
General Unified Microbiome Profiling Pipeline (GUMPP) for Large Scale, Streamlined and Reproducible Analysis of Bacterial 16S rRNA Data to Predicted Microbial Metagenomes, Enzymatic Reactions and Metabolic Pathways




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
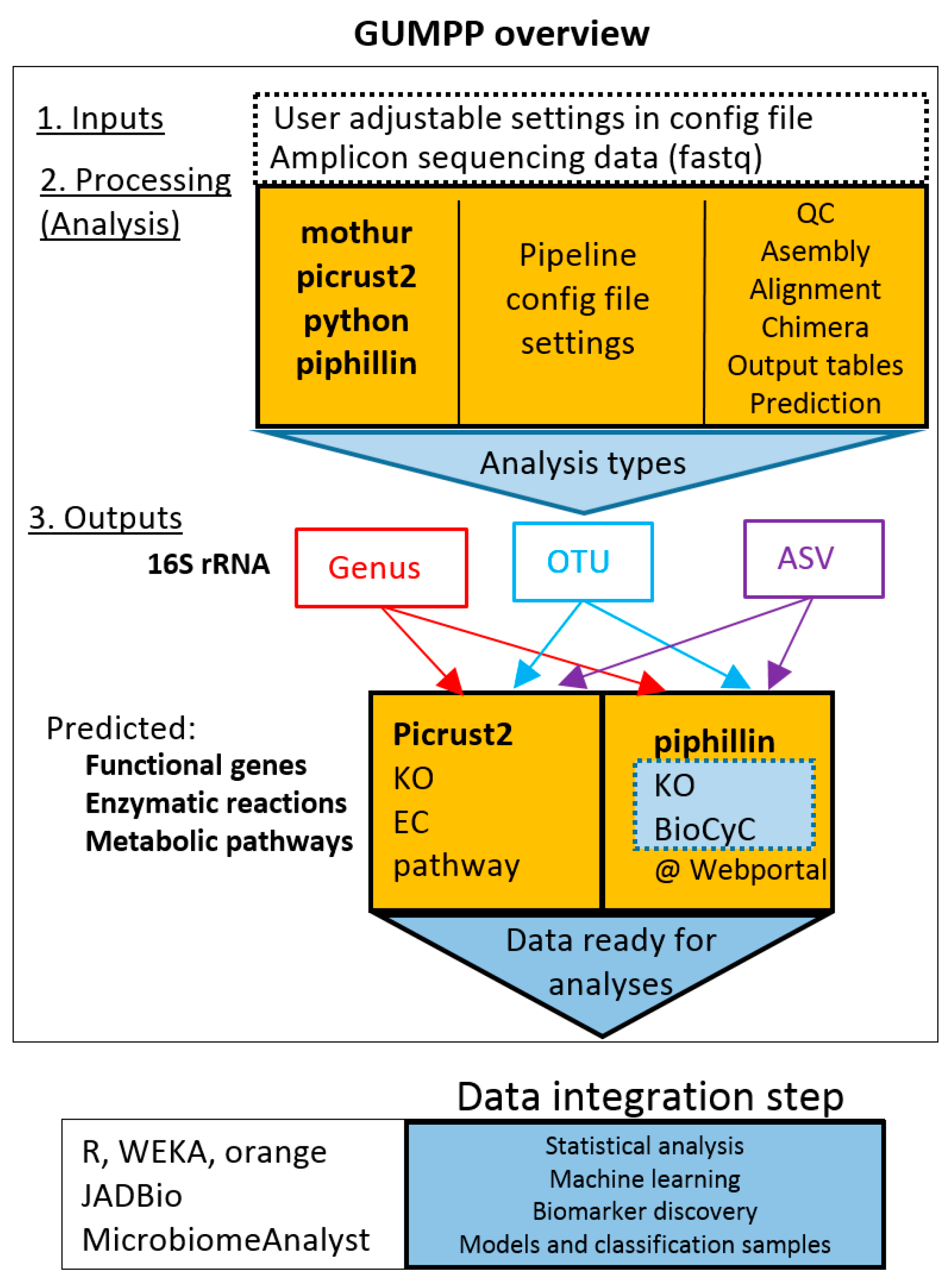
2.1. Design of GUMPP Workflow
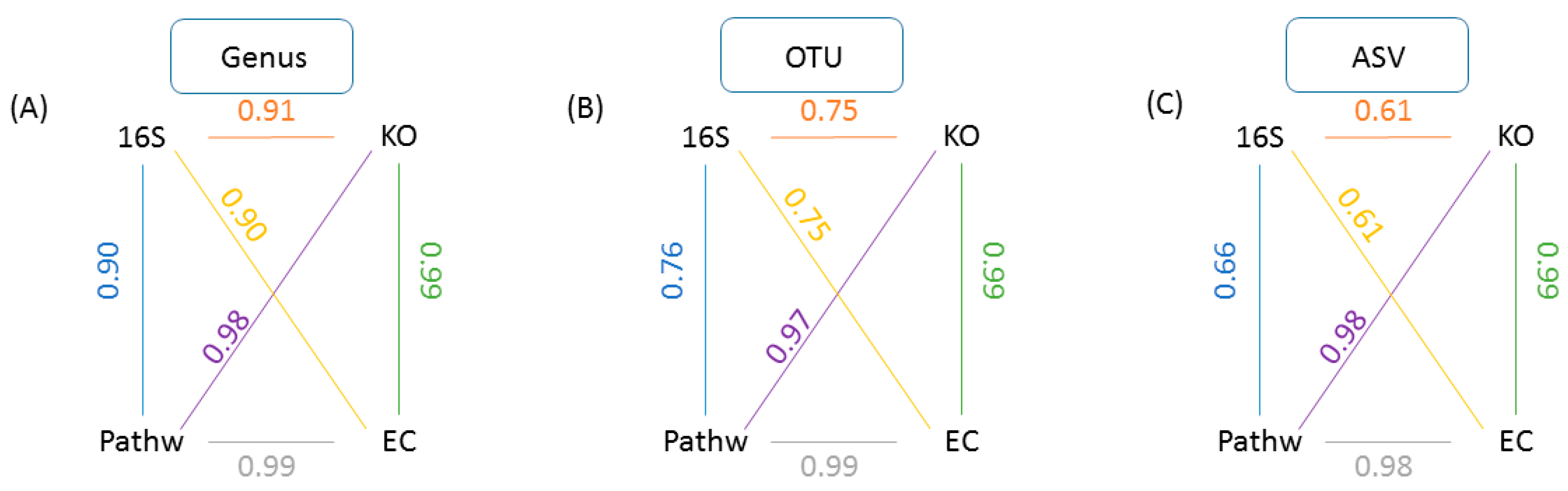
2.2. Reanalysis and Extension of Mice Gut Microbiome Data Using GUMPP: The Choice of Level of Analysis (Genus, OTU, ASV) Is far from Arbitrary
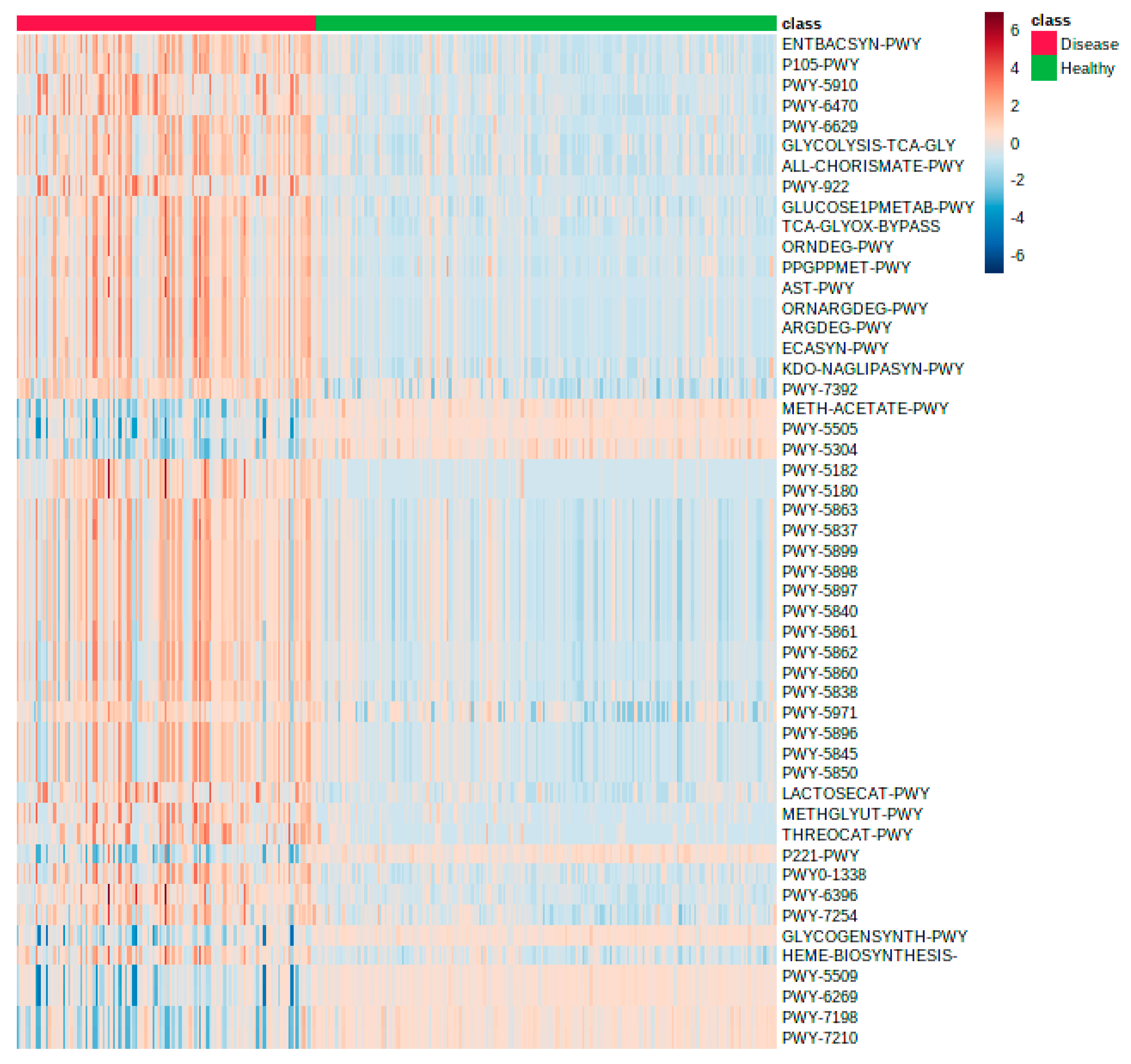
2.3. Reanalysis and Extension of Human Gut Microbiome Data Using GUMPP
3. Materials and Methods
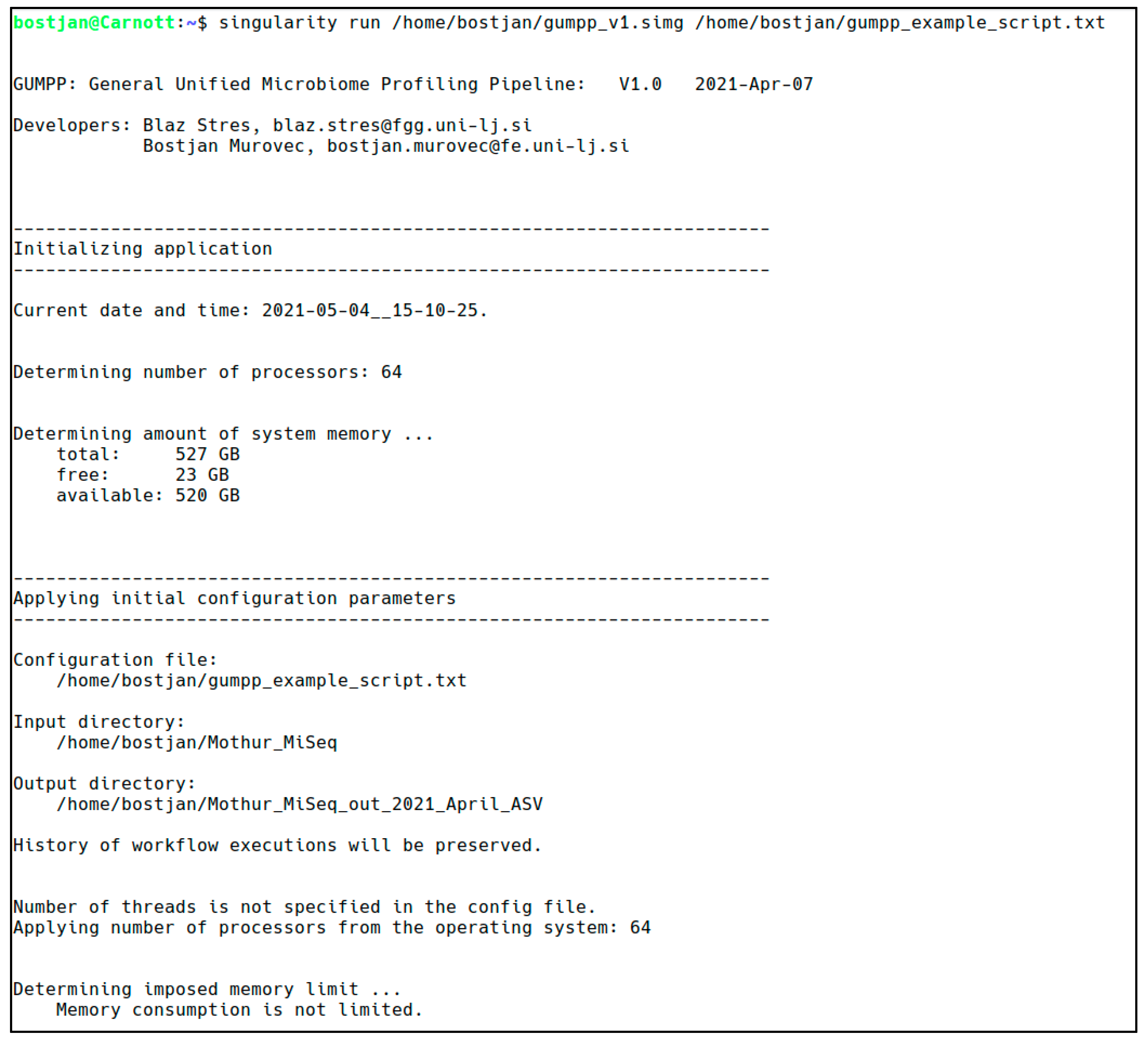
3.1. GUMPP Implementation
3.2. Sequence Data Collections
3.3. Statistical Analyses and Machine Learning
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stres, B.; Kronegger, L. Shift in the paradigm towards next-generation microbiology. FEMS Microbiol. Lett. 2019, 366. [Google Scholar] [CrossRef]
- Vernocchi, P.; Del Chierico, F.; Putignani, L. Gut Microbiota Profiling: Metabolomics Based Approach to Unravel Compounds Affecting Human Health. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef]
- Wu, J.; Wang, K.; Wang, X.; Pang, Y.; Jiang, C. The role of the gut microbiome and its metabolites in metabolic diseases. Protein Cell 2020. [Google Scholar] [CrossRef]
- Visconti, A.; Le Roy, C.I.; Rosa, F.; Rossi, N.; Martin, T.C.; Mohney, R.P.; Li, W.; de Rinaldis, E.; Bell, J.T.; Venter, J.C.; et al. Interplay between the human gut microbiome and host metabolism. Nat. Commun. 2019, 10, 4505. [Google Scholar] [CrossRef]
- Lee-Sarwar, K.A.; Lasky-Su, J.; Kelly, R.S.; Litonjua, A.A.; Weiss, S.T. Metabolome-Microbiome Crosstalk and Human Disease. Metabolites 2020, 10, 181. [Google Scholar] [CrossRef] [PubMed]
