
Revealing the Metabolic Alterations during Biofilm Development of Burkholderia cenocepacia Based on Genome-Scale Metabolic Modeling


 and
and 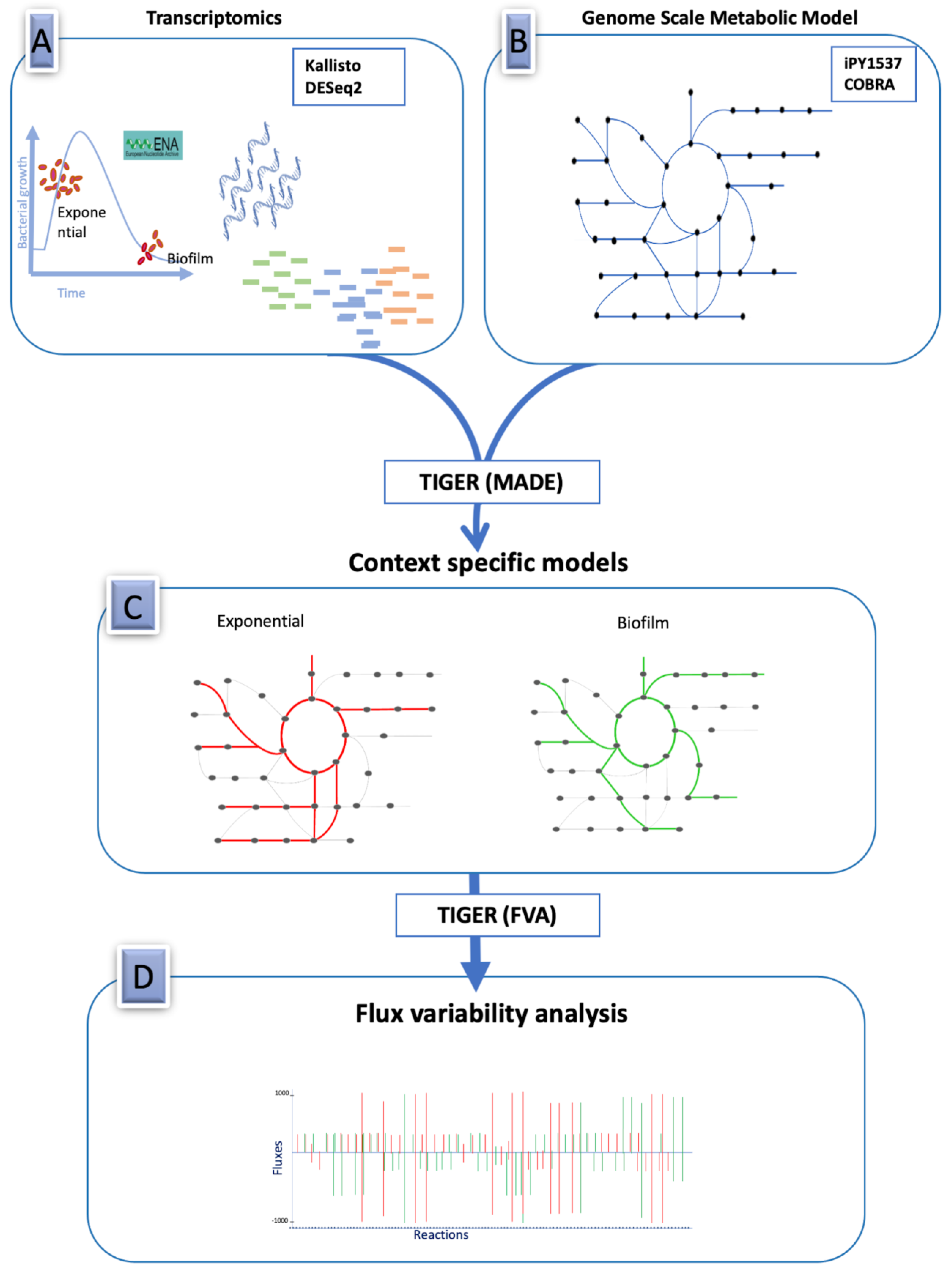{kind=link}
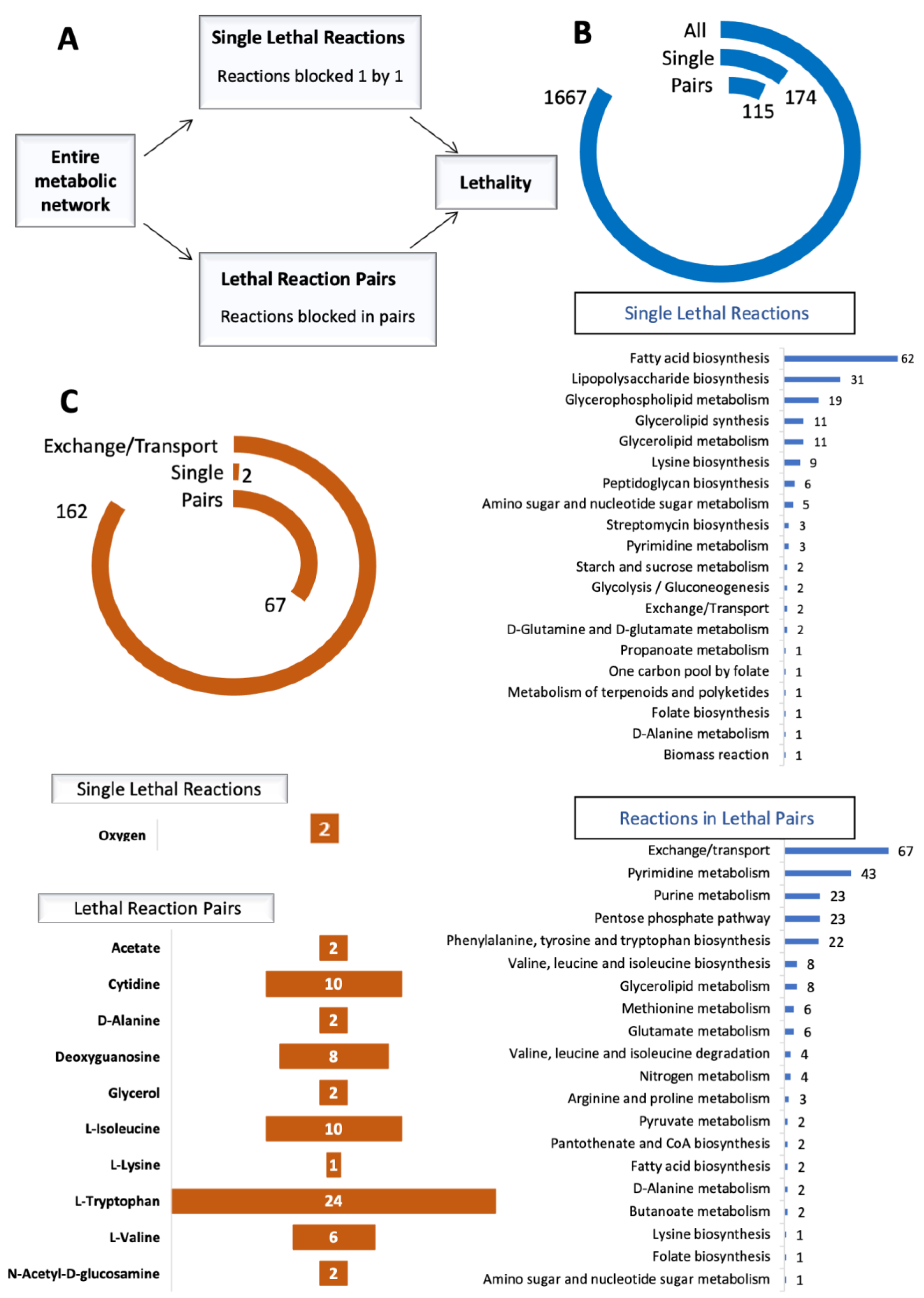{kind=link}
{kind=link}
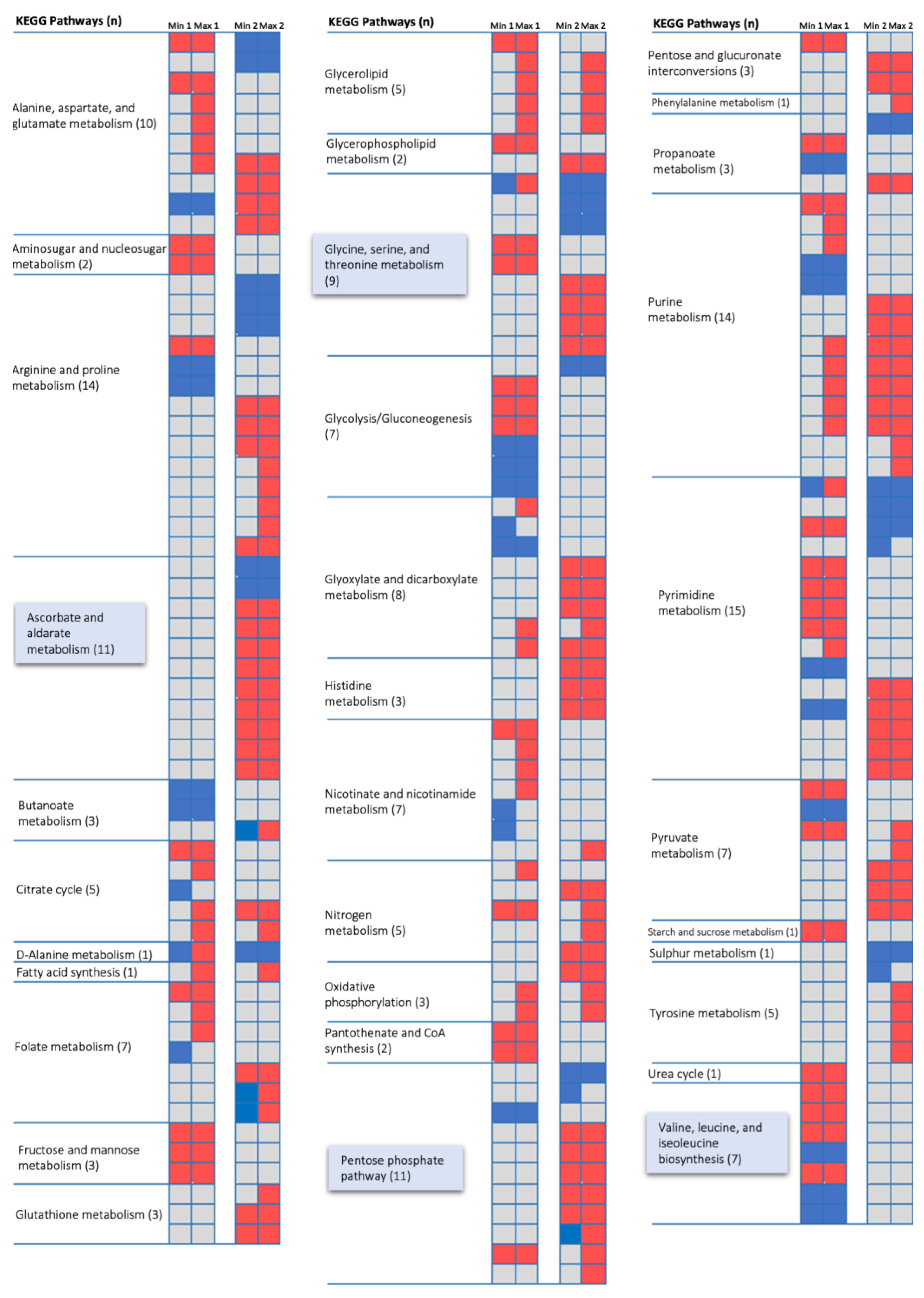{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Genome-Scale Metabolic Model of Burkholderia Cenocepacia
2.2. Synthetic Lethality Analysis
2.3. Differential Expression Analysis of RNA-Seq Data
2.4. Integrating RNA-Seq Data with the Genome-Scale Metabolic Model
3. Results
3.1. In Silico Identification of Reaction Essentialities and Affected Pathways
3.2. Condition-Dependent Metabolic Models of B. cenocepacia
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Sullivan, B.P.; Freedman, S.D. Cystic fibrosis. Lancet 2009, 373, 1891–1904. [Google Scholar] [CrossRef]
- WHO. Human Genomics in Global Health. Available online: https://www.who.int/genomics/public/geneticdiseases/en/index2.html (accessed on 23 April 2020).
