
The Plasticity of Pancreatic β-Cells

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
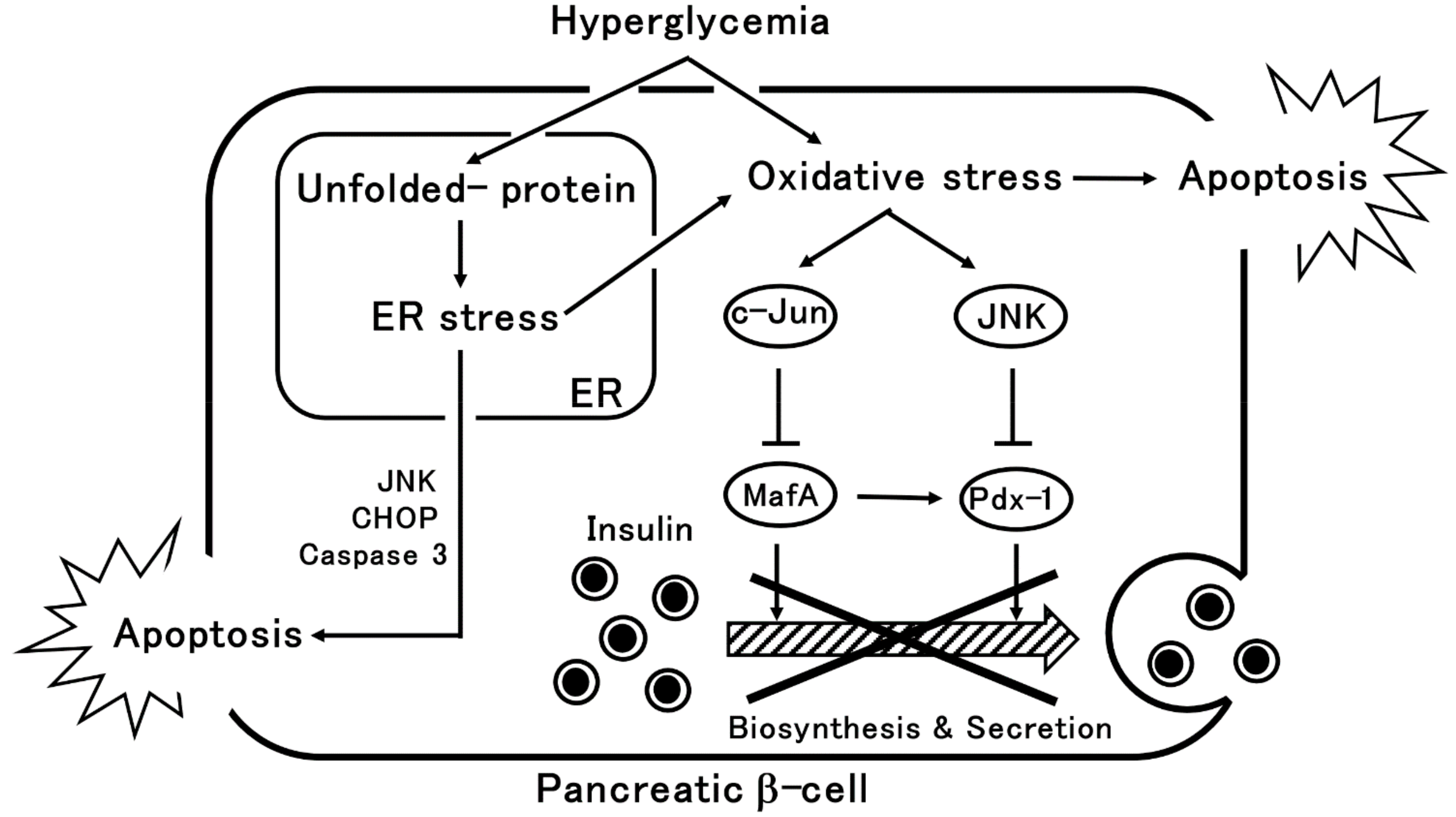
2. β-Cells Dysfunction via Oxidative Stress or ER Stress
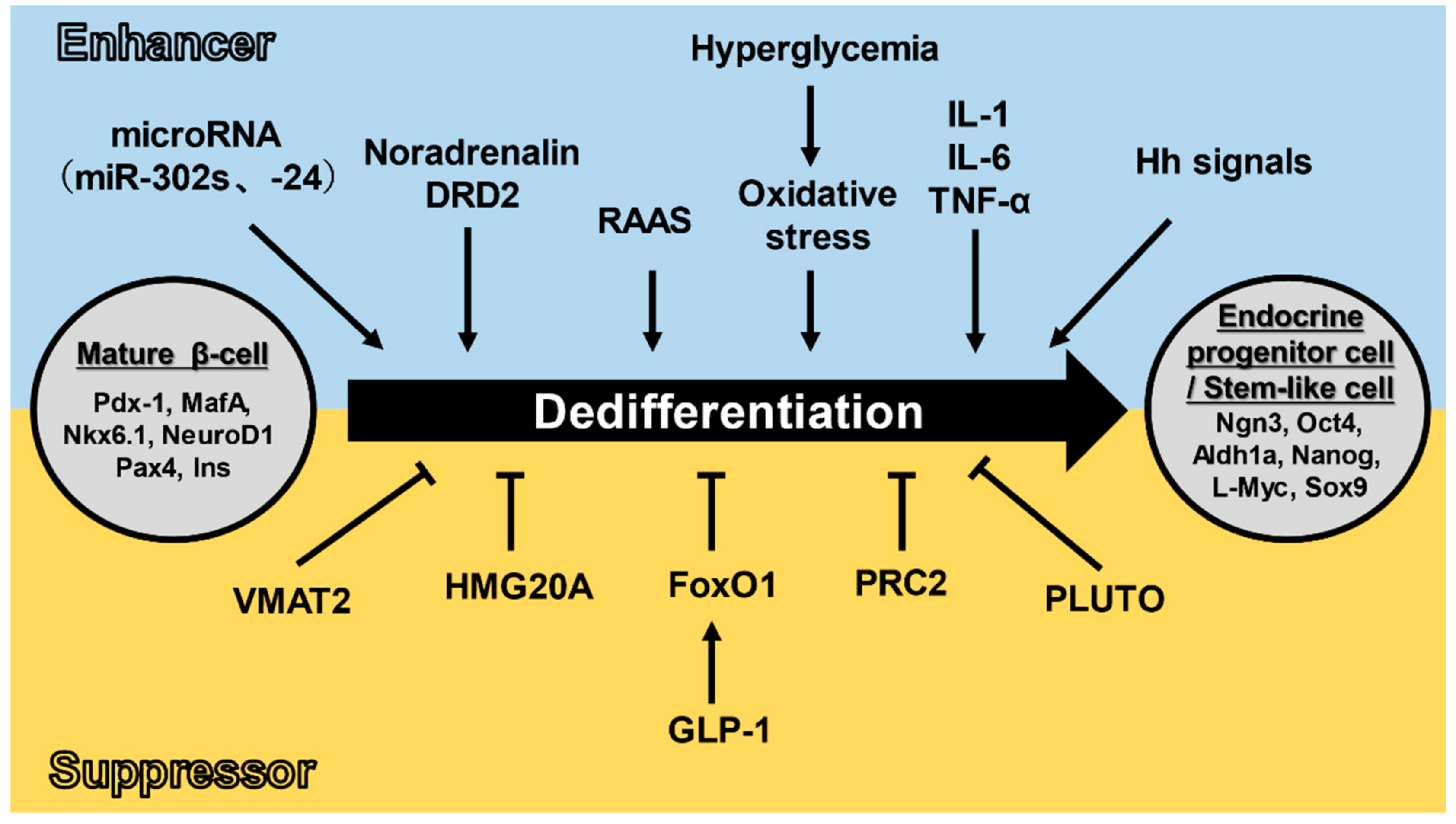
3. Pancreatic β-Cell Dedifferentiation and Transdifferentiation
3.1. The History of Dedifferentiation
3.2. Dedifferentiation Mechanisms
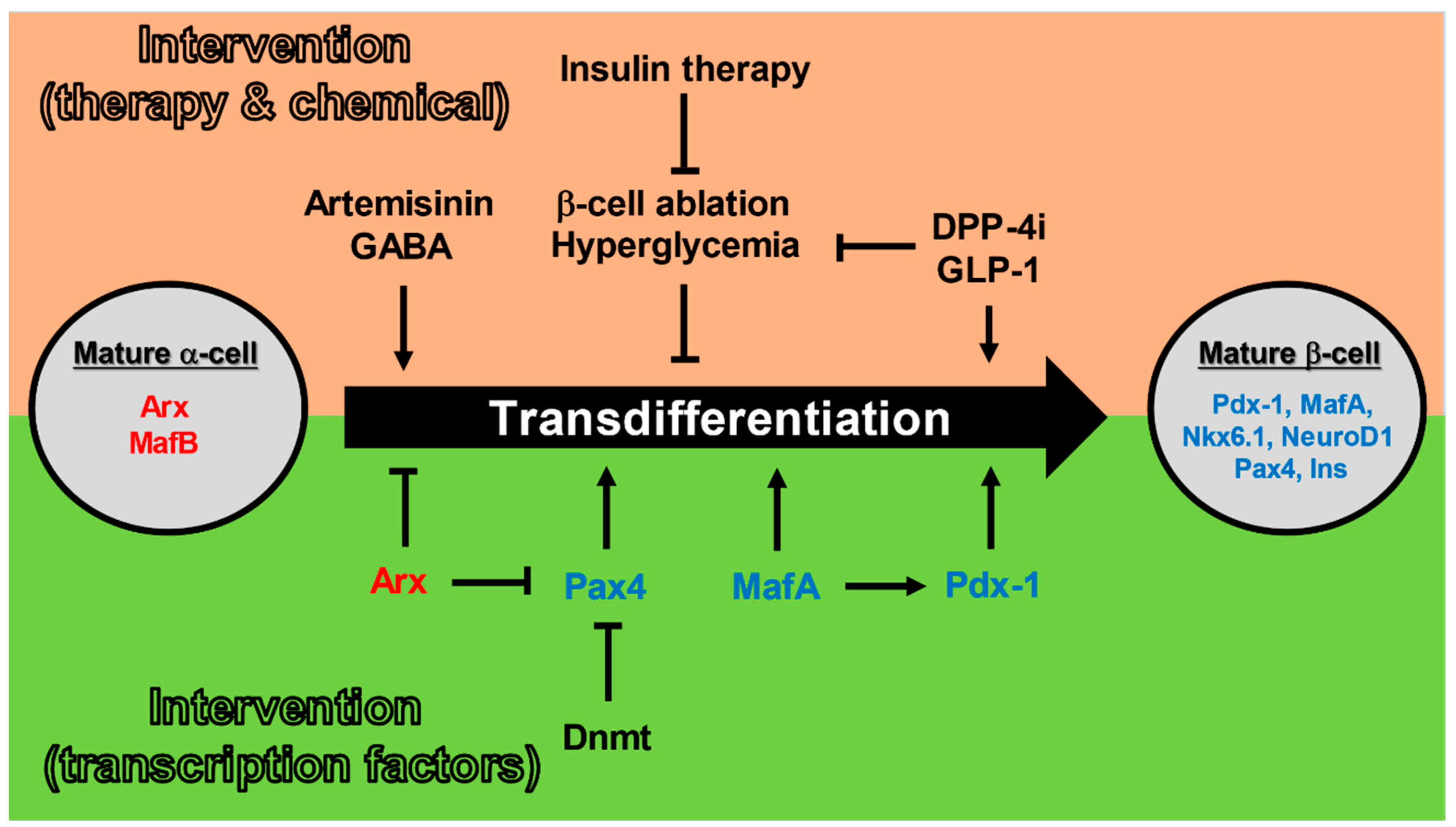
3.3. Pancreatic α-Cells to β-Cells Transdifferentiation
4. Potential Therapies Utilizing Differentiation and Transdifferentiation
5. Final Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Da Silva Xavier, G. The Cells of the Islets of Langerhans. J. Clin. Med. 2018, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Cohrs, C.M.; Stertmann, J.; Bozsak, R.; Speier, S. Human beta cell mass and function in diabetes: Recent advances in knowledge and technologies to understand disease pathogenesis. Mol. Metab. 2017, 6, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of beta-Cell Dedifferentiation in Human Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef]
- Scheuner, D.; Kaufman, R.J. The unfolded protein response: A pathway that links insulin demand with beta-cell failure and diabetes. Endocr. Rev. 2008, 29, 317–333. [Google Scholar] [CrossRef]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.Y.; Jun, H.S. Fatty Acid-Induced Lipotoxicity in Pancreatic Beta-Cells During Development of Type 2 Diabetes. Front. Endocrinol. 2018, 9, 384. [Google Scholar] [CrossRef] [PubMed]
- Hecker, M.; Wagner, A.H. Role of protein carbonylation in diabetes. J. Inherit. Metab. Dis. 2018, 41, 29–38. [Google Scholar] [CrossRef]
- Asthana, S.; Mallick, B.; Alexandrescu, A.T.; Jha, S. IAPP in type II diabetes: Basic research on structure, molecular interactions, and disease mechanisms suggests potential intervention strategies. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1765–1782. [Google Scholar] [CrossRef]
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B., Jr.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of beta Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab. 2019, 30, 129–142.e4. [Google Scholar] [CrossRef] [PubMed]
