
Pemetrexed Hinders Translation Inhibition upon Low Glucose in Non-Small Cell Lung Cancer Cells

 , , ,
, , , 
 and
and 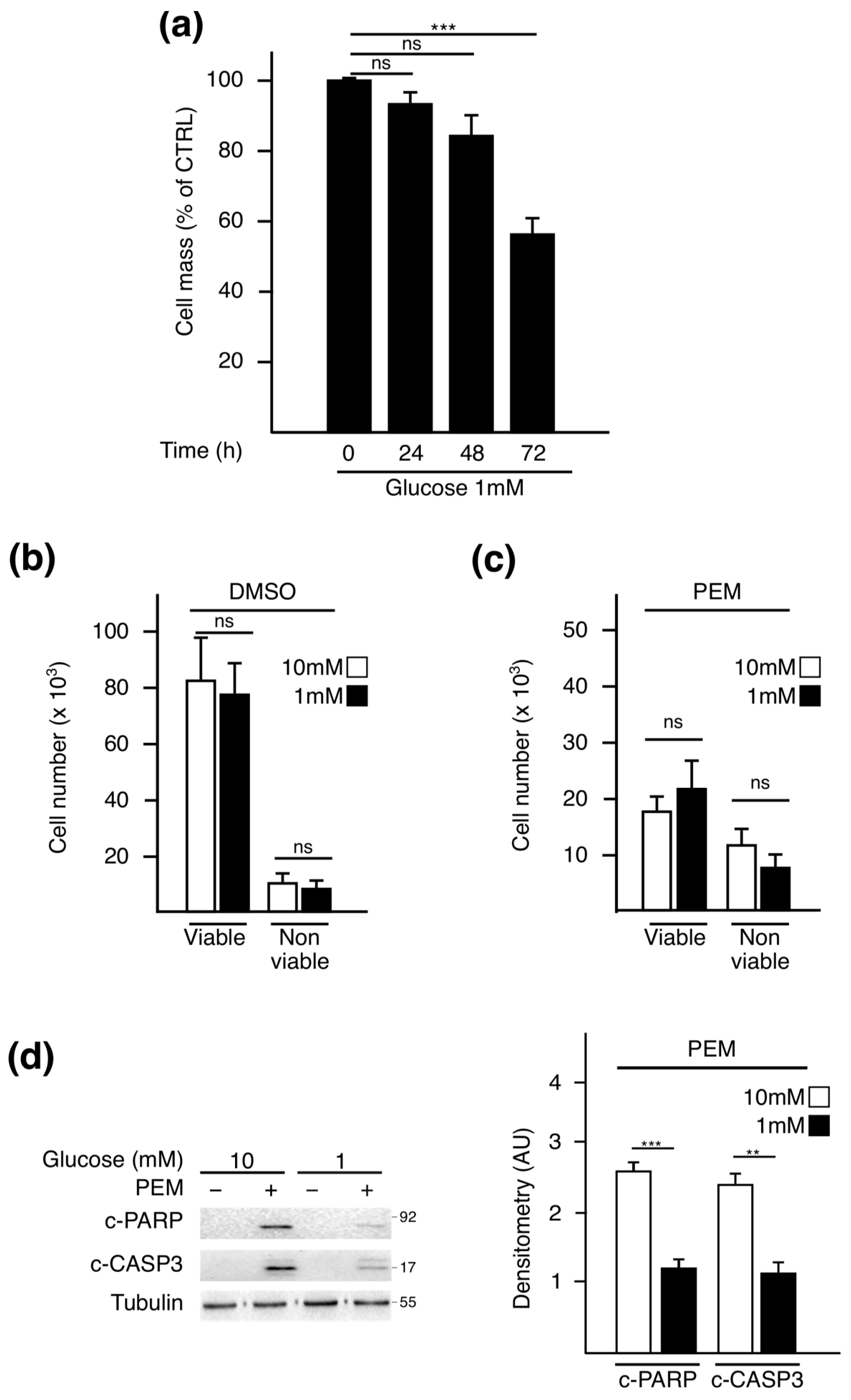{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
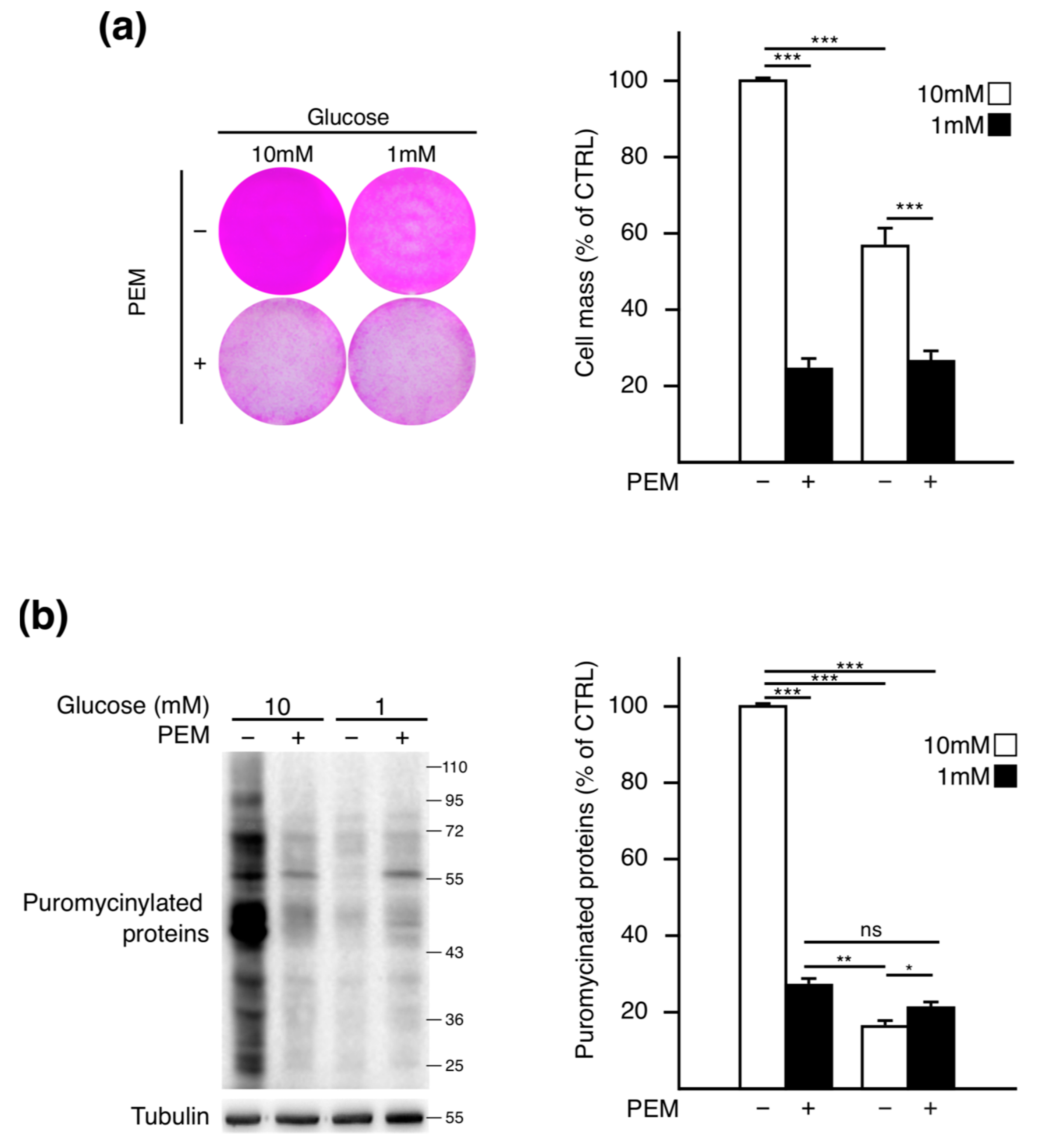
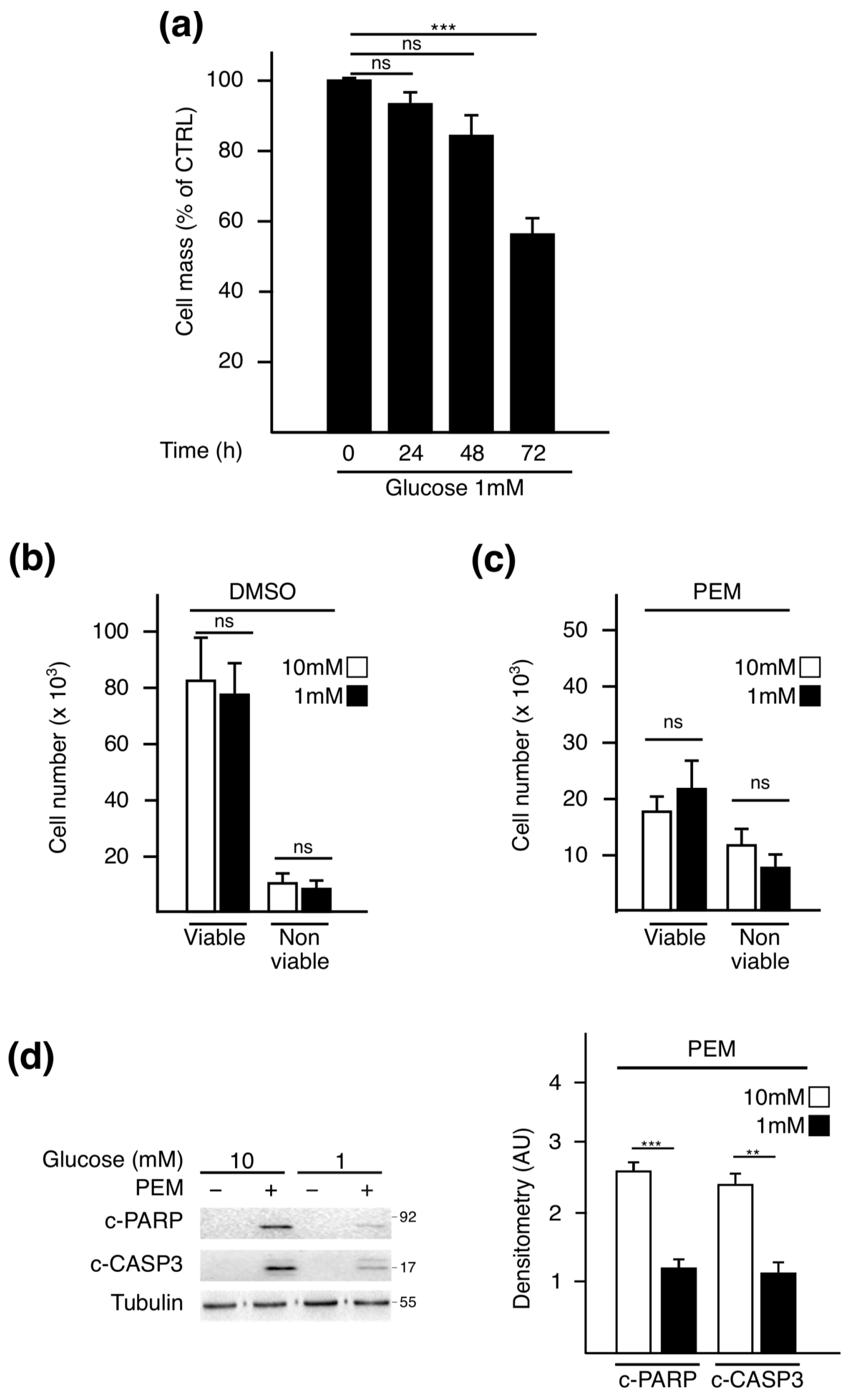
2.1. Pemetrexed Partially Reverts Low Glucose Inhibition of Protein Synthesis
2.2. Pemetrexed Treatment Alleviates UPR Induction in Low Glucose Condition
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Cell Extracts and Western Blot Analysis
4.3. SUnSET Assay
4.4. RNA Extraction and RT-qPCR
4.5. Cell Mass Assay
4.6. Cell Viability Assay
4.7. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Vazquez, A.; Liu, J.; Zhou, Y.; Oltvai, Z.N. Catabolic Efficiency of Aerobic Glycolysis: The Warburg Effect Revisited. BMC Syst. Biol. 2010, 4, 58. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond Aerobic Glycolysis: Transformed Cells Can Engage in Glutamine Metabolism That Exceeds the Requirement for Protein and Nucleotide Synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt Stimulates Aerobic Glycolysis in Cancer Cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef]
- Flier, J.S.; Mueckler, M.M.; Usher, P.; Lodish, H.F. Elevated Levels of Glucose Transport and Transporter Messenger RNA Are Induced by Ras or Src Oncogenes. Sci. New Ser. 1987, 235, 1492–1495. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.-S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. C-Myc Transactivation of LDH-A: Implications for Tumor Metabolism and Growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial Metabolism and ROS Generation Are Essential for Kras-Mediated Tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human Pancreatic Cancer Tumors Are Nutrient Poor and Tumor Cells Actively Scavenge Extracellular Protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef]