- Sanders, D.B.; Li, Z.; Laxova, A.; Rock, M.J.; Levy, H.; Collins, J.; Ferec, C.; Farrell, P.M. Risk factors for the progression of cystic fibrosis lung disease throughout childhood. Ann. Am. Thorac. Soc. 2014, 11, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Scoffone, V.C.; Chiarelli, L.R.; Trespidi, G.; Mentasti, M.; Riccardi, G.; Buroni, S. Burkholderia cenocepacia Infections in Cystic Fibrosis Patients: Drug Resistance and Therapeutic Approaches. Front. Microbiol. 2017, 8, 1592. [Google Scholar] [CrossRef] [PubMed]
- Burgel, P.R.; Lemonnier, L.; Dehillotte, C.; Sykes, J.; Stanojevic, S.; Stephenson, A.L.; Paillasseur, J.L. Cluster and CART analyses identify large subgroups of adults with cystic fibrosis at low risk of 10-year death. Eur. Respir. J. 2019, 53, 1801943. [Google Scholar] [CrossRef] [PubMed]
- Altay, O.; Nielsen, J.; Uhlen, M.; Boren, J.; Mardinoglu, A. Systems biology perspective for studying the gut microbiota in human physiology and liver diseases. EBioMedicine 2019, 49, 364–373. [Google Scholar] [CrossRef]
- Turanli, B.; Altay, O.; Borén, J.; Turkez, H.; Nielsen, J.; Uhlen, M.; Arga, K.Y.; Mardinoglu, A. Systems biology based drug repositioning for development of cancer therapy. Semin. Cancer Biol. 2019, 86, 47–58. [Google Scholar] [CrossRef]
- Henson, M.A. Genome-scale modelling of microbial metabolism with temporal and spatial resolution. Biochem. Soc. Trans. 2015, 43, 1164–1171. [Google Scholar] [CrossRef]
- Zhang, C.; Aldrees, M.; Arif, M.; Li, X.; Mardinoglu, A.; Aziz, M.A. Elucidating the reprograming of colorectal cancer metabolism using genome-scale metabolic modeling. Front. Oncol. 2019, 9, 681. [Google Scholar] [CrossRef]
- Lam, S.; Bayraktar, A.; Zhang, C.; Turkez, H.; Nielsen, J.; Boren, J.; Shoaie, S.; Uhlen, M.; Mardinoglu, A. A systems biology approach for studying neurodegenerative diseases. Drug Discov. Today 2020, 25, 1146–1159. [Google Scholar] [CrossRef]
- Papin, J. Burkholderia Cepacia Complex. Available online: https://bme.virginia.edu/csbl/Downloads1-burkholderia-cepacia.html (accessed on 29 October 2019).
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef]
- Kiekens, S.; Sass, A.; Van Nieuwerburgh, F.; Deforce, D.; Coenye, T. The Small RNA ncS35 Regulates Growth in Burkholderia cenocepacia J2315. mSphere 2018, 3. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- NCBI. Genome; NCBI. Available online: https://www.ncbi.nlm.nih.gov/genome/475?genome_assembly_id=281623 (accessed on 29 October 2019).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Jensen, P.A.; Papin, J.A. Functional integration of a metabolic network model and expression data without arbitrary thresholding. Bioinformatics 2010, 27, 541–547. [Google Scholar] [CrossRef]
- Jensen, P.A.; Lutz, K.A.; Papin, J.A. TIGER: Toolbox for integrating genome-scale metabolic models, expression data, and transcriptional regulatory networks. BMC Syst. Biol. 2011, 5, 1–12. [Google Scholar] [CrossRef]
- Kumar, B.; Cardona, S.T. Synthetic cystic fibrosis sputum medium regulates flagellar biosynthesis through the flhF gene in Burkholderia cenocepacia. Front. Cell Infect. Microbiol. 2016, 6, 65. [Google Scholar] [CrossRef]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Heath, R.J.; White, S.W.; Rock, C.O. Lipid biosynthesis as a target for antibacterial agents. Prog. Lipid. Res. 2001, 40, 467–497. [Google Scholar] [CrossRef]
- Parsons, J.B.; Rock, C.O. Is bacterial fatty acid synthesis a valid target for antibacterial drug discovery? Curr. Opin. Microbiol. 2011, 14, 544–549. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Reddy, M.C.; Ioerger, T.R.; Rothchild, A.C.; Dartois, V.; Schuster, B.M.; Trauner, A.; Wallis, D.; Galaviz, S.; Huttenhower, C.; et al. Tryptophan biosynthesis protects mycobacteria from CD4 T-cell-mediated killing. Cell 2013, 155, 1296–1308. [Google Scholar] [CrossRef] [PubMed]
