
Protein Acetylation at the Interface of Genetics, Epigenetics and Environment in Cancer


Abstract
:1. Introduction
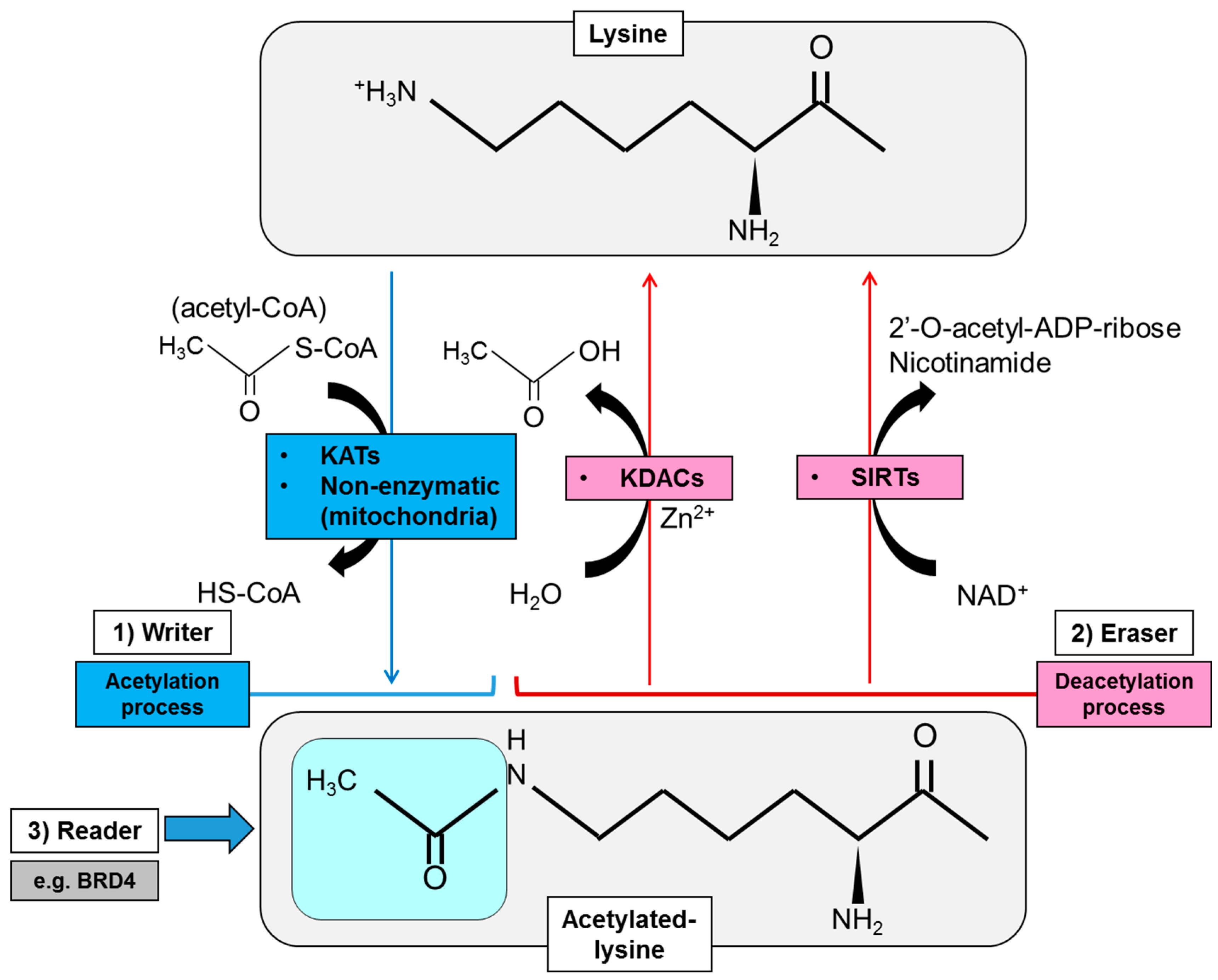
2. Regulatory Mode of Protein Lysine Acetylation
2.1. Acetylation Enzymes: Writers, Erasers and Readers of Lysine Acetylation
2.1.1. Writers of Lysine Acetylation
2.1.2. Erasers of Lysine Acetylation
2.1.3. Readers of Lysine Acetylation
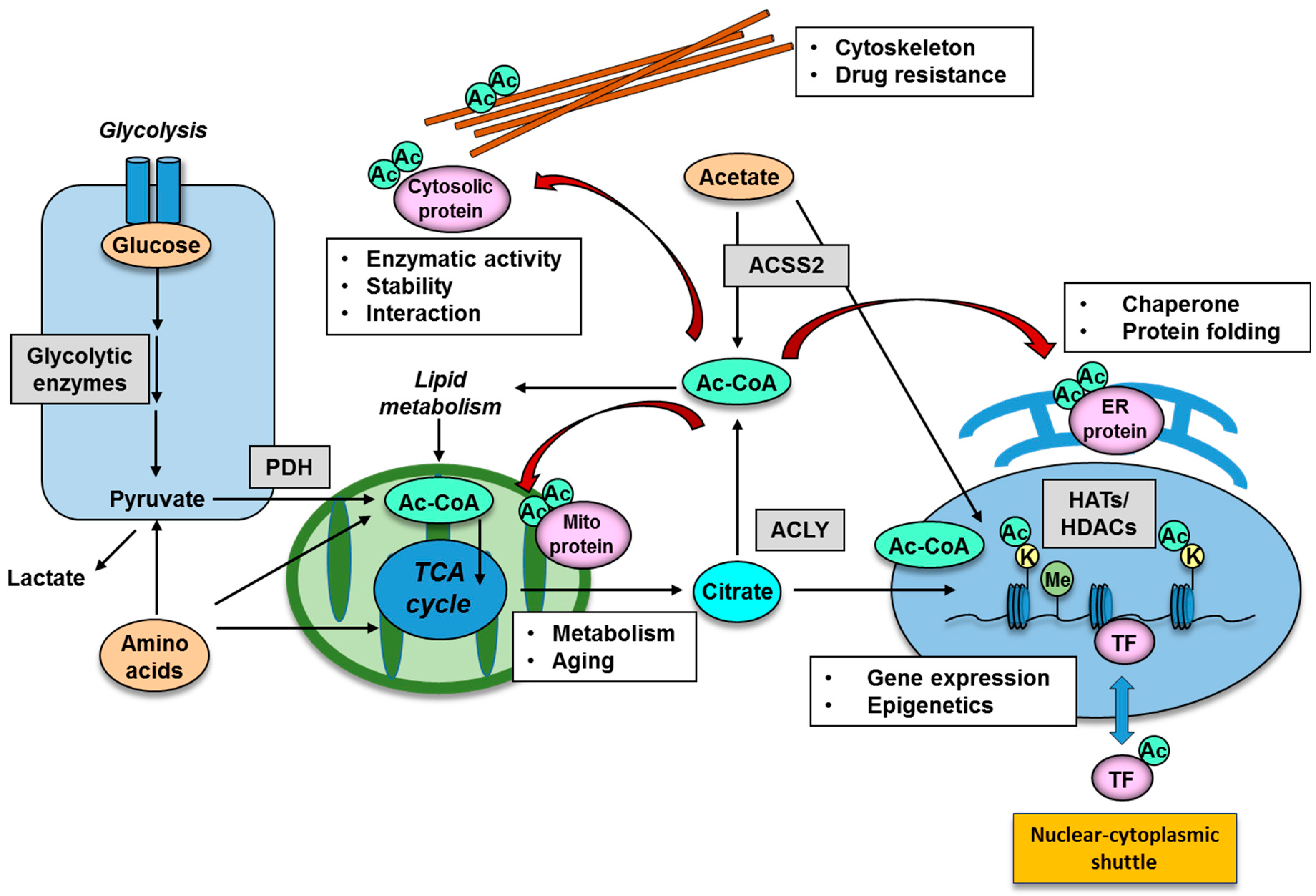
2.2. Donor Substrate for Acetylation: Production of Intermediary Metabolites for Protein Acetylation
3. Functional Significance of Lysine Acetylation in Different Cellular Organelle
3.1. Acetylation of Nuclear Proteins: Implication for Epigenetics
3.2. Acetylation of Cytosolic Proteins in Specific Organelles
4. Aberrant Protein Acetylation in the Phenotypes of Cancer Cells
4.1. Driver Mutations of Lysine Acetylation/Deacetylation Genes in Cancer
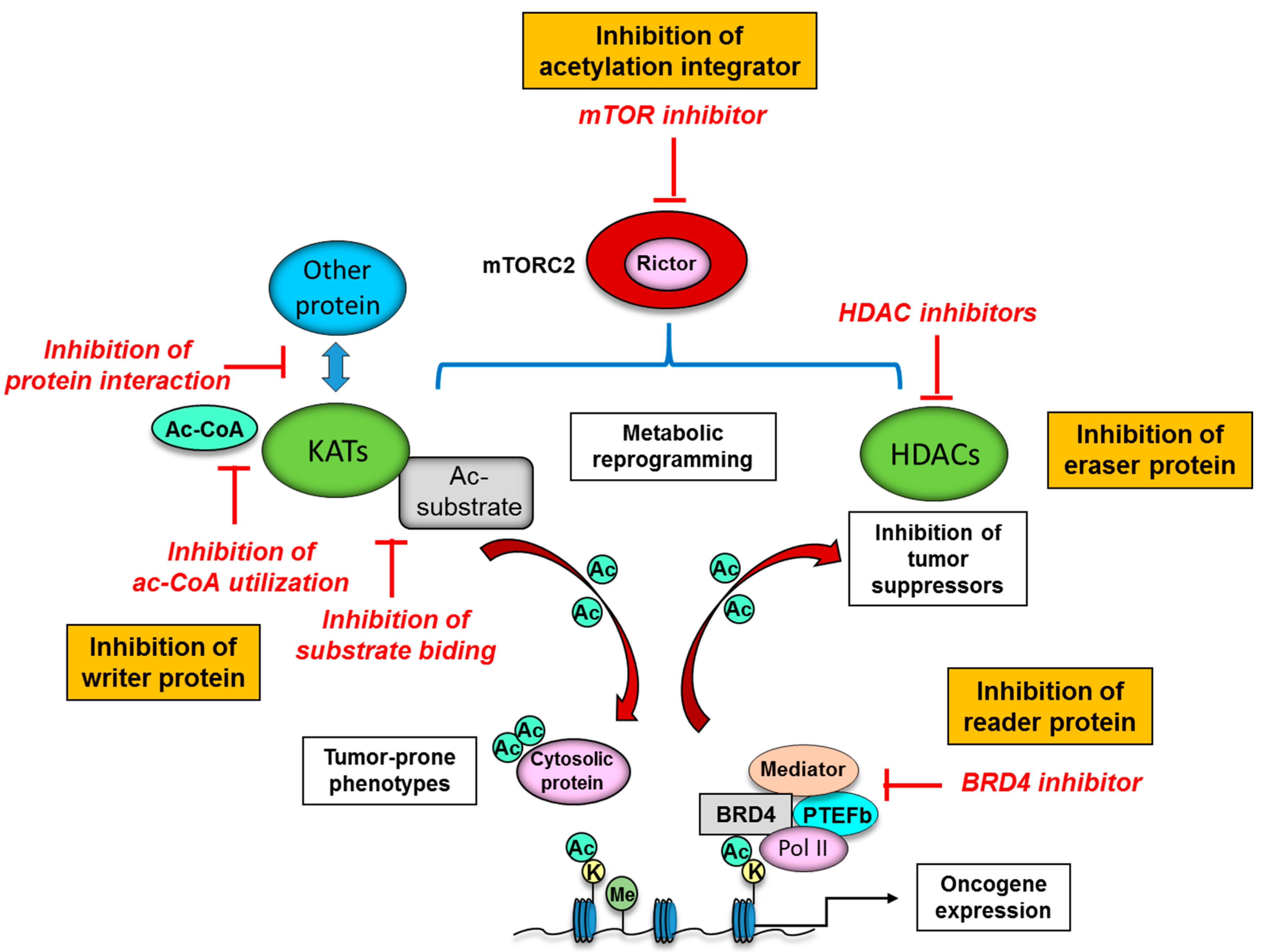
4.2. Oncogenic Signaling and Protein Acetylation: Mechanistic Target of Rapamycin Complex 2 (mTORC2) as a Strong Acetylation Driver in Cancer
5. Novel Therapeutic Strategies to Target Protein Acetylation Systems in Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KAT Inhibitors | ||
|---|---|---|
| Mechanisms | Status | Inhibitors |
| Compete with substrates | Preclinical | CPTH2, CPTH6, BF1 [138,139] |
| Inhibit Ac-CoA utilization | Preclinical | Garcinol, C646, TH1834, Lys-CoA [140,141,142] |
| Block interaction with other protein | Preclinical | Chetomin (HIF), KCN1 (HIF), ICG-001 (β-catenin), Windorphen (β-catenin) [143,144,145,146] |
| HDAC Inhibitors | ||
| Class | Status | Inhibitors (targeted HDAC) |
| Hydroxamates | FDA-approved Preclinical | * Vorinostat (SAHA) (pan-class), Belinostat (pan-class), Panobinostat (pan-class) [147,148,149] Trichostatin A (pan-class) [150] |
| Benzamides | Clinical trials | Entinostat (class I), Mocetinostat (class I, IV), Tacedinaline (class I) [151,152,153] |
| Short-chain fatty acids | Clinical trials | Valproic acid (class I, IIa), Butyric acid (class I, II), Phenylbutyrate (class I, II) [154,155,156] |
| Cyclic peptides | FDA-approved | Romidepsin (class I) [157] |
6. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.B.; Gui, D.Y.; Van Der Heiden, M.G. Altered metabolite levels in cancer: Implications for tumour biology and cancer therapy. Nat. Rev. Cancer 2016, 16, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Boroughs, L.K.; Deberardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, J.; Chowdhry, S.; Wu, S.; Zhang, W.; Masui, K.; Mischel, P.S. Altered cellular metabolism in gliomas—An emerging landscape of actionable co-dependency targets. Nat. Rev. Cancer 2020, 20, 57–70. [Google Scholar] [CrossRef]
- Chen, L.; Liu, S.; Tao, Y. Regulating tumor suppressor genes: Post-translational modifications. Signal. Transduct. Target. Ther. 2020, 5, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Dahan, P.; Lu, V.; Nguyen, R.M.T.; Kennedy, S.A.L.; Teitell, M.A. Metabolism in pluripotency: Both driver and passenger? J. Biol. Chem. 2019, 294, 5420–5429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of cellular metabolism by protein lysine acetylation. Science 2010, 327, 1000–1004. [Google Scholar] [CrossRef] [Green Version]
- Kori, Y.; Sidoli, S.; Yuan, Z.F.; Lund, P.J.; Zhao, X.; Garcia, B.A. Proteome-wide acetylation dynamics in human cells. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Di Martile, M.; Del Bufalo, D.; Trisciuoglio, D. The multifaceted role of lysine acetylation in cancer: Prognostic biomarker and therapeutic target. Oncotarget 2016, 7, 55789–55810. [Google Scholar] [CrossRef] [Green Version]
- Masui, K.; Shibata, N.; Cavenee, W.K.; Mischel, P.S. mTORC2 activity in brain cancer: Extracellular nutrients are required to maintain oncogenic signaling. BioEssays 2016, 38, 839–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Yu, T.; Zhang, N.; Chen, J.; Zhang, P.; Li, S.; Luo, L.; Cui, Z.; Qin, Y.; Liu, F. Nuclear e-cadherin acetylation promotes colorectal tumorigenesis via enhancing β-catenin activity. Mol. Cancer Res. 2019, 17, 655–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide Mapping of HATs and HDACs Reveals Distinct Functions in Active and Inactive Genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Farria, A.; Li, W.; Dent, S.Y.R. KATs in cancer: Functions and therapies. Oncogene 2015, 34, 4901–4913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, B.N.; Akhtar, A. The many lives of KATs—Detectors, integrators and modulators of the cellular environment. Nat. Rev. Genet. 2019, 20, 7–23. [Google Scholar] [CrossRef]
- McCullough, C.E.; Marmorstein, R. Molecular Basis for Histone Acetyltransferase Regulation by Binding Partners, Associated Domains, and Autoacetylation. ACS Chem. Biol. 2016, 11, 632–642. [Google Scholar] [CrossRef]
- Butler, J.S.; Koutelou, E.; Schibler, A.C.; Dent, S.Y.R. Histone-modifying enzymes: Regulators of developmental decisions and drivers of human disease. Epigenomics 2012, 4, 163–177. [Google Scholar] [CrossRef] [Green Version]