- Satoh, K.; Yachida, S.; Sugimoto, M.; Oshima, M.; Nakagawa, T.; Akamoto, S.; Tabata, S.; Saitoh, K.; Kato, K.; Sato, S.; et al. Global Metabolic Reprogramming of Colorectal Cancer Occurs at Adenoma Stage and Is Induced by MYC. Proc. Natl. Acad. Sci. USA 2017, 114, E7697–E7706. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.R.; Danai, L.V.; Lewis, C.A.; Chan, S.H.; Gui, D.Y.; Kunchok, T.; Dennstedt, E.A.; Vander Heiden, M.G.; Muir, A. Quantification of Microenvironmental Metabolites in Murine Cancers Reveals Determinants of Tumor Nutrient Availability. Elife 2019, 8. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal Integration in the Endoplasmic Reticulum Unfolded Protein Response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the Mechanisms of Apoptosis Induced by Endoplasmic Reticulum Stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The Integrated Stress Response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- Huber, A.-L.; Lebeau, J.; Guillaumot, P.; Pétrilli, V.; Malek, M.; Chilloux, J.; Fauvet, F.; Payen, L.; Kfoury, A.; Renno, T.; et al. P58IPK-Mediated Attenuation of the Proapoptotic PERK-CHOP Pathway Allows Malignant Progression upon Low Glucose. Mol. Cell 2013, 49, 1049–1059. [Google Scholar] [CrossRef]
- Van ’t Wout, E.F.A.; Hiemstra, P.S.; Marciniak, S.J. The Integrated Stress Response in Lung Disease. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1005–1009. [Google Scholar] [CrossRef]
- Muaddi, H.; Majumder, M.; Peidis, P.; Papadakis, A.I.; Holcik, M.; Scheuner, D.; Kaufman, R.J.; Hatzoglou, M.; Koromilas, A.E. Phosphorylation of EIF2α at Serine 51 Is an Important Determinant of Cell Survival and Adaptation to Glucose Deficiency. Mol. Biol. Cell 2010, 21, 3220–3231. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Bennett, B.S.; Cullinan, S.B.; Diehl, J.A. PERK and GCN2 Contribute to EIF2α Phosphorylation and Cell Cycle Arrest after Activation of the Unfolded Protein Response Pathway. Mol. Biol. Cell 2005, 16, 5493–5501. [Google Scholar] [CrossRef] [PubMed]
