
Investigation of Equine In Vivo and In Vitro Derived Metabolites of the Selective Androgen Receptor Modulator (SARM) ACP-105 for Improved Doping Control

 ,
, 
Abstract

1. Introduction
2. Results and Discussion
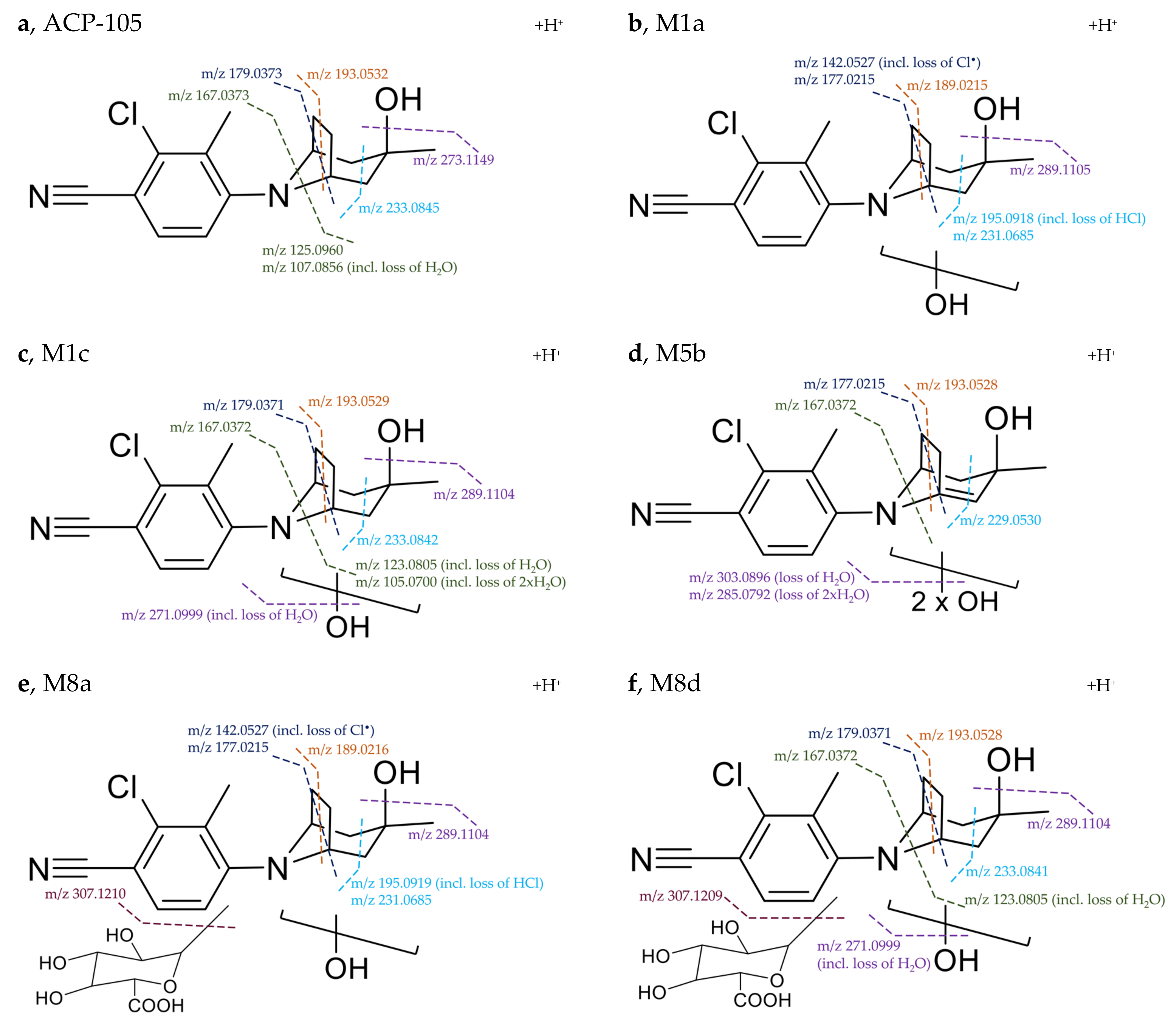
2.1. Detection of Parent ACP-105
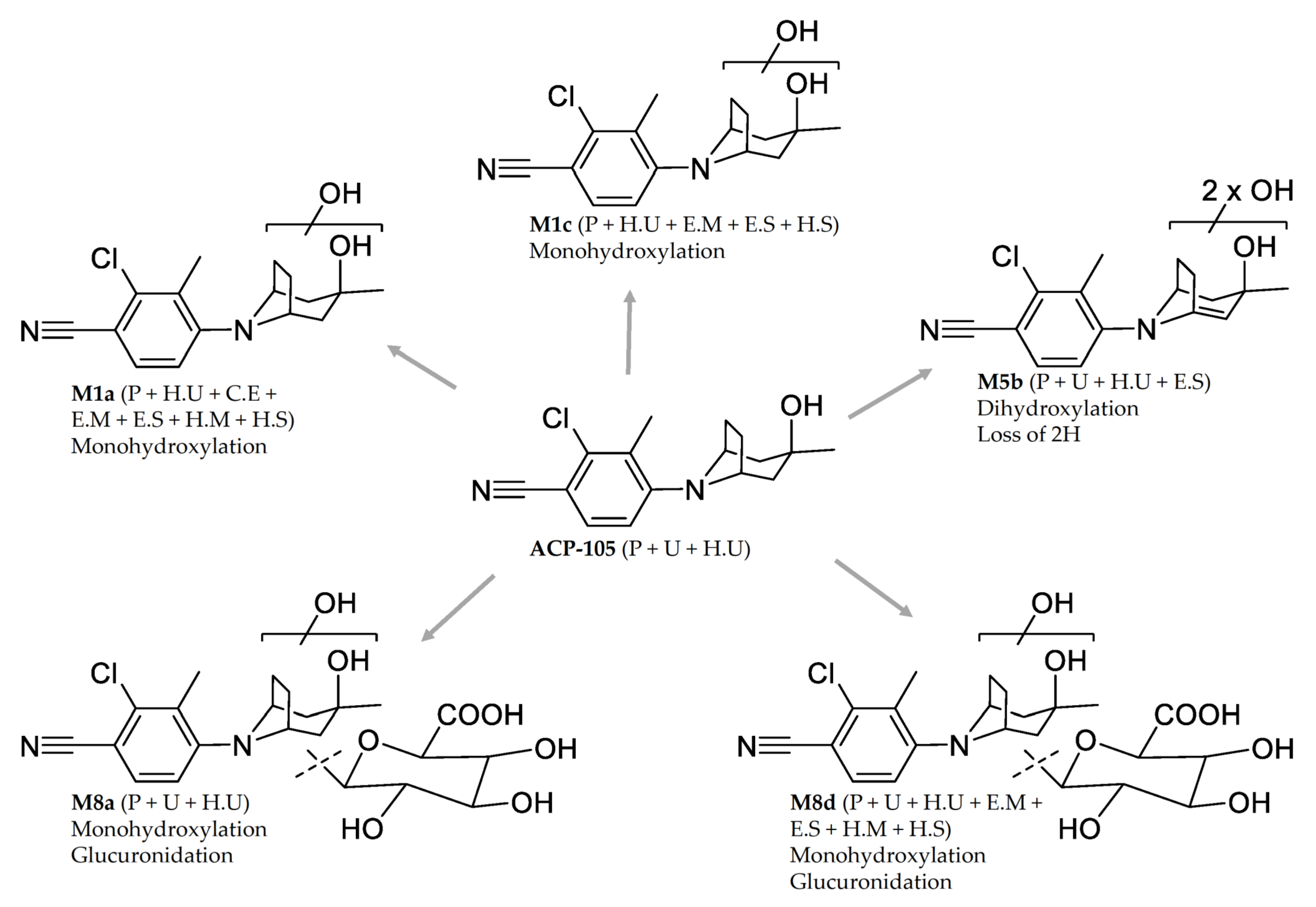
2.2. In Vivo Metabolites
Structural Elucidation of Major Metabolites
2.3. Metabolites from C. elegans Incubations
2.4. Metabolites from Microsome and S9 Fraction Incubations
2.5. Suggested Analytical Target for ACP-105 Equine Doping Analysis
3. Materials and Methods
3.1. Chemicals
3.2. Drug Administration and Sample Collection
3.3. Urine Sample Preparation
3.3.1. Sample Dilution
3.3.2. Solid-Phase Extraction with HLB
3.3.3. Hydrolysis with β-Glucuronidase
3.4. Plasma Sample Preparation
3.5. Microsomes and S9 Fraction Incubation
3.6. C. elegans Incubation
3.7. Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Elemental Composition [M+H]+ | rt (min) | Th. m/z (T) Ex. m/z (E) ppm | Fragment m/z (ppm) | Neutral Loss | Detectability after Administration | |
|---|---|---|---|---|---|---|---|
