact Assessment for Heterologous Protein Production in Streptomyces Lividans Based on Genome-Scale Metabolic Network Modeling. Math. Biosci. 2013, 246, 113–121. [Google Scholar] [CrossRef]
- Calero, P.; Nikel, P.I. Chasing Bacterial Chassis for Metabolic Engineering: A Perspective Review from Classical to Non-Traditional Microorganisms. Microb. Biotechnol. 2019, 12, 98–124. [Google Scholar] [CrossRef]
- Cusano, A.M.; Parrilli, E.; Marino, G.; Tutino, M.L. A Novel Genetic System for Recombinant Protein Secretion in the Antarctic Pseudoalteromonas Haloplanktis TAC125. Microb. Cell Factories 2006, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Unzueta, U.; Vázquez, F.; Accardi, G.; Mendoza, R.; Toledo-Rubio, V.; Giuliani, M.; Sannino, F.; Parrilli, E.; Abasolo, I.; Schwartz, S.; et al. Strategies for the Production of Difficult-to-Express Full-Length Eukaryotic Proteins Using Microbial Cell Factories: Production of Human Alpha-Galactosidase A. Appl. Microbiol. Biotechnol. 2015, 99, 5863–5874. [Google Scholar] [CrossRef] [PubMed]
- Vigentini, I.; Merico, A.; Tutino, M.L.; Compagno, C.; Marino, G. Optimization of Recombinant Human Nerve Growth Factor Production in the Psychrophilic Pseudoalteromonas Haloplanktis. J. Biotechnol. 2006, 127, 141–150. [Google Scholar] [CrossRef]
- Papa, R.; Rippa, V.; Sannia, G.; Marino, G.; Duilio, A. An Effective Cold Inducible Expression System Developed in Pseudoalteromonas Haloplanktis TAC125. J. Biotechnol. 2007, 127, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Katayama, S.; Sueyoshi, N.; Inazu, T.; Kameshita, I. Cyclin-Dependent Kinase-Like 5 (CDKL5): Possible Cellular Signalling Targets and Involvement in CDKL5 Deficiency Disorder. Neural Plast. 2020, 2020, 6970190. [Google Scholar] [CrossRef] [PubMed]
- Trazzi, S.; De Franceschi, M.; Fuchs, C.; Bastianini, S.; Viggiano, R.; Lupori, L.; Mazziotti, R.; Medici, G.; Lo Martire, V.; Ren, E.; et al. CDKL5 Protein Substitution Therapy Rescues Neurological Phenotypes of a Mouse Model of CDKL5 Disorder. Hum. Mol. Genet. 2018, 27, 1572–1592. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.; Sullivan, S.; Gray, H. Recombinant CDKL5 Proteins, Gene Therapy and Production Methods. WO/2021/087282, 6 May 2021. [Google Scholar]
- Fondi, M.; Maida, I.; Perrin, E.; Mellera, A.; Mocali, S.; Parrilli, E.; Tutino, M.L.; Liò, P.; Fani, R. Genome-Scale Metabolic Reconstruction and Constraint-Based Modelling of the Antarctic Bacterium P Seudoalteromonas Haloplanktis TAC125: Modelling of P. Haloplanktis TAC125 Metabolism. Environ. Microbiol. 2015, 17, 751–766. [Google Scholar] [CrossRef]
- Hucka, M.; Bergmann, F.T.; Chaouiya, C.; Dräger, A.; Hoops, S.; Keating, S.M.; König, M.; Novère, N.L.; Myers, C.J.; Olivier, B.G.; et al. The Systems Biology Markup Language (SBML): Language Specification for Level 3 Version 2 Core Release 2. J. Integr. Bioinform. 2019, 16, 1–183. [Google Scholar] [CrossRef]
- Olivier, B.G.; Bergmann, F.T. SBML Level 3 Package: Flux Balance Constraints Version 2. J. Integr. Bioinform. 2018, 15, 1–38. [Google Scholar] [CrossRef]
- Lachance, J.-C.; Lloyd, C.J.; Monk, J.M.; Yang, L.; Sastry, A.V.; Seif, Y.; Palsson, B.O.; Rodrigue, S.; Feist, A.M.; King, Z.A.; et al. BOFdat: Generating Biomass Objective Functions for Genome-Scale Metabolic Models from Experimental Data. PLoS Comput. Biol. 2019, 15, e1006971. [Google Scholar] [CrossRef]