- Nagy, Z.; Tora, L. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene 2007, 26, 5341–5357. [Google Scholar] [CrossRef] [Green Version]
- Özdaǧ, H.; Batley, S.J.; Försti, A.; Iyer, N.G.; Daigo, Y.; Boutell, J.; Arends, M.J.; Ponder, B.A.J.; Kouzarides, T.; Caldas, C. Mutation analysis of CBP and PCAF reveals rare inactivating mutations in cancer cell lines but not in primary tumours. Br. J. Cancer 2002, 87, 1162–1165. [Google Scholar] [CrossRef] [PubMed]
- Ladang, A.; Rapino, F.; Heukamp, L.C.; Tharun, L.; Shostak, K.; Hermand, D.; Delaunay, S.; Klevernic, I.; Jiang, Z.; Jacques, N.; et al. Elp3 drives Wnt-dependent tumor initiation and regeneration in the intestine. J. Exp. Med. 2015, 212, 2057–2075. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Li, M.; Clarkson, B.D.; Pehar, M.; Lao, P.J.; Hillmer, A.T.; Barnhart, T.E.; Christian, B.T.; Mitchell, H.A.; Bendlin, B.B.; et al. Deficient import of Acetyl-CoA into the ER lumen causes neurodegeneration and propensity to infections, inflammation, and cancer. J. Neurosci. 2014, 34, 6772–6789. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.; Johnson, M.; Shridhar, V.; Van Deursen, J.; Couch, F.J. CBP truncating mutations in ovarian cancer. J. Med Genet. 2005, 42, 514–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishimoto, M.; Kohno, T.; Okudela, K.; Otsuka, A.; Sasaki, H.; Tanabe, C.; Sakiyama, T.; Hirama, C.; Kitabayashi, I.; Minna, J.D.; et al. Mutations and Deletions of the CBP Gene in Human Lung Cancer. Clin. Cancer Res. 2005, 11, 512–519. [Google Scholar] [PubMed]
- Iyer, N.G.; Özdag, H.; Caldas, C. p300/CBP and cancer. Oncogene 2004, 23, 4225–4231. [Google Scholar] [CrossRef] [Green Version]
- Attar, N.; Kurdistani, S.K. Exploitation of EP300 and CREBBP lysine acetyltransferases by cancer. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.; Molkentine, D.; Molkentine, J.; Bridges, K.; Xie, T.; Yang, L.; Hefner, A.; Gao, M.; Frederick, M.; Seth, S.; et al. Inhibition of histone acetyltranserase function radiosensitizes CREBBP/EP300 mutants via repression of homologous recombination, potentially targeting a novel gain of function. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Gorrini, C.; Squatrito, M.; Luise, C.; Syed, N.; Perna, D.; Wark, L.; Martinato, F.; Sardella, D.; Verrecchia, A.; Bennett, S.; et al. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature 2007, 448, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Chevillard-briet, M.; Quaranta, M.; Grézy, A.; Mattera, L.; Courilleau, C.; Philippe, M.; Mercier, P.; Corpet, D.; Lough, J.; Ueda, T.; et al. Interplay between chromatin-modifying enzymes controls colon cancer progression through Wnt signaling. Hum. Mol. Genet. 2014, 23, 2120–2131. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-Awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015, 525, 206–211. [Google Scholar] [CrossRef] [Green Version]
- Borrow, J.; Stanton, V.P.; Andresen, J.M.; Becher, R.; Behm, F.G.; Chaganti, R.S.K.; Civin, C.I.; Disteche, C.; Dubé, I.; Frischauf, A.M.; et al. The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nat. Genet. 1996, 14, 33–41. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Fioretos, T.; Isaksson, M.; Samuelsson, U.; Billström, R.; Strömbeck, B.; Mitelman, F.; Johansson, B. Fusion of the MORF and CBP genes in acute myeloid leukemia with the t(10;16)(q22;p13). Hum. Mol. Genet. 2001, 10, 395–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, S.D.P.; Herrick, S.R.; Ince, T.A.; Kleinman, M.S.; Dal Cin, P.; Morton, C.C.; Quade, B.J. Uterine leiomyomata with t(10;17) disrupt the histone acetyltrasferase MORF. Cancer Res. 2004, 64, 5570–5577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duong, M.L.T.; Akli, S.; Macalou, S.; Biernacka, A.; Debeb, B.G.; Yi, M.; Hunt, K.K.; Keyomarsi, K. Hbo1 is a cyclin E/CDK2 substrate that enriches breast cancer stem-like cells. Cancer Res. 2013, 73, 5556–5568. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Zou, J.; Li, J.; Pang, Y.; Liu, Y.; Deng, C.; Chen, F.; Cui, H. MYST1/KAT8 contributes to tumor progression by activating EGFR signaling in glioblastoma cells. Cancer Med. 2019, 8, 7793–7808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, W.K.; Pearson, D.; Lee, H.W.; Kim, S. Nonenzymatic acetylation of histones with acetyl-CoA. Bba Sect. Nucleic Acids Protein Synth. 1970, 213, 513–522. [Google Scholar] [CrossRef]
- James, A.M.; Hoogewijs, K.; Logan, A.; Hall, A.R.; Ding, S.; Fearnley, I.M.; Murphy, M.P. Non-enzymatic N-acetylation of Lysine Residues by AcetylCoA Often Occurs via a Proximal S-acetylated Thiol Intermediate Sensitive to Glyoxalase II. Cell Rep. 2017, 18, 2105–2112. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.Y.; Ng, L.T.; Ng, L.F.; Inoue, T.; Tolwinski, N.S.; Hagen, T.; Gruber, J. The role of mitochondrial non-enzymatic protein acylation in ageing. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Hebert, A.S.; Dittenhafer-Reed, K.E.; Yu, W.; Bailey, D.J.; Selen, E.S.; Boersma, M.D.; Carson, J.J.; Tonelli, M.; Balloon, A.J.; Higbee, A.J.; et al. Calorie Restriction and SIRT3 Trigger Global Reprogramming of the Mitochondrial Protein Acetylome. Mol. Cell 2013, 49, 186–199. [Google Scholar] [CrossRef] [Green Version]