ACP-105 | C16H20ClN2O+ | 11.59 | T: 291.1259 E: 291.1262 −1.2 ppm | 273.1149 (1.4) 233.0845 (−2.2) 193.0532 (−2.6) 179.0373 (−1.4) 167.0373 (−1.5) 125.0960 (0.7) 107.0856 (−0.7) | H2O C3H6O C6H10O C7H12O C8H12O C8H7ClN2 C8H7ClN2, H2O | Plasma: Urine: H. urine: | 24/12 h 24/24 h 48/48 h |
M1a | Monohydroxylation C16H20ClN2O2+ | 8.65 | T: 307.1208 E: 307.1209 −0.5 ppm | 289.1105 (−1.0) 231.0685 (−0.7) 195.0918 (−0.7) 189.0215 (−0.6) 177.0215 (−0.6) 142.0527 (−1.4) 107.0856 (−0.7) | H2O C3H8O2 C3H8O2, HCl C6H14O2 C7H14O2 C7H14O2, Cl• C8H5ClN2, 2xH2O | Plasma: Urine: H. urine: | 36/24h - 96/96 h |
M1b | Monohydroxylation C16H20ClN2O2+ | 9.00 | T: 307.1208 E: 307.1210 −0.8 ppm | 289.1104 (−0.7) 271.0999 (−1.0) 233.0477 (−0.4) 193.0529 (−1.1) 179.0371 (−0.3) 142.0527 (−1.4) 105.0696 (2.6) | H2O 2xH2O C4H10O C6H10O2 C7H12O2 C7H14O2, Cl• C8H7ClN2, 2xH2O | Plasma: Urine: H. urine: | - - 48/72 h |
M1c | Monohydroxylation C16H20ClN2O2+ | 9.14 | T: 307.1208 E: 307.1209 −0.5 ppm | 289.1104 (−0.7) 271.0999 (−1.0) 233.0842 (−0.9) 193.0529 (−1.1) 179.0371 (−0.3) 167.0372 (−0.9) 123.0805 (−0.5) 105.0700 (−1.2) | H2O 2xH2O C3H6O2 C6H10O2 C7H12O2 C8H12O2 C8H7ClN2, H2O C8H7ClN2, 2xH2O | Plasma: Urine: H. urine: | 36/24 h - 96/96 h |
M2a | Dihydroxylation C16H20ClN2O3+ | 7.03 | T: 323.1157 E: 323.1159 −0.7 ppm | 305.1053 (−0.6) 231.0685 (−0.7) 195.0918 (−0.7) 189.0215 (−0.6) 177.0215 (−0.6) 142.0527 (−1.4) | H2O C3H8O3 C3H8O3, HCl C6H14O3 C7H14O3 C7H14O3, Cl• | Plasma: Urine: H. urine: | 36/48 h -/7 h 48/24 h |