- Qi, W.; Colarusso, A.; Olombrada, M.; Parrilli, E.; Patrignani, A.; Tutino, M.L.; Toll-Riera, M. New Insights on Pseudoalteromonas Haloplanktis TAC125 Genome Organization and Benchmarks of Genome Assembly Applications Using next and Third Generation Sequencing Technologies. Sci. Rep. 2019, 9, 16444. [Google Scholar] [CrossRef]
- Perrin, E.; Ghini, V.; Giovannini, M.; Di Patti, F.; Cardazzo, B.; Carraro, L.; Fagorzi, C.; Turano, P.; Fani, R.; Fondi, M. Diauxie and Co-Utilization of Carbon Sources Can Coexist during Bacterial Growth in Nutritionally Complex Environments. Nat. Commun. 2020, 11, 3135. [Google Scholar] [CrossRef]
- Colarusso, A.; Lauro, C.; Calvanese, M.; Parrilli, E.; Tutino, M.L. Improvement of Pseudoalteromonas Haloplanktis TAC125 as a Cell Factory: IPTG-Inducible Plasmid Construction and Strain Engineering. Microorganisms 2020, 8, 1466. [Google Scholar] [CrossRef]
- Sannino, F.; Giuliani, M.; Salvatore, U.; Apuzzo, G.A.; de Pascale, D.; Fani, R.; Fondi, M.; Marino, G.; Tutino, M.L.; Parrilli, E. A Novel Synthetic Medium and Expression System for Subzero Growth and Recombinant Protein Production in Pseudoalteromonas Haloplanktis TAC125. Appl. Microbiol. Biotechnol. 2017, 101, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Boyle, J. Lehninger Principles of Biochemistry (4th ed.): Nelson, D., and Cox, M. Biochem. Mol. Biol. Educ. 2005, 33, 74–75. [Google Scholar] [CrossRef]
- Carneiro, S.; Ferreira, E.C.; Rocha, I. Metabolic Responses to Recombinant Bioprocesses in Escherichia Coli. J. Biotechnol. 2013, 164, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Lee, S.Y.; Kim, T.Y.; Woo, H.M. In Silico Identification of Gene Amplification Targets for Improvement of Lycopene Production. AEM 2010, 76, 3097–3105. [Google Scholar] [CrossRef] [PubMed]
- Driouch, H.; Melzer, G.; Wittmann, C. Integration of in Vivo and in Silico Metabolic Fluxes for Improvement of Recombinant Protein Production. Metab. Eng. 2012, 14, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sun, Y.; Ramos, K.R.M.; Nisola, G.M.; Valdehuesa, K.N.G.; Lee, W.; Park, S.J.; Chung, W.-J. Combination of Entner-Doudoroff Pathway with MEP Increases Isoprene Production in Engineered Escherichia Coli. PLoS ONE 2013, 8, e83290. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ying, L.-Q.; Zhang, S.-S.; Chen, N.; Liu, W.-F.; Tao, Y. Modification of Targets Related to the Entner–Doudoroff/Pentose Phosphate Pathway Route for Methyl-d-Erythritol 4-Phosphate-Dependent Carotenoid Biosynthesis in Escherichia Coli. Microb. Cell Factories 2015, 14, 117. [Google Scholar] [CrossRef]
- Tutino, M.; Duilio, A.; Parrilli, E.; Remaut, E.; Sannia, G.; Marino, G. A Novel Replication Element from an Antarctic Plasmid as a Tool for the Expression of Proteins at Low Temperature. Extremophiles 2001, 5, 257–264. [Google Scholar] [CrossRef]
- Tascon, R.I.; Rodriguez-Ferri, E.F.; Gutierrez-Martin, C.B.; Rodriguez-Barbosa, I.; Berche, P.; Vazquez-Boland, J.A. Transposon Mutagenesis in Actinobacillus Pleuropneumoniae with a Tn10 Derivative. J. Bacteriol. 1993, 175, 5717–5722. [Google Scholar] [CrossRef]
- Marblestone, J.G. Comparison of SUMO Fusion Technology with Traditional Gene Fusion Systems: Enhanced Expression and Solubility with SUMO. Protein Sci. 2006, 15, 182–189. [Google Scholar] [CrossRef]