- Guha, P.; Kaptan, E.; Gade, P.; Kalvakolanu, D.V.; Ahmed, H. Tunicamycin Induced Endoplasmic Reticulum Stress Promotes Apoptosis of Prostate Cancer Cells by Activating MTORC1. Oncotarget 2017, 8, 68191–68207. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.-H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-Stress-Induced Transcriptional Regulation Increases Protein Synthesis Leading to Cell Death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Tan, P.; Yan, B.; Gao, R.; Zhao, J.; Wang, J.; Guo, J.; Li, N.; Ma, Z. ER Stress and Autophagy Are Involved in the Apoptosis Induced by Cisplatin in Human Lung Cancer Cells. Oncol. Rep. 2016, 35, 2606–2614. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Justice, R.; Pazdur, R. Approval Summary: Pemetrexed in the Initial Treatment of Advanced/Metastatic Non-Small Cell Lung Cancer. Oncologist 2009, 14, 930–935. [Google Scholar] [CrossRef]
- Cohen, M.H.; Cortazar, P.; Justice, R.; Pazdur, R. Approval Summary: Pemetrexed Maintenance Therapy of Advanced/Metastatic Nonsquamous, Non-Small Cell Lung Cancer (NSCLC). Oncologist 2010, 15, 1352–1358. [Google Scholar] [CrossRef]
- Tedeschi, P.M.; Johnson-Farley, N.; Lin, H.; Shelton, L.M.; Ooga, T.; Mackay, G.; Van Den Broek, N.; Bertino, J.R.; Vazquez, A. Quantification of Folate Metabolism Using Transient Metabolic Flux Analysis. Cancer Metab. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Reich, S.; Nguyen, C.D.L.; Has, C.; Steltgens, S.; Soni, H.; Coman, C.; Freyberg, M.; Bichler, A.; Seifert, N.; Conrad, D.; et al. A Multi-Omics Analysis Reveals the Unfolded Protein Response Regulon and Stress-Induced Resistance to Folate-Based Antimetabolites. Nat. Commun. 2020, 11, 2936. [Google Scholar] [CrossRef]