M2b | Dihydroxylation C16H20ClN2O3+ | 7.63 | T: 323.1157 E: 323.1161 −1.3 ppm | 305.1053 (−0.6) 287.0948 (−0.9) 247.0635 (−1.0) 229.0529 (−0.9) 194.0840 (−1.0) 179.0604 (0.0) 142.0527 (−1.4) | H2O 2xH2O C3H8O2 C3H10O3 C3H10O3, Cl• C4H12O3, HCl C7H14O3, Cl• | Plasma: Urine: H. urine: | - - 72/48 h |
M3a 1 | Loss of 2H C16H18ClN2O+ | 10.11 | T: 289.1102 E: 289.1106 −1.4 ppm | 271.1000 (−1.3) 243.0685 (−0.7) 233.0477 (−0.4) 179.0371 (−0.3) 177.0214 (0.0) 142.0526 (−0.7) 111.0805 (−0.6) | H2O C2H6O C4H8 C7H10O C7H12O C7H12O, Cl• C9H7ClN2 | Plasma: Urine: H. urine: | - - 24/24 h |
M3b 1,2 | Loss of 2H C16H18ClN2O+ | 11.56 | T: 289.1102 E: 289.1108 −2.1 ppm | 271.1000 (−1.3) 229.0523 (1.7) 203.0370 (0.2) 183.0321 (−0.8) 165.0215 (−0.7) | H2O C3H8O C5H10O C8H10 C8H12O | Plasma: Urine: H. urine: | - 24/7 h 48/48 h |
M4a 1 | Monohydroxylation + loss of 2H C16H18ClN2O2+ | 8.88 | T: 305.1051 E: 305.1054 −0.9 ppm | 287.0949 (−1.2) 245.0842 (−0.9) 229.0529 (−0.9) 194.0840 (−1.0) 179.0605 (−0.7) 142.0527 (−1.4) 123.0805 (−0.5) | H2O C2H4O2 C3H8O2 C3H8O2, Cl• C4H10O2, HCl C7H12O2, Cl• C8H5ClN2, H2O | Plasma: Urine: H. urine: | - -/7 h 48/48 h |