- Gil, J.; Ramírez-Torres, A.; Encarnación-Guevara, S. Lysine acetylation and cancer: A proteomics perspective. J. Proteom. 2017, 150, 297–309. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Wang, Z.; Liu, J. Role of HDACs in normal and malignant hematopoiesis. Mol. Cancer 2020, 19, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micelli, C.; Rastelli, G. Histone deacetylases: Structural determinants of inhibitor selectivity. Drug Discov. Today 2015, 20, 718–735. [Google Scholar] [CrossRef] [PubMed]
- Chalkiadaki, A.; Guarente, L. The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer 2015, 15, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihaylova, M.M.; Vasquez, D.S.; Ravnskjaer, K.; Denechaud, P.D.; Yu, R.T.; Alvarez, J.G.; Downes, M.; Evans, R.M.; Montminy, M.; Shaw, R.J. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell 2011, 145, 607–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masui, K.; Tanaka, K.; Akhavan, D.; Babic, I.; Gini, B.; Matsutani, T.; Iwanami, A.; Liu, F.; Villa, G.R.; Gu, Y.; et al. MTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013, 18, 726–739. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.N.; Choijookhuu, N.; Takagi, H.; Srisowanna, N.; Nguyen Nhat Huynh, M.; Yamaguchi, Y.; Synn Oo, P.; Tin Htwe Kyaw, M.; Sato, K.; Yamaguchi, R.; et al. The HDAC Inhibitor, SAHA, Prevents Colonic Inflammation by Suppressing Pro-inflammatory Cytokines and Chemokines in DSS-induced Colitis. Acta Histochem. Et Cytochem. 2018, 51, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Hanigan, C.L.; van Engeland, M.; De Bruine, A.P.; Wouters, K.A.; Weijenberg, M.P.; Eshleman, J.R.; Herman, J.G. An Inactivating Mutation in HDAC2 Leads to Dysregulation of Apoptosis Mediated by APAF. Gastroenterology 2008, 135. [Google Scholar] [CrossRef]
- Ji, H.; Zhou, Y.; Zhuang, X.; Zhu, Y.; Wu, Z.; Lu, Y.; Li, S.; Zeng, Y.; Lu, Q.R.; Huo, Y.; et al. HDAC3 deficiency promotes liver cancer through a defect in H3K9ac/H3K9me3 transition. Cancer Res. 2019, 79, 3676–3688. [Google Scholar] [CrossRef]
- Xu, G.; Zhu, H.; Zhang, M.; Xu, J. Histone deacetylase 3 is associated with gastric cancer cell growth via the miR-454-mediated targeting of Chdinternational. J. Mol. Med. 2018, 41, 155–163. [Google Scholar] [CrossRef]
- Durst, K.L.; Lutterbach, B.; Kummalue, T.; Friedman, A.D.; Hiebert, S.W. The inv(16) Fusion Protein Associates with Corepressors via a Smooth Muscle Myosin Heavy-Chain Domain. Mol. Cell. Biol. 2003, 23, 607–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjöblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Lachenmayer, A.; Toffanin, S.; Cabellos, L.; Alsinet, C.; Hoshida, Y.; Villanueva, A.; Minguez, B.; Tsai, H.W.; Ward, S.C.; Thung, S.; et al. Combination therapy for hepatocellular carcinoma: Additive preclinical efficacy of the HDAC inhibitor panobinostat with sorafenib. J. Hepatol. 2012, 56, 1343–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Liu, L.; Zhang, S.; Guo, S.; Li, X.; Wang, J.; Su, B.; Fang, Y.; Chen, X.; Ke, H.; et al. Hdac7 promotes lung tumorigenesis by inhibiting Stat3 activation. Mol. Cancer 2017, 16, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milde, T.; Oehme, I.; Korshunov, A.; Kopp-Schneider, A.; Remke, M.; Northcott, P.; Deubzer, H.E.; Lodrini, M.; Taylor, M.D.; Von Deimling, A.; et al. HDAC5 and HDAC9 in medulloblastoma: Novel markers for risk stratification and role in tumor cell growth. Clin. Cancer Res. 2010, 16, 3240–3252. [Google Scholar] [CrossRef] [Green Version]
- Bitler, B.G.; Wu, S.; Park, P.H.; Hai, Y.; Aird, K.M.; Wang, Y.; Zhai, Y.; Kossenkov, A.V.; Vara-Ailor, A.; Rauscher, F.J.; et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat. Cell Biol. 2017, 19, 962–973. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, X.; Zhu, S.; Dejene, E.A.; Peng, W.; Sepulveda, A.; Seto, E. HDAC10 regulates cancer stem-like cell properties in KRAS-driven lung adenocarcinoma. Cancer Res. 2020, 80, 3265–3278. [Google Scholar] [CrossRef]
- Deng, C.X. SIRT1, is it a tumor promoter or tumor suppressor? Int. J. Biol. Sci. 2009, 5, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, R.; Utani, K.; Thakur, B.; Ishine, K.; Aladjem, M.I.; Shimizu, N. SIRT1 stabilizes extrachromosomal gene amplification and contributes to repeat-induced gene silencing. J. Biol. Chem. 2021, 296, 100356. [Google Scholar] [CrossRef]
- Head, P.E.; Zhang, H.; Bastien, A.J.; Koyen, A.E.; Withers, A.E.; Daddacha, W.B.; Cheng, X.; Yu, D.S. Sirtuin 2 mutations in human cancers impair its function in genome maintenance. J. Biol. Chem. 2017, 292, 9919–9931. [Google Scholar] [CrossRef] [Green Version]
