
The Metano Modeling Toolbox MMTB: An Intuitive, Web-Based Toolbox Introduced by Two Use Cases

 , , and
, , and
Abstract
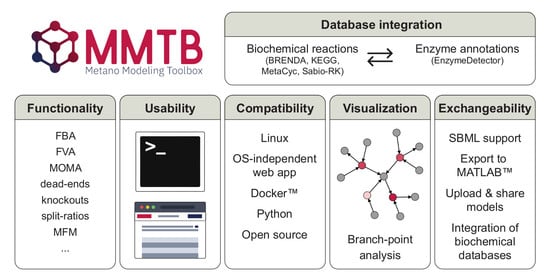
1. Introduction
2. Results
2.1. Metano Standalone Toolbox
2.2. MMTB Website
2.3. The Metabolic Model of Corynebacterium Glutamicum
2.4. The Metabolic Model iPin571 and Biological Implications
3. Discussion
4. Materials and Methods
4.1. Strains and Growth Conditions
4.2. The Metabolic Model iMG481
4.3. Reconstruction of the Metabolic Model iPin571
4.4. Algorithm
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raman, K.; Chandra, N. Flux Balance Analysis of Biological Systems: Applications and Challenges. Brief. Bioinform. 2009, 10, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What Is Flux Balance Analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- García Sánchez, C.E.; Torres Sáez, R.G. Comparison and Analysis of Objective Functions in Flux Balance Analysis. Biotechnol. Prog. 2014, 30, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Segrè, D.; Vitkup, D.; Church, G.M. Analysis of Optimality in Natural and Perturbed Metabolic Networks. Proc. Natl. Acad. Sci. USA 2002, 99, 15112–15117. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, R.; Schilling, C.H. The Effects of Alternate Optimal Solutions in Constraint-Based Genome-Scale Metabolic Models. Metab. Eng. 2003, 5, 264–276. [Google Scholar] [CrossRef]
- Kim, P.-J.; Lee, D.-Y.; Kim, T.Y.; Lee, K.H.; Jeong, H.; Lee, S.Y.; Park, S. Metabolite Essentiality Elucidates Robustness of Escherichia Coli Metabolism. Proc. Natl. Acad. Sci. USA 2007, 104, 13638–13642. [Google Scholar] [CrossRef]
- Kim, T.Y.; Kim, H.U.; Lee, S.Y. Metabolite-Centric Approaches for the Discovery of Antibacterials Using Genome-Scale Metabolic Networks. Metab. Eng. 2010, 12, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, D.C.; Filipp, F.V.; Bordbar, A.; Jensen, K.; Smith, J.W.; Herrgard, M.J.; Mo, M.L.; Palsson, B.O. Pharmacogenomic and Clinical Data Link Non-Pharmacokinetic Metabolic Dysregulation to Drug Side Effect Pathogenesis. Nat. Commun. 2015, 6, 7101. [Google Scholar] [CrossRef]
- Fong, S.S.; Marciniak, J.Y.; Palsson, B.Ø. Description and Interpretation of Adaptive Evolution of Escherichia Coli K-12 MG1655 by Using a Genome-Scale In Silico Metabolic Model. J. Bacteriol. 2003, 185, 6400–6408. [Google Scholar] [CrossRef] [PubMed]
- Drüppel, K.; Hensler, M.; Trautwein, K.; Koßmehl, S.; Wöhlbrand, L.; Schmidt-Hohagen, K.; Ulbrich, M.; Bergen, N.; Meier-Kolthoff, J.P.; Göker, M.; et al. Pathways and Substrate-Specific Regulation of Amino Acid Degradation in Phaeobacter Inhibens DSM 17395 (Archetype of the Marine Roseobacter Clade). Environ. Microbiol. 2014, 16, 218–238. [Google Scholar] [CrossRef]
- Wiegmann, K.; Hensler, M.; Wöhlbrand, L.; Ulbrich, M.; Schomburg, D.; Rabus, R. Carbohydrate Catabolism in Phaeobacter Inhibens DSM 17395, a Member of the Marine Roseobacter Clade. Appl. Environ. Microbiol. 2014, 80, 4725–4737. [Google Scholar] [CrossRef][Green Version]