- Kappel, B.A.; De Angelis, L.; Heiser, M.; Ballanti, M.; Stoehr, R.; Goettsch, C.; Mavilio, M.; Artati, A.; Paoluzi, O.A.; Adamski, J.; et al. Cross-omics analysis revealed gut microbiome-related metabolic pathways underlying atherosclerosis development after antibiotics treatment. Mol. Metab. 2020, 36. [Google Scholar] [CrossRef] [PubMed]
- Wilmanski, T.; Rappaport, N.; Earls, J.C.; Magis, A.T.; Manor, O.; Lovejoy, J.; Omenn, G.S.; Hood, L.; Gibbons, S.M.; Price, N.D. Blood metabolome predicts gut microbiome alpha-diversity in humans. Nat. Biotechnol. 2019, 37, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Armour, C.R.; Hu, C.; Mei, M.; Tian, C.; Sharpton, T.J.; Jiang, Y. Microbiome Multi-Omics Network Analysis: Statistical Considerations, Limitations, and Opportunities. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, K.; Wu, W.; Giannoulatou, E.; Ho, J.W.K.; Li, L. Host and microbiome multi-omics integration: Applications and methodologies. Biophys. Rev. 2019, 11. [Google Scholar] [CrossRef]
- Poretsky, R.; Rodriguez-R, L.M.; Luo, C.; Tsementzi, D.; Konstantinidis, K.T. Strengths and limitations of 16S rRNA gene amplicon sequencing in revealing temporal microbial community dynamics. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Pasolli, E.; Schiffer, L.; Manghi, P.; Renson, A.; Obenchain, V.; Truong, D.T.; Beghini, F.; Malik, F.; Ramos, M.; Dowd, J.B.; et al. Accessible, curated metagenomic data through ExperimentHub. Nat. Methods 2017, 14. [Google Scholar] [CrossRef]
- Rühlemann, M.C.; Hermes, B.M.; Bang, C.; Doms, S.; Moitinho-Silva, L.; Thingholm, L.B.; Frost, F.; Degenhardt, F.; Wittig, M.; Kässens, J.; et al. Genome-wide association study in 8,956 German individuals identifies influence of ABO histo-blood groups on gut microbiome. Nat. Genet. 2021, 53. [Google Scholar] [CrossRef] [PubMed]