- Dor, Y.; Glaser, B. β-cell dedifferentiation and type 2 diabetes. N. Engl. J. Med. 2013, 368, 572–573. [Google Scholar] [CrossRef]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef]
- Xiao, X.; Guo, P.; Shiota, C.; Zhang, T.; Coudriet, G.M.; Fischbach, S.; Prasadan, K.; Fusco, J.; Ramachandran, S.; Witkowski, P.; et al. Endogenous Reprogramming of Alpha Cells into Beta Cells, Induced by Viral Gene Therapy, Reverses Autoimmune Diabetes. Cell Stem Cell 2018, 22, 78–90.e74. [Google Scholar] [CrossRef] [PubMed]
- Tiedge, M.; Lortz, S.; Drinkgern, J.; Lenzen, S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 1997, 46, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Taka-aki Matsuoka, Y.K.; Watada, H.; Kaneto, H.; Kishimoto, M.; Umayahara, Y.; Fujitani, Y.; Kamada, T.; Kawamori, R.; Yamasaki, Y. Glycation-dependent, Reactive Oxygen Species–mediated Suppression of the Insulin Gene Promoter Activity in HIT Cells. J. Clin. Investig. 1997, 99, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Kajimoto, Y.; Miyagawa, J.; Matsuoka, T.; Fujitani, Y.; Umayahara, Y.; Hanafusa, T.; Matsuzawa, Y.; Yamasaki, Y.; Hori, M. Beneficial effects of antioxidants in diabetes: Possible protection of pancreatic beta-cells against glucose toxicity. Diabetes 1999, 48, 2398–2406. [Google Scholar] [CrossRef]
- Tanaka, Y.; Gleason, C.E.; Tran, P.O.T.; Harmon, J.S.; Robertson, R.P. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc. Natl. Acad. Sci. USA 1999, 96, 10857–10862. [Google Scholar] [CrossRef]
- Kitamura, Y.I.; Kitamura, T.; Kruse, J.P.; Raum, J.C.; Stein, R.; Gu, W.; Accili, D. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005, 2, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Polonsky, K.S. Pdx1 and other factors that regulate pancreatic beta-cell survival. Diabetes Obes. Metab. 2009, 11 (Suppl. 4), 30–37. [Google Scholar] [CrossRef]
- Kawamori, D.; Kaneto, H.; Nakatani, Y.; Matsuoka, T.A.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. The forkhead transcription factor Foxo1 bridges the JNK pathway and the transcription factor PDX-1 through its intracellular translocation. J. Biol. Chem. 2006, 281, 1091–1098. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Miyatsuka, T.; Sasaki, S.; Miyashita, K.; Kubo, F.; Shimo, N.; Takebe, S.; Watada, H.; Kaneto, H.; Matsuoka, T.A.; et al. Preserving expression of Pdx1 improves beta-cell failure in diabetic mice. Biochem. Biophys. Res. Commun. 2017, 483, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, T.A.; Kaneto, H.; Miyatsuka, T.; Yamamoto, T.; Yamamoto, K.; Kato, K.; Shimomura, I.; Stein, R.; Matsuhisa, M. Regulation of MafA expression in pancreatic beta-cells in db/db mice with diabetes. Diabetes 2010, 59, 1709–1720. [Google Scholar] [CrossRef]
- Matsuoka, T.A.; Kaneto, H.; Kawashima, S.; Miyatsuka, T.; Tochino, Y.; Yoshikawa, A.; Imagawa, A.; Miyazaki, J.; Gannon, M.; Stein, R.; et al. Preserving Mafa expression in diabetic islet beta-cells improves glycemic control in vivo. J. Biol. Chem. 2015, 290, 7647–7657. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, T.A.; Kaneto, H.; Stein, R.; Miyatsuka, T.; Kawamori, D.; Henderson, E.; Kojima, I.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. MafA regulates expression of genes important to islet beta-cell function. Mol. Endocrinol. 2007, 21, 2764–2774. [Google Scholar] [CrossRef]
- Wali, J.A.; Masters, S.L.; Thomas, H.E. Linking metabolic abnormalities to apoptotic pathways in Beta cells in type 2 diabetes. Cells 2013, 2, 266–283. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.G.; Gromada, J.; Urano, F. Endoplasmic reticulum stress and pancreatic beta-cell death. Trends Endocrinol. Metab. 2011, 22, 266–274. [Google Scholar] [CrossRef]
- Ghosh, R.; Colon-Negron, K.; Papa, F.R. Endoplasmic reticulum stress, degeneration of pancreatic islet beta-cells, and therapeutic modulation of the unfolded protein response in diabetes. Mol. Metab. 2019, 27S, S60–S68. [Google Scholar] [CrossRef] [PubMed]
- Gershengorn, M.C.; Hardikar, A.A.; Wei, C.; Geras-Raaka, E.; Marcus-Samuels, B.; Raaka, B.M. Epithelial-to-Mesenchymal Transition Generates Proliferative Human Islet Precursor Cells. Science 2004, 306, 2261–2264. [Google Scholar] [CrossRef]
- Weinberg, N.; Ouziel-Yahalom, L.; Knoller, S.; Efrat, S.; Dor, Y. Lineage tracing evidence for in vitro dedifferentiation but rare proliferation of mouse pancreatic beta-cells. Diabetes 2007, 56, 1299–1304. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; York, N.W.; Nichols, C.G.; Remedi, M.S. Pancreatic beta cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab. 2014, 19, 872–882. [Google Scholar] [CrossRef]