- Rasheva, V.I.; Domingos, P.M. Cellular Responses to Endoplasmic Reticulum Stress and Apoptosis. Apoptosis 2009, 14, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Uppala, J.K.; Gani, A.R.; Ramaiah, K.V.A. Chemical Chaperone, TUDCA Unlike PBA, Mitigates Protein Aggregation Efficiently and Resists ER and Non-ER Stress Induced HepG2 Cell Death. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Goodwin, J.; Neugent, M.L.; Lee, S.Y.; Choe, J.H.; Choi, H.; Jenkins, D.M.R.; Ruthenborg, R.J.; Robinson, M.W.; Jeong, J.Y.; Wake, M.; et al. The Distinct Metabolic Phenotype of Lung Squamous Cell Carcinoma Defines Selective Vulnerability to Glycolytic Inhibition. Nat. Commun. 2017, 8, 15503. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic Reticulum Stress Signalling—From Basic Mechanisms to Clinical Applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed]
- Brush, M.H.; Weiser, D.C.; Shenolikar, S. Growth Arrest and DNA Damage-Inducible Protein GADD34 Targets Protein Phosphatase 1α to the Endoplasmic Reticulum and Promotes Dephosphorylation of the α Subunit of Eukaryotic Translation Initiation Factor 2. Mol. Cell. Biol. 2003, 23, 1292–1303. [Google Scholar] [CrossRef]
- Yan, W.; Frank, C.L.; Korth, M.J.; Sopher, B.L.; Novoa, I.; Ron, D.; Katze, M.G. Control of PERK EIF2alpha Kinase Activity by the Endoplasmic Reticulum Stress-Induced Molecular Chaperone P58IPK. Proc. Natl. Acad. Sci. USA 2002, 99, 15920–15925. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.C.; Kuhnt, D.; Shih, C.; Rinzel, S.M.; Grindey, G.B.; Barredo, J.; Jannatipour, M.; Moran, R.G. A Dideazatetrahydrofolate Analog Lacking a Chiral Center at C-6: N-[4-[2-(2-Amino-3,4-Dihydro-4-Oxo-7H-Pyrrolo[2,3-d]Pyrimidin-5yl)Ethyl[Benzoyl]-L-Glutamic Acid Is an Inhibitor of Thymidylate Synthase. J. Med. Chem. 1992, 35, 4450–4454. [Google Scholar] [CrossRef]