- Zech, H.; Hensler, M.; Koßmehl, S.; Drüppel, K.; Wöhlbrand, L.; Trautwein, K.; Colby, T.; Schmidt, J.; Reinhardt, R.; Schmidt-Hohagen, K.; et al. Dynamics of Amino Acid Utilization in Phaeobacter Inhibens DSM 17395. Proteomics 2013, 13, 2869–2885. [Google Scholar] [CrossRef]
- Zech, H.; Hensler, M.; Koßmehl, S.; Drüppel, K.; Wöhlbrand, L.; Trautwein, K.; Hulsch, R.; Maschmann, U.; Colby, T.; Schmidt, J.; et al. Adaptation of Phaeobacter Inhibens DSM 17395 to Growth with Complex Nutrients. Proteomics 2013, 13, 2851–2868. [Google Scholar] [CrossRef]
- Zech, H.; Thole, S.; Schreiber, K.; Kalhöfer, D.; Voget, S.; Brinkhoff, T.; Simon, M.; Schomburg, D.; Rabus, R. Growth Phase-Dependent Global Protein and Metabolite Profiles of Phaeobacter Gallaeciensis Strain DSM 17395, a Member of the Marine Roseobacter-Clade. Proteomics 2009, 9, 3677–3697. [Google Scholar] [CrossRef] [PubMed]
- Thole, S.; Kalhoefer, D.; Voget, S.; Berger, M.; Engelhardt, T.; Liesegang, H.; Wollherr, A.; Kjelleberg, S.; Daniel, R.; Simon, M.; et al. Phaeobacter Gallaeciensis Genomes from Globally Opposite Locations Reveal High Similarity of Adaptation to Surface Life. ISME J. 2012, 6, 2229–2244. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.; Frank, O.; Göker, M.; Pradella, S. Extrachromosomal, Extraordinary and Essential--the Plasmids of the Roseobacter Clade. Appl. Microbiol. Biotechnol. 2013, 97, 2805–2815. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Neumann, A.; Schulz, S.; Simon, M.; Brinkhoff, T. Tropodithietic Acid Production in Phaeobacter Gallaeciensis Is Regulated by N-Acyl Homoserine Lactone-Mediated Quorum Sensing. J. Bacteriol. 2011, 193, 6576–6585. [Google Scholar] [CrossRef]
- Brock, N.L.; Nikolay, A.; Dickschat, J.S. Biosynthesis of the Antibiotic Tropodithietic Acid by the Marine Bacterium Phaeobacter Inhibens. Chem. Commun. Camb. Engl. 2014, 50, 5487–5489. [Google Scholar] [CrossRef] [PubMed]
- Trautwein, K.; Will, S.E.; Hulsch, R.; Maschmann, U.; Wiegmann, K.; Hensler, M.; Michael, V.; Ruppersberg, H.; Wünsch, D.; Feenders, C.; et al. Native Plasmids Restrict Growth of Phaeobacter Inhibens DSM 17395: Energetic Costs of Plasmids Assessed by Quantitative Physiological Analyses. Environ. Microbiol. 2016, 18, 4817–4829. [Google Scholar] [CrossRef]
- Will, S.E.; Neumann-Schaal, M.; Heydorn, R.L.; Bartling, P.; Petersen, J.; Schomburg, D. The Limits to Growth—Energetic Burden of the Endogenous Antibiotic Tropodithietic Acid in Phaeobacter Inhibens DSM 17395. PLoS ONE 2017, 12, e0177295. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.Z.; Wang, R.; Gitai, Z.; Seyedsayamdost, M.R. Mode of Action and Resistance Studies Unveil New Roles for Tropodithietic Acid as an Anticancer Agent and the γ-Glutamyl Cycle as a Proton Sink. Proc. Natl. Acad. Sci. USA 2016, 113, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, S.; Nakayama, K.; Akita, S. Taxonomical Study of Glutamic Acid Accumulating Bacteria, Micrococcus Glutamicus Nov. Sp. Bull. Agric. Chem. Soc. Jpn. 1958, 22, 176–185. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Na, Y.-A.; Kim, E.; Lee, H.S.; Kim, P. The Actinobacterium Corynebacterium Glutamicum, an Industrial Workhorse. J. Microbiol. Biotechnol. 2016, 26, 807–822. [Google Scholar] [CrossRef]