- Hunter, C.S.; Stein, R.W. Evidence for Loss in Identity, De-Differentiation, and Trans-Differentiation of Islet beta-Cells in Type 2 Diabetes. Front. Genet. 2017, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Han, X. Death versus dedifferentiation: The molecular bases of beta cell mass reduction in type 2 diabetes. Semin. Cell Dev. Biol. 2020, 103, 76–82. [Google Scholar] [CrossRef]
- Guo, S.; Dai, C.; Guo, M.; Taylor, B.; Harmon, J.S.; Sander, M.; Robertson, R.P.; Powers, A.C.; Stein, R. Inactivation of specific beta cell transcription factors in type 2 diabetes. J. Clin. Investig. 2013, 123, 3305–3316. [Google Scholar] [CrossRef] [PubMed]
- Kjørholt, C.; Mia, C.Å.; Trevor, J.B.; Laybutt, A.D.R. Chronic Hyperglycemia, Independent of Plasma Lipid Levels, Is Sufficient for the Loss of β-Cell Differentiation and Secretory Function in the db/db Mouse Model of Diabetes. Diabetes 2005, 54, 2755–2763. [Google Scholar] [CrossRef] [PubMed]
- Neelankal John, A.; Ram, R.; Jiang, F.X. RNA-Seq Analysis of Islets to Characterise the Dedifferentiation in Type 2 Diabetes Model Mice db/db. Endocr. Pathol. 2018, 29, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, T.M.; Dror, E.; Schulze, F.; Traub, S.; Berishvili, E.; Barbieux, C.; Boni-Schnetzler, M.; Donath, M.Y. The Role of Inflammation in beta-cell Dedifferentiation. Sci. Rep. 2017, 7, 6285. [Google Scholar] [CrossRef]
- Burke, S.J.; Batdorf, H.M.; Burk, D.H.; Martin, T.M.; Mendoza, T.; Stadler, K.; Alami, W.; Karlstad, M.D.; Robson, M.J.; Blakely, R.D.; et al. Pancreatic deletion of the interleukin-1 receptor disrupts whole body glucose homeostasis and promotes islet beta-cell de-differentiation. Mol. Metab. 2018, 14, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Landsman, L.; Parent, A.; Hebrok, M. Elevated Hedgehog/Gli signaling causes beta-cell dedifferentiation in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 17010–17015. [Google Scholar] [CrossRef]