- Mysara, M.; Vandamme, P.; Props, R.; Kerckhof, F.M.; Leys, N.; Boon, N.; Raes, J.; Monsieurs, P. Reconciliation between operational taxonomic units and species boundaries. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D. Amplicon sequence variants artificially split bacterial genomes into separate clusters. bioRxiv 2021. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11. [Google Scholar] [CrossRef]
- Nilakanta, H.; Drews, K.L.; Firrell, S.; Foulkes, M.A.; Jablonski, K.A. A review of software for analyzing molecular sequences. BMC Res. Notes 2014, 7, 1–9. [Google Scholar] [CrossRef]
- Pollock, J.; Glendinning, L.; Wisedchanwet, T.; Watson, M. The Madness of Microbiome: Attempting To Find Consensus “Best Practice” for 16S Microbiome Studies. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef]
- Schloss, P.D. Reintroducing mothur: 10 Years Later. Appl. Environ. Microbiol. 2020, 86, e02343-19. [Google Scholar] [CrossRef]
- López-García, A.; Pineda-Quiroga, C.; Atxaerandio, R.; Pérez, A.; Hernández, I.; García-Rodríguez, A.; González-Recio, O. Comparison of Mothur and QIIME for the Analysis of Rumen Microbiota Composition Based on 16S rRNA Amplicon Sequences. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Winand, R.; Bogaerts, B.; Hoffman, S.; Lefevre, L.; Delvoye, M.; Braekel, J.V.; Fu, Q.; Roosens, N.H.; Keersmaecker, S.C.; Vanneste, K. Targeting the 16S rrna gene for bacterial identification in complex mixed samples: Comparative evaluation of second (illumina) and third (oxford nanopore technologies) generation sequencing technologies. Int. J. Mol. Sci. 2019, 21, 298. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449. [Google Scholar] [CrossRef] [PubMed]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the MiSeq Illumina Sequencing Platform. Appl. Environ. Microbiol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Prodan, A.; Tremaroli, V.; Brolin, H.; Zwinderman, A.H.; Nieuwdorp, M.; Levin, E. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 2020, 15. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38. [Google Scholar] [CrossRef]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015, 31. [Google Scholar] [CrossRef]