- Bhalla, K.; Jaber, S.; Reagan, K.; Hamburg, A.; Underwood, K.F.; Jhajharia, A.; Singh, M.; Bhandary, B.; Bhat, S.; Nanaji, N.M.; et al. SIRT3, a metabolic target linked to ataxia-telangiectasia mutated (ATM) gene deficiency in diffuse large B-cell lymphoma. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Finley, L.W.S.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.I.; Cardoso, S.M.; Clish, C.B.; et al. SIRT3 Opposes Reprogramming of Cancer Cell Metabolism through HIF1α Destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Yan, Y.; Principe, D.R.; Zou, X.; Vassilopoulos, A.; Gius, D. SIRT3 and SIRT4 are mitochondrial tumor suppressor proteins that connect mitochondrial metabolism and carcinogenesis. Cancer Metab. 2014, 2, 15. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Q.; Wang, H.L.; Xu, J.; Tan, J.; Fu, L.N.; Wang, J.L.; Zou, T.H.; Sun, D.F.; Gao, Q.Y.; Chen, Y.X.; et al. Sirtuin5 contributes to colorectal carcinogenesis by enhancing glutaminolysis in a deglutarylation-dependent manner. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Lerrer, B.; Gertler, A.A.; Cohen, H.Y. The complex role of SIRT6 in carcinogenesis. Carcinogenesis 2015, 37, 108–118. [Google Scholar] [CrossRef]
- Malik, S.; Villanova, L.; Tanaka, S.; Aonuma, M.; Roy, N.; Berber, E.; Pollack, J.R.; Michishita-Kioi, E.; Chua, K.F. SIRT7 inactivation reverses metastatic phenotypes in epithelial and mesenchymal tumors. Sci. Rep. 2015, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Yue, L.; Sharma, V.; Horvat, N.P.; Akuffo, A.A.; Beatty, M.S.; Murdun, C.; Colin, C.; Billington, J.M.R.; Goodheart, W.E.; Sahakian, E.; et al. HDAC11 deficiency disrupts oncogene-induced hematopoiesis in myeloproliferative neoplasms. Blood 2020, 135, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Vakoc, C.R. The Mechanisms behind the Therapeutic Activity of BET Bromodomain Inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef] [Green Version]
- Moon, K.J.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [Green Version]
- Rahnamoun, H.; Lee, J.; Sun, Z.; Lu, H.; Ramsey, K.M.; Komives, E.A.; Lauberth, S.M. RNAs interact with BRD4 to promote enhanced chromatin engagement and transcription activation. Nat. Struct. Mol. Biol. 2018, 25, 687–697. [Google Scholar] [CrossRef]
- Kaelin, W.G.; McKnight, S.L. Influence of metabolism on epigenetics and disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.Q.; Lowman, X.H.; Kong, M. Molecular pathways: Metabolic control of histone methylation and gene expression in cancer. Clin. Cancer Res. 2017, 23, 4004–4009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Saha, N.; Gajan, A.; Saadat, N.; Gupta, S.V.; Pile, L.A. A complex interplay between SAM synthetase and the epigenetic regulator SIN3 controls metabolism and transcription. J. Biol. Chem. 2020, 295, 375–389. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [Green Version]
- Sutendra, G.; Kinnaird, A.; Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Hashimoto, K.; Zhang, N.; Flaim, E.; Michelakis, E.D. A nuclear pyruvate dehydrogenase complex is important for the generation of Acetyl-CoA and histone acetylation. Cell 2014, 158, 84–97. [Google Scholar] [CrossRef] [Green Version]
- Masui, K.; Harachi, M.; Ikegami, S.; Yang, H.; Onizuka, H.; Yong, W.H.; Cloughesy, T.F.; Muragaki, Y.; Kawamata, T.; Arai, N.; et al. MTORC2 links growth factor signaling with epigenetic regulation of iron metabolism in glioblastoma. J. Biol. Chem. 2019, 294, 19740–19751. [Google Scholar] [CrossRef]
- Eguchi, K.; Nakayama, K. Prolonged hypoxia decreases nuclear pyruvate dehydrogenase complex and regulates the gene expression. Biochem. Biophys. Res. Commun. 2019, 520, 128–135. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [Green Version]
- Migita, T.; Narita, T.; Nomura, K.; Miyagi, E.; Inazuka, F.; Matsuura, M.; Ushijima, M.; Mashima, T.; Seimiya, H.; Satoh, Y.; et al. ATP citrate lyase: Activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 2008, 68, 8547–8554. [Google Scholar] [CrossRef] [Green Version]
- Masui, K.; Tanaka, K.; Ikegami, S.; Villa, G.R.; Yang, H.; Yong, W.H.; Cloughesy, T.F.; Yamagata, K.; Arai, N.; Cavenee, W.K.; et al. Glucose-dependent acetylation of Rictor promotes targeted cancer therapy resistance. Proc. Natl. Acad. Sci. USA 2015, 112, 9406–9411. [Google Scholar] [CrossRef] [Green Version]
- Schug, Z.T.; Vande Voorde, J.; Gottlieb, E. The metabolic fate of acetate in cancer. Nat. Rev. Cancer 2016, 16, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmugam, M.K.; Arfuso, F.; Arumugam, S.; Chinnathambi, A.; Jinsong, B.; Warrier, S.; Wang, L.Z.; Kumar, A.P.; Ahn, K.S.; Sethi, G.; et al. Role of novel histone modifications in cancer. Oncotarget 2018, 9, 11414–11426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simone, C.; Peserico, A. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J. Biomed. Biotechnol. 2010, 2011. [Google Scholar] [CrossRef] [Green Version]