- Chen, H.; Zhou, W.; Ruan, Y.; Yang, L.; Xu, N.; Chen, R.; Yang, R.; Sun, J.; Zhang, Z. Reversal of angiotensin ll-induced beta-cell dedifferentiation via inhibition of NF-kappab signaling. Mol. Med. 2018, 24, 43. [Google Scholar] [CrossRef]
- Xuan, X.; Gao, F.; Ma, X.; Huang, C.; Wang, Y.; Deng, H.; Wang, S.; Li, W.; Yuan, L. Activation of ACE2/angiotensin (1-7) attenuates pancreatic β cell dedifferentiation in a high-fat-diet mouse model. Metabolism 2018, 81, 83–96. [Google Scholar] [CrossRef]
- Sebastiani, G.; Grieco, G.E.; Brusco, N.; Ventriglia, G.; Formichi, C.; Marselli, L.; Marchetti, P.; Dotta, F. MicroRNA Expression Analysis of In Vitro Dedifferentiated Human Pancreatic Islet Cells Reveals the Activation of the Pluripotency-Related MicroRNA Cluster miR-302s. Int. J. Mol. Sci. 2018, 19, 1170. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Sun, Y.; Zhou, Y.; Zhang, Y.; Zhang, T.; Li, Y.; You, W.; Chang, X.; Yuan, L.; Han, X. MicroRNA-24 promotes pancreatic beta cells toward dedifferentiation to avoid endoplasmic reticulum stress-induced apoptosis. J. Mol. Cell. Biol. 2019, 11, 747–760. [Google Scholar] [CrossRef]
- Cinti, F.; Mezza, T.; Severi, I.; Suleiman, M.; Cefalo, C.M.A.; Sorice, G.P.; Moffa, S.; Impronta, F.; Quero, G.; Alfieri, S.; et al. Noradrenergic fibers are associated with beta-cell dedifferentiation and impaired beta-cell function in humans. Metabolism 2021, 114, 154414. [Google Scholar] [CrossRef] [PubMed]
- Sakano, D.; Choi, S.; Kataoka, M.; Shiraki, N.; Uesugi, M.; Kume, K.; Kume, S. Dopamine D2 Receptor-Mediated Regulation of Pancreatic beta Cell Mass. Stem Cell Rep. 2016, 7, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.T.; Heyne, S.; Dror, E.; Casas, E.; Leonhardt, L.; Boenke, T.; Yang, C.H.; Sagar; Arrigoni, L.; Dalgaard, K.; et al. The Polycomb-Dependent Epigenome Controls beta Cell Dysfunction, Dedifferentiation, and Diabetes. Cell Metab. 2018, 27, 1294–1308. [Google Scholar] [CrossRef]
- Mellado-Gil, J.M.; Fuente-Martin, E.; Lorenzo, P.I.; Cobo-Vuilleumier, N.; Lopez-Noriega, L.; Martin-Montalvo, A.; Gomez, I.G.H.; Ceballos-Chavez, M.; Gomez-Jaramillo, L.; Campos-Caro, A.; et al. The type 2 diabetes-associated HMG20A gene is mandatory for islet beta cell functional maturity. Cell Death Dis. 2018, 9, 279. [Google Scholar] [CrossRef] [PubMed]