- Shih, C.; Chen, V.J.; Gossett, L.S.; Gates, S.B.; MacKellar, W.C.; Habeck, L.L.; Shackelford, K.A.; Mendelsohn, L.G.; Soose, D.J.; Patel, V.F.; et al. LY231514, a Pyrrolo[2,3-d]Pyrimidine-Based Antifolate That Inhibits Multiple Folate-Requiring Enzymes. Cancer Res. 1997, 57, 1116–1123. [Google Scholar] [PubMed]
- Chen, V.J.; Bewley, J.R.; Andis, S.L.; Schultz, R.M.; Iversen, P.W.; Shih, C.; Mendelsohn, L.G.; Seitz, D.E.; Tonkinson, J.L. Preclinical Cellular Pharmacology of LY231514 (MTA): A Comparison with Methotrexate, LY309887 and Raltitrexed for Their Effects on Intracellular Folate and Nucleoside Triphosphate Pools in CCRF-CEM Cells. Br. J. Cancer 1998, 78, 27–34. [Google Scholar] [CrossRef][Green Version]
- Racanelli, A.C.; Rothbart, S.B.; Heyer, C.L.; Moran, R.G. Therapeutics by Cytotoxic Metabolite Accumulation: Pemetrexed Causes ZMP Accumulation, AMPK Activation, and MTOR Inhibition. Cancer Res. 2009, 69, 5467–5474. [Google Scholar] [CrossRef]
- Lu, C.-S.; Lin, C.-W.; Chang, Y.-H.; Chen, H.-Y.; Chung, W.-C.; Lai, W.-Y.; Ho, C.-C.; Wang, T.-H.; Chen, C.-Y.; Yeh, C.-L.; et al. Antimetabolite Pemetrexed Primes a Favorable Tumor Microenvironment for Immune Checkpoint Blockade Therapy. J. Immunother. Cancer 2020, 8, e001392. [Google Scholar] [CrossRef]
- Lindqvist, L.M.; Tandoc, K.; Topisirovic, I.; Furic, L. Cross-Talk between Protein Synthesis, Energy Metabolism and Autophagy in Cancer. Curr. Opin. Genet. Dev. 2018, 48, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Rothbart, S.B.; Racanelli, A.C.; Moran, R.G. Pemetrexed Indirectly Activates the Metabolic Kinase AMPK in Human Carcinomas. Cancer Res. 2010, 70, 10299–10309. [Google Scholar] [CrossRef] [PubMed]
- Brunn, G.J.; Hudson, C.C.; Sekulić, A.; Williams, J.M.; Hosoi, H.; Houghton, P.J.; Lawrence, J.C.; Abraham, R.T. Phosphorylation of the Translational Repressor PHAS-I by the Mammalian Target of Rapamycin. Science 1997, 277, 99–101. [Google Scholar] [CrossRef] [PubMed]