- Wemheuer, F.; Taylor, J.A.; Daniel, R.; Johnston, E.; Meinicke, P.; Thomas, T.; Wemheuer, B. Tax4Fun2: Prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene sequences. Environ. Microb. 2020, 15. [Google Scholar] [CrossRef]
- Narayan, N.R.; Weinmaier, T.; Laserna-Mendieta, E.J.; Claesson, M.J.; Shanahan, F.; Dabbagh, K.; Iwai, S.; DeSantis, T.Z. Piphillin predicts metagenomic composition and dynamics from DADA2-corrected 16S rDNA sequences. BMC Genom. 2020, 21. [Google Scholar] [CrossRef]
- Iwai, S.; Weinmaier, T.; Schmidt, B.L.; Albertson, D.G.; Poloso, N.J.; Dabbagh, K.; DeSantis, T.Z. Piphillin: Improved Prediction of Metagenomic Content by Direct Inference from Human Microbiomes. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Jones, R.B.; Fodor, A.A. Inference-based accuracy of metagenome prediction tools varies across sample types and functional categories. Microbiome 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Salosensaari, A.; Laitinen, V.; Havulinna, A.S.; Meric, G.; Cheng, S.; Perola, M.; Valsta, L.; Alfthan, G.; Inouye, M.; Watrous, J.D.; et al. Taxonomic signatures of cause-specific mortality risk in human gut microbiome. Nat. Commun. 2021, 12, 2671. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Kurtzer, G.M.; Sochat, V.; Bauer, M.W. Singularity: Scientific containers for mobility of compute. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457. [Google Scholar] [CrossRef]
- Stoddard, S.F.; Smith, B.J.; Hein, R.; Roller, B.R.; Schmidt, T.M. rrnDB: Improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res. 2015, 43. [Google Scholar] [CrossRef]
- Větrovský, T.; Baldrian, P. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.P.; Warnow, T.; Pop, M.; White, B. A perspective on 16S rRNA operational taxonomic unit clustering using sequence similarity. NPJ Biofilms Microb. 2016, 2, 16004. [Google Scholar] [CrossRef]