- Akerman, I.; Tu, Z.; Beucher, A.; Rolando, D.M.Y.; Sauty-Colace, C.; Benazra, M.; Nakic, N.; Yang, J.; Wang, H.; Pasquali, L.; et al. Human Pancreatic beta Cell lncRNAs Control Cell-Specific Regulatory Networks. Cell Metab. 2017, 25, 400–411. [Google Scholar] [CrossRef]
- You, L.; Wang, N.; Yin, D.; Wang, L.; Jin, F.; Zhu, Y.; Yuan, Q.; De, W. Downregulation of Long Noncoding RNA Meg3 Affects Insulin Synthesis and Secretion in Mouse Pancreatic Beta Cells. J. Cell Physiol. 2016, 231, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Sakano, D.; Uefune, F.; Tokuma, H.; Sonoda, Y.; Matsuura, K.; Takeda, N.; Nakagata, N.; Kume, K.; Shiraki, N.; Kume, S. VMAT2 Safeguards beta-Cells against Dopamine Cytotoxicity Under High-Fat Diet-Induced Stress. Diabetes 2020, 69, 2377–2391. [Google Scholar] [CrossRef] [PubMed]
- Rattanaamnuaychai, P.; Roshorm, Y.M.; Wilasrusmee, C.; Proprom, N.; Ongphiphadhanakul, B.; Talchai, S.C. Direct suppression of human islet dedifferentiation, progenitor genes, but not epithelial to mesenchymal transition by liraglutide. Heliyon 2020, 6, e04951. [Google Scholar] [CrossRef]
- Kitamura, T.; Kitamura, Y.I.; Funahashi, Y.; Shawber, C.J.; Castrillon, D.H.; Kollipara, R.; DePinho, R.A.; Kitajewski, J.; Accili, D. A Foxo/Notch pathway controls myogenic differentiation and fiber type specification. J. Clin. Investig. 2007, 117, 2477–2485. [Google Scholar] [CrossRef]
- Basile, G.; Kulkarni, R.N.; Morgan, N.G. How, When, and Where Do Human beta-Cells Regenerate? Curr. Diab. Rep. 2019, 19, 48. [Google Scholar] [CrossRef]
- Thorel, F.; Nepote, V.; Avril, I.; Kohno, K.; Desgraz, R.; Chera, S.; Herrera, P.L. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 2010, 464, 1149–1154. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lee, C.; Choung, J.S.; Jung, H.S.; Jun, H.S. Glucagon-Like Peptide 1 Increases beta-Cell Regeneration by Promoting alpha- to beta-Cell Transdifferentiation. Diabetes 2018, 67, 2601–2614. [Google Scholar] [CrossRef] [PubMed]
- Tanday, N.; Flatt, P.R.; Irwin, N.; Moffett, R.C. Liraglutide and sitagliptin counter beta- to alpha-cell transdifferentiation in diabetes. J. Endocrinol. 2020, 245, 53–64. [Google Scholar] [CrossRef]