- Burnett, P.E.; Barrow, R.K.; Cohen, N.A.; Snyder, S.H.; Sabatini, D.M. RAFT1 Phosphorylation of the Translational Regulators P70 S6 Kinase and 4E-BP1. Proc. Natl. Acad. Sci. USA 1998, 95, 1432–1437. [Google Scholar] [CrossRef] [PubMed]
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. MTOR and S6K1 Mediate Assembly of the Translation Preinitiation Complex through Dynamic Protein Interchange and Ordered Phosphorylation Events. Cell 2005, 123, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Reid, M.A.; Lowman, X.H.; Kulkarni, R.P.; Tran, T.Q.; Liu, X.; Yang, Y.; Hernandez-Davies, J.E.; Rosales, K.K.; Li, H.; et al. Regional Glutamine Deficiency in Tumours Promotes De-Differentiation through Inhibition of Histone Demethylation. Nat. Cell Biol. 2016, 18, 1090–1101. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G. Regulation of MTOR Function in Response to Hypoxia by REDD1 and the TSC1/TSC2 Tumor Suppressor Complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [PubMed]
- Jewer, M.; Lee, L.; Leibovitch, M.; Zhang, G.; Liu, J.; Findlay, S.D.; Vincent, K.M.; Tandoc, K.; Dieters-Castator, D.; Quail, D.F.; et al. Translational Control of Breast Cancer Plasticity. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Markova, B.; Hähnel, P.S.; Kasper, S.; Herbertz, S.; Schuler, M.; Breitenbuecher, F. Pharmacologic Inhibition of MTOR Antagonizes the Cytotoxic Activity of Pemetrexed in Non-Small Cell Lung Cancer. J. Cancer Res. Clin. Oncol. 2012, 138, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Golub, T.R.; Sabatini, D.M. The Immunosuppressant Rapamycin Mimics a Starvation-Like Signal Distinct from Amino Acid and Glucose Deprivation. Mol. Cell. Biol. 2002, 22, 5575–5584. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible Factors: Coupling Glucose Metabolism and Redox Regulation with Induction of the Breast Cancer Stem Cell Phenotype. EMBO J. 2017, 36, 252–259. [Google Scholar] [CrossRef]