- Flinterman, M.; Farzaneh, F.; Habib, N.; Malik, F.; Gäken, J.; Tavassoli, M. Delivery of Therapeutic Proteins as Secretable TAT Fusion Products. Mol. Ther. 2009, 17, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Takis, P.G.; Ghini, V.; Tenori, L.; Turano, P.; Luchinat, C. Uniqueness of the NMR Approach to Metabolomics. TrAC Trends Anal. Chem. 2019, 120, 115300. [Google Scholar] [CrossRef]
- Vignoli, A.; Ghini, V.; Meoni, G.; Licari, C.; Takis, P.G.; Tenori, L.; Turano, P.; Luchinat, C. High-Throughput Metabolomics by 1D NMR. Angew. Chem. Int. Ed. 2019, 58, 968–994. [Google Scholar] [CrossRef] [PubMed]
- Lieven, C.; Beber, M.E.; Olivier, B.G.; Bergmann, F.T.; Ataman, M.; Babaei, P.; Bartell, J.A.; Blank, L.M.; Chauhan, S.; Correia, K.; et al. MEMOTE for Standardized Genome-Scale Metabolic Model Testing. Nat. Biotechnol. 2020, 38, 272–276. [Google Scholar] [CrossRef]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and Analysis of Biochemical Constraint-Based Models Using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef]
- Wang, H.; Marcišauskas, S.; Sánchez, B.J.; Domenzain, I.; Hermansson, D.; Agren, R.; Nielsen, J.; Kerkhoven, E.J. RAVEN 2.0: A Versatile Toolbox for Metabolic Network Reconstruction and a Case Study on Streptomyces Coelicolor. PLoS Comput. Biol. 2018, 14, e1006541. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Model Code | Subsystem | Reaction Name |
|---|---|---|
| rxn00789 | Histidine metabolism | 1-(5-Phospho-d-ribosyl)-ATP:pyrophosphate phosphoribosyl-transferase |
| rxn00863 | Histidine metabolism | l-histidinal:NAD + oxidoreductase |
| rxn02159 | Histidine metabolism | l-histidinol:NAD + oxidoreductase |
| rxn02160 | Histidine metabolism | l-histidinol-phosphate phosphohydrolase |
| rxn02320 | Histidine metabolism | 5-Amino-2-oxopentanoate:2-oxoglutarate aminotransferase |
| rxn02473 | Histidine metabolism | d-erythro-1-(imidazol-4-yl)glycerol 3-phosphate hydrolyase |
| rxn02834 | Histidine metabolism | Phosphoribosyl-ATP pyrophosphohydrolase |
| rxn02835 | Histidine metabolism | 1-(5-Phospho-d-ribosyl)-AMP 1,6-hydrolase |
| rxn03135 | Histidine metabolism | Imidazole-glycerol-3-phosphate synthase |
| rxn03175 | Histidine metabolism | N-(5′-phospho-d-ribosylformimino)-5-amino-1- |
| rxn05466 | Ammonia transport | Ammonia transport via diffusion |
| Reaction Model Code | Subsystem | Reaction Name |
|---|---|---|
| rxn05466 | Ammonium transporter | Ammonia transport via diffusion |
| rxn02160 | Histidine metabolism | l-histidinol-phosphate phosphohydrolase |
| rxn00863 | Histidine metabolism | l-histidinal:NAD + oxidoreductase |
| rxn02159 | Histidine metabolism | l-histidinol:NAD + oxidoreductase |
| rxn02834 | Histidine metabolism | Phosphoribosyl-ATP pyrophosphohydrolase |
| rxn03175 | Histidine metabolism | N-(5′-phospho-d-ribosylformimino)-5-amino-1- |
| rxn02473 | Histidine metabolism | d-erythro-1-(imidazol-4-yl)glycerol 3-phosphate hydro-lyase |
| rxn02508 | Phenylalanine, tyrosine and tryptophan biosynthesis | N-(5-phospho-beta-d-ribosyl)anthranilate ketol-isomerase |
| rxn02320 | Histidine metabolism | 5-Amino-2-oxopentanoate:2-oxoglutarate aminotransferase |
| rxn02507 | Phenylalanine, tyrosine and tryptophan biosynthesis | 1-(2-Carboxyphenylamino)-1-deoxy-d-ribulose-5-phosphate |