- Ben-Othman, N.; Vieira, A.; Courtney, M.; Record, F.; Gjernes, E.; Avolio, F.; Hadzic, B.; Druelle, N.; Napolitano, T.; Navarro-Sanz, S.; et al. Long-Term GABA Administration Induces Alpha Cell-Mediated Beta-like Cell Neogenesis. Cell 2017, 168, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Casteels, T.; Frogne, T.; Ingvorsen, C.; Honore, C.; Courtney, M.; Huber, K.V.M.; Schmitner, N.; Kimmel, R.A.; Romanov, R.A.; et al. Artemisinins Target GABAA Receptor Signaling and Impair alpha Cell Identity. Cell 2017, 168, 86–100.e115. [Google Scholar] [CrossRef]
- Hirotaka Watada, Y.K.; Miyagawa, J.; Hanafusa, T.; Hamaguchi, K.; Matsuoka, T.; Yamamoto, K.; Matsuzawa, Y.; Kawamori, R.; Yamasaki, Y. PDX-1 Induces Insulin and Glucokinase Gene Expressions in aTCl Clone 6 Cells in the Presence of Betacellulin. Diabetes 1996, 45, 1826–1831. [Google Scholar]
- Collombat, P.; Xu, X.; Ravassard, P.; Sosa-Pineda, B.; Dussaud, S.; Billestrup, N.; Madsen, O.D.; Serup, P.; Heimberg, H.; Mansouri, A. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell 2009, 138, 449–462. [Google Scholar] [CrossRef]
- Al-Hasani, K.; Pfeifer, A.; Courtney, M.; Ben-Othman, N.; Gjernes, E.; Vieira, A.; Druelle, N.; Avolio, F.; Ravassard, P.; Leuckx, G.; et al. Adult duct-lining cells can reprogram into beta-like cells able to counter repeated cycles of toxin-induced diabetes. Dev. Cell 2013, 26, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Courtney, M.; Gjernes, E.; Druelle, N.; Ravaud, C.; Vieira, A.; Ben-Othman, N.; Pfeifer, A.; Avolio, F.; Leuckx, G.; Lacas-Gervais, S.; et al. The inactivation of Arx in pancreatic alpha-cells triggers their neogenesis and conversion into functional beta-like cells. PLoS Genet. 2013, 9, e1003934. [Google Scholar] [CrossRef]
- Chakravarthy, H.; Gu, X.; Enge, M.; Dai, X.; Wang, Y.; Damond, N.; Downie, C.; Liu, K.; Wang, J.; Xing, Y.; et al. Converting Adult Pancreatic Islet alpha Cells into beta Cells by Targeting Both Dnmt1 and Arx. Cell Metab. 2017, 25, 622–634. [Google Scholar] [CrossRef]