- Schellenberger, J.; Que, R.; Fleming, R.M.T.; Thiele, I.; Orth, J.D.; Feist, A.M.; Zielinski, D.C.; Bordbar, A.; Lewis, N.E.; Rahmanian, S.; et al. Quantitative Prediction of Cellular Metabolism with Constraint-Based Models: The COBRA Toolbox v2.0. Nat. Protoc. 2011, 6, 1290–1307. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, A.; Hoffmann, S.; Gerasch, A.; Gille, C.; Holzhütter, H.-G. FASIMU: Flexible Software for Flux-Balance Computation Series in Large Metabolic Networks. BMC Bioinform. 2011, 12, 28. [Google Scholar] [CrossRef]
- Rocha, I.; Maia, P.; Evangelista, P.; Vilaca, P.; Soares, S.; Pinto, J.P.; Nielsen, J.; Patil, K.R.; Ferreira, E.C.; Rocha, M. OptFlux: An Open-Source Software Platform for in Silico Metabolic Engineering. BMC Syst. Biol. 2010, 4, 45. [Google Scholar] [CrossRef]
- Boele, J.; Olivier, B.G.; Teusink, B. FAME, the Flux Analysis and Modeling Environment. BMC Syst. Biol. 2012, 6, 8. [Google Scholar] [CrossRef]
- Ebrahim, A.; Lerman, J.A.; Palsson, B.O.; Hyduke, D.R. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst. Biol. 2013, 7, 74. [Google Scholar] [CrossRef]
- Riemer, S.A.; Rex, R.; Schomburg, D. A Metabolite-Centric View on Flux Distributions in Genome-Scale Metabolic Models. BMC Syst. Biol. 2013, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, S.; Thiele, I. Computationally Efficient Flux Variability Analysis. BMC Bioinformatics 2010, 11, 489. [Google Scholar] [CrossRef] [PubMed]
- Ulas, T.; Riemer, S.A.; Zaparty, M.; Siebers, B.; Schomburg, D. Genome-Scale Reconstruction and Analysis of the Metabolic Network in the Hyperthermophilic Archaeon Sulfolobus Solfataricus. PLoS ONE 2012, 7, e43401. [Google Scholar] [CrossRef] [PubMed]
- Rex, R.; Bill, N.; Schmidt-Hohagen, K.; Schomburg, D. Swimming in Light: A Large-Scale Computational Analysis of the Metabolism of Dinoroseobacter Shibae. PLoS Comput. Biol. 2013, 9, e1003224. [Google Scholar] [CrossRef] [PubMed]
- Stark, H.; Wolf, J.; Albersmeier, A.; Pham, T.K.; Hofmann, J.D.; Siebers, B.; Kalinowski, J.; Wright, P.C.; Neumann-Schaal, M.; Schomburg, D. Oxidative Stickland Reactions in an Obligate Aerobic Organism—Amino Acid Catabolism in the Crenarchaeon Sulfolobus Solfataricus. FEBS J. 2017, 1–18. [Google Scholar] [CrossRef]
- Wolf, J.; Stark, H.; Fafenrot, K.; Albersmeier, A.; Meyer, B.H.; Hoffmann, L.; Shen, L.; Albaum, S.P.; Kouril, T.; Schmidt-Hohagen, K.; et al. A Systems Biology Approach Reveals Major Metabolic Changes in the Thermoacidophilic Archaeon Sulfolobus Solfataricus in Response to the Carbon Source L-Fucose versus D-Glucose. Mol. Microbiol. 2016, 102, 882–908. [Google Scholar] [CrossRef]