- Lott, J.S. The tryptophan biosynthetic pathway is essential for Mycobacterium tuberculosis to cause disease. Biochem. Soc. Trans. 2020, 48, 2029–2037. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.B.; Rock, C.O. Bacterial lipids: Metabolism and membrane homeostasis. Prog. Lipid Res. 2013, 52, 249–276. [Google Scholar] [CrossRef] [PubMed]
- Neis, E.P.J.G.; Dejong, C.H.C.; Rensen, S.S. The role of microbial amino acid metabolism in host metabolism. Nutrients 2015, 7, 2930–2946. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Koo, H. Structural organization and dynamics of exopolysaccharide matrix and microcolonies formation by Streptococcus mutans in biofilms. J. Appl. Microbiol. 2010, 108, 2103–2113. [Google Scholar] [CrossRef]
- Bodini, S.; Nunziangeli, L.; Santori, F. Influence of amino acids on low-density Escherichia coli responses to nutrient downshifts. J. Bacteriol. 2007, 189, 3099–3105. [Google Scholar] [CrossRef]
- Okamura, E.; Hirai, M.Y. Novel regulatory mechanism of serine biosynthesis associated with 3-phosphoglycerate dehydrogenase in Arabidopsis thaliana. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Greenwich, J.; Reverdy, A.; Gozzi, K.; Di Cecco, G.; Tashjian, T.; Godoy-Carter, V.; Chai, Y. A decrease in serine levels during growth transition triggers biofilm formation in bacillus subtilis. J. Bacteriol. 2019, 201, e00155-19. [Google Scholar] [CrossRef]
- Prüss, B.M.; Nelms, J.M.; Park, C.; Wolfe, A.J. Mutations in NADH:ubiquinone oxidoreductase of Escherichia coli affect growth on mixed amino acids. J. Bacteriol. 1994, 176, 2143–2150. [Google Scholar] [CrossRef]
- Zhang, X.; Newman, E. Deficiency in l-serine deaminase results in abnormal growth and cell division of Escherichia coli K-12. Mol. Microbiol. 2008, 69, 870–881. [Google Scholar] [CrossRef]
- Sezonov, G.; Joseleau-Petit, D.; Ari, R. Escherichia coli Physiology in Luria-Bertani Broth. J. Bacteriol. 2007, 189, 8746. [Google Scholar] [CrossRef]
- Hofmann, J.D.; Otto, A.; Berges, M.; Biedendieck, R.; Michel, A.M.; Becher, D.; Jahn, D.; Neumann-Schaal, M. Metabolic reprogramming of clostridioides difficile during the stationary phase with the induction of toxin production. Front. Microbiol. 2018, 9, 1970. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altay, O.; Zhang, C.; Turkez, H.; Nielsen, J.; Uhlén, M.; Mardinoglu, A. Revealing the Metabolic Alterations during Biofilm Development of Burkholderia cenocepacia Based on Genome-Scale Metabolic Modeling. Metabolites 2021, 11, 221. https://doi.org/10.3390/metabo11040221
Altay O, Zhang C, Turkez H, Nielsen J, Uhlén M, Mardinoglu A. Revealing the Metabolic Alterations during Biofilm Development of Burkholderia cenocepacia Based on Genome-Scale Metabolic Modeling. Metabolites. 2021; 11(4):221. https://doi.org/10.3390/metabo11040221
Chicago/Turabian StyleAltay, Ozlem, Cheng Zhang, Hasan Turkez, Jens Nielsen, Mathias Uhlén, and Adil Mardinoglu. 2021. "Revealing the Metabolic Alterations during Biofilm Development of Burkholderia cenocepacia Based on Genome-Scale Metabolic Modeling" Metabolites 11, no. 4: 221. https://doi.org/10.3390/metabo11040221
APA StyleAltay, O., Zhang, C., Turkez, H., Nielsen, J., Uhlén, M., & Mardinoglu, A. (2021). Revealing the Metabolic Alterations during Biofilm Development of Burkholderia cenocepacia Based on Genome-Scale Metabolic Modeling. Metabolites, 11(4), 221. https://doi.org/10.3390/metabo11040221