- Dekker, J.; Mirny, L. The 3D Genome as Moderator of Chromosomal Communication. Cell 2016, 164, 1110–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.W.; Battenhouse, A.M.; Shivram, H.; Morris, A.R.; Cowperthwaite, M.C.; Shpak, M.; Iyer, V.R. Bivalent chromatin domains in glioblastoma reveal a subtype-specific signature of glioma stem cells. Cancer Res. 2018, 78, 2463–2474. [Google Scholar] [CrossRef] [Green Version]
- Rice, J.C.; Allis, C.D. Histone methylation versus histone acetylation: New insights into epigenetic regulation. Curr. Opin. Cell Biol. 2001, 13, 263–273. [Google Scholar] [CrossRef]
- Masui, K.; Harachi, M.; Cavenee, W.K.; Mischel, P.S.; Shibata, N. Codependency of metabolism and epigenetics drives cancer progression: A review. Acta Histochem. Et Cytochem. 2020, 53, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.W.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Z.; Song, J.; Wang, Y.; Zhao, Y.; Guda, K.; Yang, S.; Kao, H.Y.; Xu, Y.; Willis, J.; Markowitz, S.D.; et al. DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Sci. Signal. 2010, 3, ra80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlev, N.A.; Liu, L.; Chehab, N.H.; Mansfield, K.; Harris, K.G.; Halazonetis, T.D.; Berger, S.L. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell 2001, 8, 1243–1254. [Google Scholar] [CrossRef]
- Rg Vervoorts, J.; Lü Scher-Firzlaff, J.; Lü Scher, B. The Ins and Outs of MYC Regulation by Posttranslational Mechanisms. J. Biol. Chem. 2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.M.; Jo, S.H.; Kim, M.Y.; Kim, T.H.; Ahn, Y.H. Role of transcription factor acetylation in the regulation of metabolic homeostasis. Protein Cell 2015, 6, 804–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaki, H.; Daitoku, H.; Hatta, M.; Aoyama, H.; Yoshimochi, K.; Fukamizu, A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc. Natl. Acad. Sci. USA 2005, 102, 11278–11283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Xu, Y.; Roy, K.; Price, B.D. DNA Damage-Induced Acetylation of Lysine 3016 of ATM Activates ATM Kinase Activity. Mol. Cell. Biol. 2007, 27, 8502–8509. [Google Scholar] [CrossRef] [Green Version]
- Spange, S.; Wagner, T.; Heinzel, T.; Krämer, O.H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198. [Google Scholar]
- Sadoul, K.; Wang, J.; Diagouraga, B.; Khochbin, S. The tale of protein lysine acetylation in the cytoplasm. J. Biomed. Biotechnol. 2010, 2011. [Google Scholar] [CrossRef]
- Eshun-Wilson, L.; Zhang, R.; Portran, D.; Nachury, M.V.; Toso, D.B.; Löhr, T.; Vendruscolo, M.; Bonomi, M.; Fraser, J.S.; Nogales, E. Effects of α-tubulin acetylation on microtubule structure and stability. Proc. Natl. Acad. Sci. USA 2019, 116, 10366–10371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shida, T.; Cueva, J.G.; Xu, Z.; Goodman, M.B.; Nachury, M.V. The major α-tubulin K40 acetyltransferase αTAT1 promotes rapid ciliogenesis and efficient mechanosensation. Proc. Natl. Acad. Sci. USA 2010, 107, 21517–21522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, K.M.; Aldana-Masangkay, G.I. The role of HDAC6 in cancer. J. Biomed. Biotechnol. 2010, 2011. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Omoto, Y.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hayashi, S.-I.; Iwase, H. HDAC6 Expression Is Correlated with Better Survival in Breast Cancer. Clin. Cancer Res. 2004, 10, 6962–6968. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.C.; Sprung, R.; Chen, Y.; Xu, Y.; Ball, H.; Pei, J.; Cheng, T.; Kho, Y.; Xiao, H.; Xiao, L.; et al. Substrate and Functional Diversity of Lysine Acetylation Revealed by a Proteomics Survey. Mol. Cell 2006, 23, 607–618. [Google Scholar] [CrossRef]
- Anderson, K.A.; Hirschey, M.D. Mitochondrial protein acetylation regulates metabolism. Essays Biochem. 2012, 52, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.S.; Koves, T.R.; Davidson, M.T.; Crown, S.B.; Fisher-Wellman, K.H.; Torres, M.J.; Draper, J.A.; Narowski, T.M.; Slentz, D.H.; Lantier, L.; et al. Disruption of Acetyl-Lysine Turnover in Muscle Mitochondria Promotes Insulin Resistance and Redox Stress without Overt Respiratory Dysfunction. Cell Metab. 2020, 31, 131–147.e11. [Google Scholar] [CrossRef]
- Wagner, G.R.; Payne, R.M. Mitochondrial acetylation and diseases of aging. J. Aging Res. 2011, 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pehar, M.; Lehnus, M.; Karst, A.; Puglielli, L. Proteomic Assessment Shows That Many Endoplasmic Reticulum (ER)-resident Proteins Are Targeted by N-Lysine Acetylation in the Lumen of the Organelle and Predicts Broad Biological Impact. J. Biol. Chem. 2012, 287, 22436–22440. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.K.; Puglielli, L. Two Endoplasmic Reticulum (ER)/ER golgi intermediate compartment-based lysine acetyltransferases post-translationally regulate BACE1 levels. J. Biol. Chem. 2009, 284, 2482–2492. [Google Scholar] [CrossRef] [Green Version]