| rxn00789 | Histidine metabolism | 1-(5-Phospho-d-ribosyl)-ATP:pyrophosphate phosphoribosyl-transferase |
| rxn03135 | Histidine metabolism | Imidazole-glycerol-3-phosphate synthase |
| rxn00791 | Phenylalanine, tyrosine and tryptophan biosynthesis | N-(5-phospho-d-ribosyl)anthranilate:pyrophosphate |
| rxn02835 | Histidine metabolism | 1-(5-Phospho-d-ribosyl)-AMP 1,6-hydrolase |
| rxn00392 | Riboflavin metabolism | ATP:riboflavin 5′-phosphotransferase |
| Reaction Model Code | Subsystem | Reaction Name | Formula |
|---|---|---|---|
| rxn05937 | NA | Ferredoxin:NADP+ oxidoreductase | NADP + H+ + reduced ferredoxin => NADPH + oxidised ferredoxin |
| rxn12822 | Glyoxylate and dicarboxylate metabolism | l-glutamateferredoxin oxidoreductase (transaminating) | 2 l-glutamate + 2 oxidised ferredoxin => 2-oxoglutarate + l-glutamine + 2 H+ + 2 reduced ferredoxin |
| rxn01477 | PPP | 6-Phospho-d-gluconate hydro-lyase (edd) | 6-Phospho-d-gluconate => H2O + 2-keto-3-deoxy-6-phosphogluconate |
| rxn03884 | PPP | 2-Dehydro-3-deoxy-d-gluconate-6-phosphate d-glyceraldehyde-3-phosphate-lyase (eda) | 2-Keto-3-deoxy-6-phosphogluconate => pyruvate + glyceraldehyde-3-phosphate |
| rxn01476 | PPP | 6-Phospho-d-glucono-1,5-lactone lactonohydrolase (AgaI) | H2O + 6-phospho-d-glucono-1-5-lactone => H+ + 6-phospho-d-gluconate |
| rxn00260 | Alanine, aspartate and glutamate metabolism | l-aspartate2-oxoglutarate aminotransferase | 2-Oxoglutarate + l-aspartate <= l-glutamate + oxaloacetate |
| rxn00337 | Glycine, serine and threonine metabolism | ATPL-aspartate 4-phosphotransferase | ATP + l-aspartate => ADP + 4-phospho-l-aspartate |
| rxn01643 | Glycine, serine and threonine-cysteine and methionine-lysine metabolism | l-aspartate-4-semialdehyde:NADP+ oxidoreductase (phosphorylating) | NADP + phosphate + l-aspartate-4-semialdehyde <= NADPH + 4-phospho-l-aspartate |
| rxn00285 | Citrate cycle (TCA cycle) | Succinate-CoA ligase (ADP forming) | ATP + CoA + succinate => ADP + phosphate + succinyl-CoA |
| rxn00423 | Cysteine and methionine metabolism | Serine O-acetyltransferase | Acetyl-CoA + l-serine <= CoA + O-acetyl-l-serine |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fondi, M.; Gonzi, S.; Dziurzynski, M.; Turano, P.; Ghini, V.; Calvanese, M.; Colarusso, A.; Lauro, C.; Parrilli, E.; Tutino, M.L. Modelling hCDKL5 Heterologous Expression in Bacteria. Metabolites 2021, 11, 491. https://doi.org/10.3390/metabo11080491
Fondi M, Gonzi S, Dziurzynski M, Turano P, Ghini V, Calvanese M, Colarusso A, Lauro C, Parrilli E, Tutino ML. Modelling hCDKL5 Heterologous Expression in Bacteria. Metabolites. 2021; 11(8):491. https://doi.org/10.3390/metabo11080491
Chicago/Turabian StyleFondi, Marco, Stefano Gonzi, Mikolaj Dziurzynski, Paola Turano, Veronica Ghini, Marzia Calvanese, Andrea Colarusso, Concetta Lauro, Ermenegilda Parrilli, and Maria Luisa Tutino. 2021. "Modelling hCDKL5 Heterologous Expression in Bacteria" Metabolites 11, no. 8: 491. https://doi.org/10.3390/metabo11080491
APA StyleFondi, M., Gonzi, S., Dziurzynski, M., Turano, P., Ghini, V., Calvanese, M., Colarusso, A., Lauro, C., Parrilli, E., & Tutino, M. L. (2021). Modelling hCDKL5 Heterologous Expression in Bacteria. Metabolites, 11(8), 491. https://doi.org/10.3390/metabo11080491