- Pei, A.Y.; Oberdorf, W.E.; Nossa, C.W.; Agarwal, A.; Chokshi, P.; Gerz, E.A.; Jin, Z.; Lee, P.; Yang, L.; Poles, M.; et al. Diversity of 16S rRNA genes within individual prokaryotic genomes. Appl. Environ. Microbiol. 2010, 76. [Google Scholar] [CrossRef]
- Sun, D.L.; Jiang, X.; Wu, Q.L.; Zhou, N.Y. Intragenomic heterogeneity of 16S rRNA genes causes overestimation of prokaryotic diversity. Appl. Environ. Microbiol. 2013, 79. [Google Scholar] [CrossRef] [PubMed]
- Huse, S.M.; Dethlefsen, L.; Huber, J.A.; Mark Welch, D.; Relman, D.A.; Sogin, M.L. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 2008, 4. [Google Scholar] [CrossRef]
- Soriano-Lerma, A.; Pérez-Carrasco, V.; Sánchez-Marañón, M.; Ortiz-González, M.; Sánchez-Martín, V.; Gijón, J.; Navarro-Mari, J.M.; García-Salcedo, J.A.; Soriano, M. Influence of 16S rRNA target region on the outcome of microbiome studies in soil and saliva samples. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Joos, L.; Beirinckx, S.; Haegeman, A.; Debode, J.; Vandecasteele, B.; Baeyen, S.; Goormachtig, S.; Clement, L.; De Tender, C. Daring to be differential: Metabarcoding analysis of soil and plant-related microbial communities using amplicon sequence variants and operational taxonomical units. BMC Genom. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Mahnic, A.; Breskvar, M.; Dzeroski, S.; Skok, P.; Pintar, S.; Rupnik, M. Distinct Types of Gut Microbiota Dysbiosis in Hospitalized Gastroenterological Patients Are Disease Non-related and Characterized With the Predominance of Either Enterobacteriaceae or Enterococcus. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Mustafa, A.; Rahimi Azghadi, M. Automated Machine Learning for Healthcare and Clinical Notes Analysis. Computers 2021, 10, 24. [Google Scholar] [CrossRef]