- Kitabayashi, I.; Aikawa, Y.; Yokoyama, A.; Hosoda, F.; Nagai, M.; Kakazu, N.; Abe, T.; Ohki, M. Fusion of MOZ and p300 histone acetyltransferases in acute monocytic leukemia with a t(8;22)(p11;q13) chromosome translocation. Leukemia 2001, 15, 89–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond transcriptional regulation. Mol. Cancer 2018, 17, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Alekseyenko, A.A.; Walsh, E.M.; Wang, X.; Grayson, A.R.; Hsi, P.T.; Kharchenko, P.V.; Kuroda, M.I.; French, C.A. The oncogenic BRD4-NUT chromatin regulator drives aberrant transcription within large topological domains. Genes Dev. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhyasen, G.W.; Yao, Y.; Zhang, J.; Dulak, A.; Castriotta, L.; Jacques, K.; Zhao, W.; Gharahdaghi, F.; Hattersley, M.M.; Lyne, P.D.; et al. BRD4 amplification facilitates an oncogenic gene expression program in high-grade serous ovarian cancer and confers sensitivity to BET inhibitors. PLoS ONE 2018, 13, e0200826. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Yin, J.; Fang, Y.; Chen, J.; Jeong, K.J.; Chen, X.; Vellano, C.P.; Ju, Z.; Zhao, W.; Zhang, D.; et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell 2018, 33, 401–416.e8. [Google Scholar] [CrossRef] [Green Version]
- Harachi, M.; Masui, K.; Okamura, Y.; Tsukui, R.; Mischel, P.S.; Shibata, N. mTOR complexes as a nutrient sensor for driving cancer progression. Int. J. Mol. Sci. 2018, 19, 3267. [Google Scholar] [CrossRef] [Green Version]
- Masui, K.; Harachi, M.; Cavenee, W.K.; Mischel, P.S.; Shibata, N. mTOR complex 2 is an integrator of cancer metabolism and epigenetics. Cancer Lett. 2020, 478, 1–7. [Google Scholar] [CrossRef]
- Glidden, E.J.; Gray, L.G.; Vemuru, S.; Li, D.; Harris, T.E.; Mayo, M.W. Multiple site acetylation of rictor stimulates mammalian target of rapamycin complex 2 (mTORC2)-dependent phosphorylation of Akt protein. J. Biol. Chem. 2012, 287, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Martinez Calejman, C.; Trefely, S.; Entwisle, S.W.; Luciano, A.; Jung, S.M.; Hsiao, W.; Torres, A.; Hung, C.M.; Li, H.; Snyder, N.W.; et al. mTORC2-AKT signaling to ATP-citrate lyase drives brown adipogenesis and de novo lipogenesis. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.; Habib, A.; Laor, D.; Yadav, S.; Kupiec, M.; Weisman, R. TOR complex 2 in fission yeast is required for chromatin-mediated gene silencing and assembly of heterochromatic domains at subtelomeres. J. Biol. Chem. 2018, 293, 8138–8150. [Google Scholar] [CrossRef] [Green Version]
- Vadla, R.; Haldar, D. Mammalian target of rapamycin complex 2 (mTORC2) controls glycolytic gene expression by regulating Histone H3 Lysine 56 acetylation. Cell Cycle 2018, 17, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Santos-Rosa, H.; Valls, E.; Kouzarides, T.; Martínez-Balbás, M. Mechanisms of P/CAF auto-acetylation. Nucleic Acids Res. 2003, 31, 4285–4292. [Google Scholar] [CrossRef]
- Schrump, D.S. Cytotoxicity mediated by histone deacetylase inhibitors in cancer cells: Mechanisms and potential clinical implications. Clin. Cancer Res. 2009, 15, 3947–3957. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsoff, J.N.; Owens, D.A.; Lopez, A.; Rodriguez, D.A.; Chee, N.T.; Kurtenbach, S.; Bilbao, D.; Roberts, E.R.; Volmar, C.H.; Wahlestedt, C.; et al. Dual screen for efficacy and toxicity identifies HDAC inhibitor with distinctive activity spectrum for BAP1-mutant uveal melanoma. Mol. Cancer Res. 2021, 19, 215–222. [Google Scholar] [CrossRef]
- Kalac, M.; Scotto, L.; Marchi, E.; Amengual, J.; Seshan, V.E.; Bhagat, G.; Ulahannan, N.; Leshchenko, V.V.; Temkin, A.M.; Parekh, S.; et al. HDAC inhibitors and decitabine are highly synergistic and associated with unique gene-expression and epigenetic profiles in models of DLBCL. Blood 2011, 118, 5506–5516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyaw, M.T.H.; Yamaguchi, Y.; Choijookhuu, N.; Yano, K.; Takagi, H.; Takahashi, N.; Oo, P.S.; Sato, K.; Hishikawa, Y. The HDAC inhibitor, SAHA, combined with cisplatin synergistically induces apoptosis in alpha-fetoprotein-producing hepatoid adenocarcinoma cells. Acta Histochem. Et Cytochem. 2019, 52, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mwakwari, S.C.; Patil, V.; Guerrant, W.; Oyelere, A.K. Macrocyclic histone deacetylase inhibitors. Curr. Top. Med. Chem. 2010, 10, 1423–1440. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Ungerstedt, J.S.; Sowa, Y.; Xu, W.S.; Shao, Y.; Dokmanovic, M.; Perez, G.; Ngo, L.; Holmgren, A.; Jiang, X.; Marks, P.A. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 673–678. [Google Scholar] [CrossRef] [Green Version]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Picaud, S.; Da Costa, D.; Thanasopoulou, A.; Filippakopoulos, P.; Fish, P.V.; Philpott, M.; Fedorov, O.; Brennan, P.; Bunnage, M.E.; Owen, D.R.; et al. PFI-1, a highly selective protein interaction inhibitor, targeting BET bromodomains. Cancer Res. 2013, 73, 3336–3346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segura, M.F.; Fontanals-Cirera, B.; Gaziel-Sovran, A.; Guijarro, M.V.; Hanniford, D.; Zhang, G.; González-Gomez, P.; Morante, M.; Jubierre, L.; Zhang, W.; et al. BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res. 2013, 73, 6264–6276. [Google Scholar] [CrossRef] [Green Version]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Kowalsky, A.H.; Namkoong, S.; Mettetal, E.; Park, H.W.; Kazyken, D.; Fingar, D.C.; Lee, J.H. The GATOR2-mTORC2 axis mediates Sestrin2-induced AKT Ser/Thr kinase activation. J. Biol. Chem. 2020, 295, 1769–1780. [Google Scholar] [CrossRef] [PubMed]