- Dannheim, H.; Will, S.E.; Schomburg, D.; Neumann-Schaal, M. Clostridioides Difficile 630Δ Erm in Silico and in Vivo—Quantitative Growth and Extensive Polysaccharide Secretion. FEBS Open Bio 2017, 7, 602–615. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Koblitz, J.; Albersmeier, A.; Kalinowski, J.; Siebers, B.; Schomburg, D.; Neumann-Schaal, M. Utilization of Phenol as Carbon Source by the Thermoacidophilic Archaeon Saccharolobus Solfataricus P2 Is Limited by Oxygen Supply and the Cellular Stress Response. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Duarte, N.C.; Herrgård, M.J.; Palsson, B.Ø. Reconstruction and Validation of Saccharomyces Cerevisiae IND750, a Fully Compartmentalized Genome-Scale Metabolic Model. Genome Res. 2004, 14, 1298–1309. [Google Scholar] [CrossRef]
- Hucka, M.; Hucka, M.; Bergmann, F.; Hoops, S.; Keating, S.; Sahle, S.; Schaff, J.; Smith, L.; Wilkinson, D. The Systems Biology Markup Language (SBML): Language Specification for Level 3 Version 1 Core. Nat. Preced. 2010. [Google Scholar] [CrossRef]
- Lang, M.; Stelzer, M.; Schomburg, D. BKM-React, an Integrated Biochemical Reaction Database. BMC Biochem. 2011, 12, 42. [Google Scholar] [CrossRef]
- Quester, S.; Schomburg, D. EnzymeDetector: An Integrated Enzyme Function Prediction Tool and Database. BMC Bioinformatics 2011, 12, 376. [Google Scholar] [CrossRef]
- Chang, A.; Jeske, L.; Ulbrich, S.; Hofmann, J.; Koblitz, J.; Schomburg, I.; Neumann-Schaal, M.; Jahn, D.; Schomburg, D. BRENDA, the ELIXIR Core Data Resource in 2021: New Developments and Updates. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New Perspectives on Genomes, Pathways, Diseases and Drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Caspi, R.; Foerster, H.; Fulcher, C.A.; Kaipa, P.; Krummenacker, M.; Latendresse, M.; Paley, S.; Rhee, S.Y.; Shearer, A.G.; Tissier, C.; et al. The MetaCyc Database of Metabolic Pathways and Enzymes and the BioCyc Collection of Pathway/Genome Databases. Nucleic Acids Res. 2008, 36, D623–D631. [Google Scholar] [CrossRef]
- Wittig, U.; Kania, R.; Golebiewski, M.; Rey, M.; Shi, L.; Jong, L.; Algaa, E.; Weidemann, A.; Sauer-Danzwith, H.; Mir, S.; et al. SABIO-RK--Database for Biochemical Reaction Kinetics. Nucleic Acids Res. 2012, 40, D790–D796. [Google Scholar] [CrossRef]
- Graf, M.; Zieringer, J.; Haas, T.; Nieß, A.; Blombach, B.; Takors, R. Physiological Response of Corynebacterium Glutamicum to Increasingly Nutrient-Rich Growth Conditions. Front. Microbiol. 2018, 9, 2058. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Brock, N.L.; Liesegang, H.; Dogs, M.; Preuth, I.; Simon, M.; Dickschat, J.S.; Brinkhoff, T. Genetic Analysis of the Upper Phenylacetate Catabolic Pathway in the Production of Tropodithietic Acid by Phaeobacter Gallaeciensis. Appl. Environ. Microbiol. 2012, 78, 3539–3551. [Google Scholar] [CrossRef] [PubMed]
- Zelle, E.; Nöh, K.; Wiechert, W. Growth and Production Capabilities of Corynebacterium glutamicum: Interrogating a Genome-scale Metabolic Network Model; Caister Academic Press: Poole, UK, 2015. [Google Scholar]
- Kjeldsen, K.R.; Nielsen, J. In Silico Genome-Scale Reconstruction and Validation of the Corynebacterium Glutamicum Metabolic Network. Biotechnol. Bioeng. 2009, 102, 583–597. [Google Scholar] [CrossRef]