- Tsamardinos, I.; Charonyktakis, P.; Lakiotaki, K.; Borboudakis, G.; Zenklusen, J.C.; Juhl, H.; Chatzaki, E.; Lagani, V. Just Add Data: Automated Predictive Modeling and BioSignature Discovery. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hörmannsperger, G.; von Schillde, M.A.; Haller, D. Lactocepin as a protective microbial structure in the context of IBD. Gut Microbes 2013, 4. [Google Scholar] [CrossRef]
- von Schillde, M.A.; Hörmannsperger, G.; Weiher, M.; Alpert, C.A.; Hahne, H.; Bäuerl, C.; van Huynegem, K.; Steidler, L.; Hrncir, T.; Pérez-Martínez, G.; et al. Lactocepin secreted by Lactobacillus exerts anti-inflammatory effects by selectively degrading proinflammatory chemokines. Cell Host Microbe 2012, 11. [Google Scholar] [CrossRef]
- Osman, D.; Patterson, C.J.; Bailey, K.; Fisher, K.; Robinson, N.J.; Rigby, S.E.; Cavet, J.S. The copper supply pathway to a Salmonella Cu,Zn-superoxide dismutase (SodCII) involves P(1B)-type ATPase copper efflux and periplasmic CueP. Mol. Microbiol. 2013, 87. [Google Scholar] [CrossRef]
- Subramani, S.; Perdreau-Dahl, H.; Morth, J.P. The magnesium transporter A is activated by cardiolipin and is highly sensitive to free magnesium in vitro. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Joyet, P.; Mokhtari, A.; Riboulet-Bisson, E.; Blancato, V.S.; Espariz, M.; Magni, C.; Hartke, A.; Deutscher, J.; Sauvageot, N. Enzymes Required for Maltodextrin Catabolism in Enterococcus faecalis Exhibit Novel Activities. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Yin, W.; Fang, X.; Guo, J.; Shi, H. Characteristics of a water-forming NADH oxidase from Methanobrevibacter smithii, an archaeon in the human gut. Biosci. Rep. 2016, 36. [Google Scholar] [CrossRef]
- Yoshii, K.; Hosomi, K.; Sawane, K.; Kunisawa, J. Metabolism of Dietary and Microbial Vitamin B Family in the Regulation of Host Immunity. Front. Nutr. 2019, 6. [Google Scholar] [CrossRef]