- Benavides-Serrato, A.; Lee, J.; Holmes, B.; Landon, K.A.; Bashir, T.; Jung, M.E.; Lichtenstein, A.; Gera, J. Specific blockade of Rictor-mTOR association inhibits mTORC2 activity and is cytotoxic in glioblastoma. PLoS ONE 2017, 12, e0176599. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Proud, C.G. Crosstalk between mTor Complexes. Nat. Cell Biol. 2013, 15, 1263–1265. [Google Scholar] [CrossRef] [PubMed]
- Harachi, M.; Masui, K.; Honda, H.; Muragaki, Y.; Kawamata, T.; Cavenee, W.K.; Mischel, P.S.; Shibata, N. Dual Regulation of Histone Methylation by mTOR Complexes Controls Glioblastoma Tumor Cell Growth via EZH2 and SAM. Mol. Cancer Res. Mcr. 2020, 18, 1142–1152. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, F.; Bizzarri, B.; Maccioni, E.; Secci, D.; Bolasco, A.; Chimenti, P.; Fioravanti, R.; Granese, A.; Carradori, S.; Tosi, F.; et al. A novel histone acetyltransferase inhibitor modulating Gcn5 network: Cyclopentylidene-[4-(4′-chlorophenyl)thiazol-2-yl)hydrazone. J. Med. Chem. 2009, 52, 530–536. [Google Scholar] [CrossRef]
- Trisciuoglio, D.; Ragazzoni, Y.; Pelosi, A.; Desideri, M.; Carradori, S.; Gabellini, C.; Maresca, G.; Nescatelli, R.; Secci, D.; Bolasco, A.; et al. CPTH6, a thiazole derivative, induces histone hypoacetylation and apoptosis in human leukemia cells. Clin. Cancer Res. 2012, 18, 475–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.; Bourke, E.; Scobie, M.; Famme, M.A.; Koolmeister, T.; Helleday, T.; Eriksson, L.A.; Lowndes, N.F.; Brown, J.A.L. Rational design and validation of a Tip60 histone acetyltransferase inhibitor. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef]
- Gao, X.N.; Lin, J.; Ning, Q.Y.; Gao, L.; Yao, Y.S.; Zhou, J.H.; Li, Y.H.; Wang, L.L.; Yu, L. A Histone Acetyltransferase p300 Inhibitor C646 Induces Cell Cycle Arrest and Apoptosis Selectively in AML1-ETO-Positive AML Cells. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramanyam, K.; Altaf, M.; Varier, R.A.; Swaminathan, V.; Ravindran, A.; Sadhale, P.P.; Kundu, T.K. Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. J. Biol. Chem. 2004, 279, 33716–33726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, J.; Ao, A.; Zhou, L.; Murphy, C.K.; Frist, A.Y.; Keel, J.J.; Thorne, C.A.; Kim, K.; Lee, E.; Hong, C.C. Selective small molecule targeting β-catenin function discovered by in vivo chemical genetic screen. Cell Rep. 2013, 4, 898–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Li, Y.; Shu, M.; Tang, J.; Huang, Y.; Zhou, Y.; Liang, Y.; Yan, G. Hypoxia-inducible factor-1α blocks differentiation of malignant gliomas. Febs J. 2009, 276, 7291–7304. [Google Scholar] [CrossRef]
- Yin, S.; Kaluz, S.; Devi, N.S.; Jabbar, A.A.; De Noronha, R.G.; Mun, J.; Zhang, Z.; Boreddy, P.R.; Wang, W.; Wang, Z.; et al. Arylsulfonamide KCN1 Inhibits In Vivo Glioma Growth and Interferes with HIF Signaling by Disrupting HIF-1a Interaction with Cofactors p300/CBP. Clin. Cancer Res. 2012. [Google Scholar] [CrossRef] [Green Version]
- Emami, K.H.; Nguyen, C.; Ma, H.; Kim, D.H.; Jeong, K.W.; Eguchi, M.; Moon, R.T.; Teo, J.L.; Oh, S.W.; Kim, H.Y.; et al. A small molecule inhibitor of β-catenin/cyclic AMP response element-binding protein transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 12682–12687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- McDermott, J.; Jimeno, A. Belinostat for the treatment of peripheral T-cell lymphomas. Drugs Today 2014, 50, 337–345. [Google Scholar] [CrossRef]
- Richardson, P.G.; Laubach, J.P.; Lonial, S.; Moreau, P.; Yoon, S.S.; Hungria, V.T.; Dimopoulos, M.A.; Beksac, M.; Alsina, M.; San-Miguel, J.F. Panobinostat: A novel pan-deacetylase inhibitor for the treatment of relapsed or relapsed and refractory multiple myeloma. Expert Rev. Anticancer. Ther. 2015, 15, 737–748. [Google Scholar] [CrossRef]
- Mariadason, J.M.; Corner, G.A.; Augenlicht, L.H. Genetic reprogramming in pathways of colonic cell maturation induced by short chain fatty acids: Comparison with trichostatin A, sulindac, and curcumin and implications for chemoprevention of colon cancer. Cancer Res. 2000, 60, 4561–4572. [Google Scholar]
- Knipstein, J.; Gore, L. Entinostat for treatment of solid tumors and hematologic malignancies. Expert Opin. Investig. Drugs 2011, 20, 1455–1467. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Oki, Y.; Bociek, R.G.; Kuruvilla, J.; Fanale, M.; Neelapu, S.; Copeland, A.; Buglio, D.; Galal, A.; Besterman, J.; et al. Mocetinostat for relapsed classical Hodgkin’s lymphoma: An open-label, single-arm, phase 2 trial. Lancet Oncol. 2011, 12, 1222–1228. [Google Scholar] [CrossRef] [Green Version]