- Matsuoka, T.A.; Kawashima, S.; Miyatsuka, T.; Sasaki, S.; Shimo, N.; Katakami, N.; Kawamori, D.; Takebe, S.; Herrera, P.L.; Kaneto, H.; et al. Mafa Enables Pdx1 to Effectively Convert Pancreatic Islet Progenitors and Committed Islet alpha-Cells Into beta-Cells In Vivo. Diabetes 2017, 66, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Kenneth, S.; Zaret, M.F.W. Extreme makeover of pancreatic α-cells. Nature 2010, 464, 1132–1133. [Google Scholar]
- Pagliuca, F.W.; Millman, J.R.; Gurtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic beta cells in vitro. Cell 2014, 159, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Engin, F.; Yermalovich, A.; Nguyen, T.; Hummasti, S.; Fu, W.; Eizirik, D.L.; Mathis, D.; Hotamisligil, G.S. Restoration of the unfolded protein response in pancreatic beta cells protects mice against type 1 diabetes. Sci. Transl. Med. 2013, 5, 211ra156. [Google Scholar] [CrossRef]
- Vial, G.; Chauvin, M.A.; Bendridi, N.; Durand, A.; Meugnier, E.; Madec, A.M.; Bernoud-Hubac, N.; Pais de Barros, J.P.; Fontaine, E.; Acquaviva, C.; et al. Imeglimin normalizes glucose tolerance and insulin sensitivity and improves mitochondrial function in liver of a high-fat, high-sucrose diet mice model. Diabetes 2015, 64, 2254–2264. [Google Scholar] [CrossRef] [PubMed]
- Hallakou-Bozec, S.; Kergoat, M.; Moller, D.E.; Bolze, S. Imeglimin preserves islet β-cell mass in Type 2 diabetic ZDF rats. Endocrinol. Diabetes Metab. 2020. [Google Scholar] [CrossRef]
- Hallakou-Bozec, S.; Vial, G.; Kergoat, M.; Fouqueray, P.; Bolze, S.; Borel, A.L.; Fontaine, E.; Moller, D.E. Mechanism of action of Imeglimin: A novel therapeutic agent for type 2 diabetes. Diabetes Obes. Metab. 2021, 23, 664–673. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honzawa, N.; Fujimoto, K. The Plasticity of Pancreatic β-Cells. Metabolites 2021, 11, 218. https://doi.org/10.3390/metabo11040218
Honzawa N, Fujimoto K. The Plasticity of Pancreatic β-Cells. Metabolites. 2021; 11(4):218. https://doi.org/10.3390/metabo11040218
Chicago/Turabian StyleHonzawa, Norikiyo, and Kei Fujimoto. 2021. "The Plasticity of Pancreatic β-Cells" Metabolites 11, no. 4: 218. https://doi.org/10.3390/metabo11040218
APA StyleHonzawa, N., & Fujimoto, K. (2021). The Plasticity of Pancreatic β-Cells. Metabolites, 11(4), 218. https://doi.org/10.3390/metabo11040218