- LeBlanc, J.G.; Milani, C.; de Giori, G.S.; Sesma, F.; van Sinderen, D.; Ventura, M. Bacteria as vitamin suppliers to their host: A gut microbiota perspective. Curr. Opin. Biotechnol. 2013, 24. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, D.A.; Arzamasov, A.A.; Khoroshkin, M.S.; Iablokov, S.N.; Leyn, S.A.; Peterson, S.N.; Novichkov, P.S.; Osterman, A.L. Micronutrient Requirements and Sharing Capabilities of the Human Gut Microbiome. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef]
- Henke, M.T.; Kenny, D.J.; Cassilly, C.D.; Vlamakis, H.; Xavier, R.J.; Clardy, J. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc. Natl. Acad. Sci. USA 2019, 116. [Google Scholar] [CrossRef]
- O’Callaghan, A.; van Sinderen, D. Bifidobacteria and Their Role as Members of the Human Gut Microbiota. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef]
- Mahnic, A.; Rupnik, M. Different host factors are associated with patterns in bacterial and fungal gut microbiota in Slovenian healthy cohort. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Murovec, B.; Deutsch, L.; Stres, B. Computational Framework for High-Quality Production and Large-Scale Evolutionary Analysis of Metagenome Assembled Genomes. Mol. Biol. Evol. 2020, 37. [Google Scholar] [CrossRef]
- Tsamardinos, I.; Rakhshani, A.; Lagani, V. Performance-Estimation Properties of Cross-Validation-Based Protocols with Simultaneous Hyper-Parameter Optimization|SpringerLink. In Artificial Intelligence: Methods and Applications; Likas, A., Blekas, K., Kalles, D., Eds.; Springer International Publishing: Cham, Switzerland, 2014. [Google Scholar]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46. [Google Scholar] [CrossRef] [PubMed]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Hammer, O.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 1, 9. [Google Scholar]
- Proffitt, C.; Bidkhori, G.; Moyes, D.; Shoaie, S. Disease, Drugs and Dysbiosis: Understanding Microbial Signatures in Metabolic Disease and Medical Interventions. Microorganisms 2020, 8, 1381. [Google Scholar] [CrossRef] [PubMed]