- Pauer, L.R.; Olivares, J.; Cunningham, C.; Williams, A.; Grove, W.; Kraker, A.; Olson, S.; Nemunaitis, J. Phase I study of oral CI-994 in combination with carboplatin and paclitaxel in the treatment of patients with advanced solid tumors. Cancer Investig. 2004, 22, 886–896. [Google Scholar] [CrossRef]
- Bilen, M.A.; Fu, S.; Falchook, G.S.; Ng, C.S.; Wheler, J.J.; Abdelrahim, M.; Erguvan-Dogan, B.; Hong, D.S.; Tsimberidou, A.M.; Kurzrock, R.; et al. Phase i trial of valproic acid and lenalidomide in patients with advanced cancer. Cancer Chemother. Pharmacol. 2015, 75, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Rowinsky, E.K.; Villalona, M.A.; Hammond, L.A.; Britten, C.D.; Siu, L.L.; Goetz, A.; Felton, S.A.; Burton, S.; Valone, F.H.; et al. A Phase I study of pivaloyloxymethyl butyrate, a prodrug of the differentiating agent butyric acid, in patients with advanced solid malignancies. Clin. Cancer Res. 2002, 8, 2142–2148. [Google Scholar]
- Iannitti, T.; Palmieri, B. Clinical and experimental applications of sodium phenylbutyrate. Drugs R D 2011, 11, 227–249. [Google Scholar] [CrossRef]
- Frye, R.; Myers, M.; Axelrod, K.C.; Ness, E.A.; Piekarz, R.L.; Bates, S.E.; Booher, S. Romidepsin: A new drug for the treatment of cutaneous T-cell lymphoma. Clin. J. Oncol. Nurs. 2012, 16, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Chowdhry, S.; Zanca, C.; Rajkumar, U.; Koga, T.; Diao, Y.; Raviram, R.; Liu, F.; Turner, K.; Yang, H.; Brunk, E.; et al. NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nature 2019, 569, 570–575. [Google Scholar] [CrossRef]
- Weinstein, I.B. Cancer: Addiction to oncogenes—The Achilles heal of cancer. Science 2002, 297, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Gini, B.; Wykosky, J.; Zanca, C.; Mischel, P.S.; Furnari, F.B.; Cavenee, W.K. A tale of two approaches: Complementary mechanisms of cytotoxic and targeted therapy resistance may inform next-generation cancer treatments. Carcinogenesis 2013, 34, 725–738. [Google Scholar] [CrossRef] [PubMed]

| Family | Nomenclature | Mutation/Aberration in Cancer |
|---|---|---|
| GNAT family | KAT1 | |
| GCN5 (KAT2A) | Cancerous transformation [18] | |
| PCAF (KAT2B) | Missense alteration in cancer [19] | |
| ELP3 (KAT9) | Wnt-driven intestinal tumor initiation [20] | |
| ATAT-1 | ||
| AT-1 | Neurodegenerative features, inflammation and cancer [21] | |
| AT-2 | ||
| P300/CBP family | CBP (KAT3A) | Truncating mutations in ovarian cancer [22] Mutations and deletions in human lung cancer [23] Hematological malignancies [24] |
| P300 (KAT3B) | Tumor suppressor and driver [25] Poor outcome in HNSCC [26] | |
| MYST family | Tip60 (KAT5) | Melanoma and colon cancer [27,28] |
| MOZ (KAT6A) | Association with gain-of-function p53 mutant [29] MOZ-CBP in leukemia [30] | |
| MORF (KAT6B) | AML [31] Leiomyoma [32] | |
| HBO1 (KAT7) | CSC phenotype [33] | |
| MOF (KAT8) | Tumor promoter in GBM [34] |
| Family | Nomenclature | Mutation/Aberration in Cancer |
|---|---|---|
| Class I | HDAC1 | Mutation and CNA in DLBCL [41] |
| HDAC2 | MSI colon cancer [48] | |
| HDAC3 | Liver cancer [49] gastric caner [50] | |
| HDAC8 | Association with inv(16) fusion protein [51] | |
| Class IIa | HDAC4 | Mutation in breast cancer [52] |
| HDAC5 | CNA in HCC [53] | |
| HDAC7 | Lung tumorigenesis [54] | |
| HDAC9 | Medulloblastoma stratification [55] | |
| Class IIb | HDAC6 | Association with ARID1A-mutated ovarian cancers [56] |
| HDAC10 | Lung CSC phenotypes [57] | |
| Class III (sirtuins) | SIRT1 | Tumor promoter or tumor suppressor [58] Stabilization of extrachromosomal amplicons [59] |
| SIRT2 | DNA-damage response proteins by impairing SIRT2 catalytic activity or protein levels [60] | |
| SIRT3 | Linked to ataxia-telangiectasia mutated (ATM) gene deficiency in DLBCL [61] HIF1α destabilization [62] | |
| SIRT4 | Mitochondrial tumor suppressor [63] | |
| SIRT5 | Overexpression in colorectal cancer [64] | |
| SIRT6 | Tumor suppressor including gliomas [65] | |
| SIRT7 | Metastatic phenotypes [66] | |
| Class IV | HDAC11 | Oncogene-induced hematopoiesis in myeloproliferative neoplasms [67] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harachi, M.; Masui, K.; Cavenee, W.K.; Mischel, P.S.; Shibata, N. Protein Acetylation at the Interface of Genetics, Epigenetics and Environment in Cancer. Metabolites 2021, 11, 216. https://doi.org/10.3390/metabo11040216
Harachi M, Masui K, Cavenee WK, Mischel PS, Shibata N. Protein Acetylation at the Interface of Genetics, Epigenetics and Environment in Cancer. Metabolites. 2021; 11(4):216. https://doi.org/10.3390/metabo11040216
Chicago/Turabian StyleHarachi, Mio, Kenta Masui, Webster K. Cavenee, Paul S. Mischel, and Noriyuki Shibata. 2021. "Protein Acetylation at the Interface of Genetics, Epigenetics and Environment in Cancer" Metabolites 11, no. 4: 216. https://doi.org/10.3390/metabo11040216
APA StyleHarachi, M., Masui, K., Cavenee, W. K., Mischel, P. S., & Shibata, N. (2021). Protein Acetylation at the Interface of Genetics, Epigenetics and Environment in Cancer. Metabolites, 11(4), 216. https://doi.org/10.3390/metabo11040216