- Mackie, A.; Keseler, I.M.; Nolan, L.; Karp, P.D.; Paulsen, I.T. Dead End Metabolites—Defining the Known Unknowns of the E. Coli Metabolic Network. PLoS ONE 2013, 8, e75210. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Feature | Metano | COBRA | FASIMU | OptFlux | FAME |
|---|---|---|---|---|---|
| FBA | + | + | + | + | + |
| FVA | + | + | + | + | + |
| MOMA | + | + | + | + | - |
| ROOM | - | - | + | + | - |
| MFM | + | - | - | - | - |
| Batch computation | + | - | + | - | - |
| OptKnock | - | + | - | + | - |
| Command line | + | + | + | - | - |
| GUI for modeling | - | - | - | + | - |
| Independence of commercial software | + | - | + | + | + |
| Dynamic visualization GUI | + | - | - | - | - |
| Static image | + | + | - | + | + |
| External visualization software | - | - | + | - | - |
| SBML import | + | + | + | + | + |
| SBML export | + | + | + | + | + |
| Text file import | + | - | + | + | - |
| MATLAB export | + | + | - | - | - |
| Linux | + | - | + | + | * |
| MacOS | + | + | + | - | * |
| Windows | - | + | - | + | * |
| Pathway import from relational database | + | - | - | - | + |
| Model verification | + | + | - | - | - |
| Automated gap filling | - | + | + | - | - |
| Amino Acid | Strain | Uptake Rate [mmol gCDW−1 h−1] | Growth Rate [h−1] | |
|---|---|---|---|---|
| Experiment | Model | |||
| Alanine | WT | 5.7 | 0.160 | 0.239 |
| Δ262 | 5.6 | 0.235 | 0.231 | |
| tdaE | 8.2 | 0.333 | 0.342 | |
| Phenylalanine | WT | 1.4 | 0.102 | 0.159 |
| Δ262 | 1.4 | 0.170 | 0.161 | |
| tdaE | 1.1 | 0.120 | 0.116 | |
| Leucine | WT | 0.9 | 0.063 | 0.062 |
| Δ262 | 1.4 | 0.104 | 0.103 | |
| tdaE | 1.5 | 0.123 | 0.114 | |
| Amino Acid | Strain | % Cell Dry Weight | % CO2 | % TDA |
|---|---|---|---|---|
| Alanine | WT | 34.1 | 51.1 | 14.8 |
| Δ262 | 51.0 | 49.0 | 0 | |
| tdaE | 52.5 | 47.5 | 0 | |
| Phenylalanine | WT | 31.6 | 49.5 | 18.9 |
| Δ262 | 49.5 | 50.5 | 0 | |
| tdaE | 49.6 | 50.4 | 0 | |
| Leucine | WT | 45.5 | 54.5 | 0 |
| Δ262 | 51.0 | 49.0 | 0 | |
| tdaE | 50.7 | 49.3 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koblitz, J.; Will, S.E.; Riemer, S.A.; Ulas, T.; Neumann-Schaal, M.; Schomburg, D. The Metano Modeling Toolbox MMTB: An Intuitive, Web-Based Toolbox Introduced by Two Use Cases. Metabolites 2021, 11, 113. https://doi.org/10.3390/metabo11020113
Koblitz J, Will SE, Riemer SA, Ulas T, Neumann-Schaal M, Schomburg D. The Metano Modeling Toolbox MMTB: An Intuitive, Web-Based Toolbox Introduced by Two Use Cases. Metabolites. 2021; 11(2):113. https://doi.org/10.3390/metabo11020113
Chicago/Turabian StyleKoblitz, Julia, Sabine Eva Will, S. Alexander Riemer, Thomas Ulas, Meina Neumann-Schaal, and Dietmar Schomburg. 2021. "The Metano Modeling Toolbox MMTB: An Intuitive, Web-Based Toolbox Introduced by Two Use Cases" Metabolites 11, no. 2: 113. https://doi.org/10.3390/metabo11020113
APA StyleKoblitz, J., Will, S. E., Riemer, S. A., Ulas, T., Neumann-Schaal, M., & Schomburg, D. (2021). The Metano Modeling Toolbox MMTB: An Intuitive, Web-Based Toolbox Introduced by Two Use Cases. Metabolites, 11(2), 113. https://doi.org/10.3390/metabo11020113