- Rosario, D.; Boren, J.; Uhlen, M.; Proctor, G.; Aarsland, D.; Mardinoglu, A.; Shoaie, S. Systems Biology Approaches to Understand the Host-Microbiome Interactions in Neurodegenerative Diseases. Front. Neurosci. 2020, 14. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Lehto, S.M.; Harty, S.; Dinan, T.G.; Cryan, J.F.; Burnet, P.W.J. Psychobiotics and the Manipulation of Bacteria-Gut-Brain Signals. Trends Neurosci. 2016, 39. [Google Scholar] [CrossRef]
- Vijay, A.; Valdes, A.M. The Metabolomic Signatures of Weight Change. Metabolites 2019, 9, 67. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murovec, B.; Deutsch, L.; Stres, B. General Unified Microbiome Profiling Pipeline (GUMPP) for Large Scale, Streamlined and Reproducible Analysis of Bacterial 16S rRNA Data to Predicted Microbial Metagenomes, Enzymatic Reactions and Metabolic Pathways. Metabolites 2021, 11, 336. https://doi.org/10.3390/metabo11060336
Murovec B, Deutsch L, Stres B. General Unified Microbiome Profiling Pipeline (GUMPP) for Large Scale, Streamlined and Reproducible Analysis of Bacterial 16S rRNA Data to Predicted Microbial Metagenomes, Enzymatic Reactions and Metabolic Pathways. Metabolites. 2021; 11(6):336. https://doi.org/10.3390/metabo11060336
Chicago/Turabian StyleMurovec, Boštjan, Leon Deutsch, and Blaž Stres. 2021. "General Unified Microbiome Profiling Pipeline (GUMPP) for Large Scale, Streamlined and Reproducible Analysis of Bacterial 16S rRNA Data to Predicted Microbial Metagenomes, Enzymatic Reactions and Metabolic Pathways" Metabolites 11, no. 6: 336. https://doi.org/10.3390/metabo11060336
APA StyleMurovec, B., Deutsch, L., & Stres, B. (2021). General Unified Microbiome Profiling Pipeline (GUMPP) for Large Scale, Streamlined and Reproducible Analysis of Bacterial 16S rRNA Data to Predicted Microbial Metagenomes, Enzymatic Reactions and Metabolic Pathways. Metabolites, 11(6), 336. https://doi.org/10.3390/metabo11060336