- Raz, S.; Sheban, D.; Gonen, N.; Stark, M.; Berman, B.; Assaraf, Y.G. Severe Hypoxia Induces Complete Antifolate Resistance in Carcinoma Cells Due to Cell Cycle Arrest. Cell Death Dis. 2014, 5, e1067. [Google Scholar] [CrossRef] [PubMed]
- Ghaddar, N.; Wang, S.; Woodvine, B.; Krishnamoorthy, J.; Darini, C.; Van Hoef, V.; Popper, H.; Topisirovic, I.; Larsson, O.; Le Quesne, J.; et al. The Integrated Stress Response Exposes a Therapeutic Vulnerability in KRAS Driven Lung Cancer. Eur. J. Cancer 2020, 138, S23. [Google Scholar] [CrossRef]
- Schmidt, E.K.; Clavarino, G.; Ceppi, M.; Pierre, P. SUnSET, a Nonradioactive Method to Monitor Protein Synthesis. Nat. Methods 2009, 6, 275–277. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piecyk, M.; Triki, M.; Laval, P.-A.; Dragic, H.; Cussonneau, L.; Fauvre, J.; Duret, C.; Aznar, N.; Renno, T.; Manié, S.N.; et al. Pemetrexed Hinders Translation Inhibition upon Low Glucose in Non-Small Cell Lung Cancer Cells. Metabolites 2021, 11, 198. https://doi.org/10.3390/metabo11040198
Piecyk M, Triki M, Laval P-A, Dragic H, Cussonneau L, Fauvre J, Duret C, Aznar N, Renno T, Manié SN, et al. Pemetrexed Hinders Translation Inhibition upon Low Glucose in Non-Small Cell Lung Cancer Cells. Metabolites. 2021; 11(4):198. https://doi.org/10.3390/metabo11040198
Chicago/Turabian StylePiecyk, Marie, Mouna Triki, Pierre-Alexandre Laval, Helena Dragic, Laura Cussonneau, Joelle Fauvre, Cédric Duret, Nicolas Aznar, Toufic Renno, Serge N. Manié, and et al. 2021. "Pemetrexed Hinders Translation Inhibition upon Low Glucose in Non-Small Cell Lung Cancer Cells" Metabolites 11, no. 4: 198. https://doi.org/10.3390/metabo11040198
APA StylePiecyk, M., Triki, M., Laval, P.-A., Dragic, H., Cussonneau, L., Fauvre, J., Duret, C., Aznar, N., Renno, T., Manié, S. N., Chaveroux, C., & Ferraro-Peyret, C. (2021). Pemetrexed Hinders Translation Inhibition upon Low Glucose in Non-Small Cell Lung Cancer Cells. Metabolites, 11(4), 198. https://doi.org/10.3390/metabo11040198