M4b 1 | Monohydroxylation + loss of 2H C16H18ClN2O2+ | 9.83 | T: 305.1051 E: 305.1053 −0.6 ppm | 223.0270 (−0.6) 177.0215 (−0.6) 142.0527 (−1.4) 111.0441 (−0.5) | C6H10 C7H12O2 C7H12O2, Cl• C10H11ClN2 | Plasma: Urine: H. urine: | - - 72/48 h |
M5a 1,2 | Dihydroxylation + loss of 2H C16H18ClN2O3+ | 9.17 | T: 321.1000 E: 321.1005 −1.6 ppm | 303.0897 (−0.8) 285.0793 (−1.4) 229.0530 (−1.3) 179.0375 (−2.6) | H2O 2xH2O C3H8O3 C7H10O3 | Plasma: Urine: H. urine: | - 24/7 h 24/24 h |
M5b 1 | Dihydroxylation + loss of 2H C16H18ClN2O3+ | 9.57 | T: 321.1000 E: 321.1003 −0.9 ppm | 303.0896 (−0.5) 285.0792 (−1.0) 275.0949 (−1.3) 229.0530 (−1.3) 217.0529 (−1.0) 193.0528 (−0.6) 177.0215 (−0.6) 167.0372 (−0.9) 142.0527 (−1.4) | H2O 2xH2O C1H2O2 C3H8O3 C4H8O3 C6H8O3 C7H12O3 C8H10O3 C7H12O3, Cl• | Plasma: Urine: H. urine: | 36/36 h 96/96 h 96/96 h |
M6a 1 | Trihydroxylation + loss of 2H C16H18ClN2O4+ | 7.21 | T: 337.0950 E: 337.0953 −1.1 ppm | 319.0847 (−1.0) 179.0372 (−0.9) 142.0527 (−1.4) 105.0336 (−1.1) | H2O C7H10O4 C7H12O4, Cl• C9H7ClN2, 3xH2O | Plasma: Urine: H. urine: | - 72/24 h 72/48 h |
M6b 1 | Trihydroxylation + loss of 2H C16H18ClN2O4+ | 7.47 | T: 337.0950 E: 337.0951 −0.5 ppm | 319.0845 (−0.4) 231.0686 (−1.1) 195.0918 (−0.7) 189.0215 (−0.6) 177.0215 (−0.6) 142.0527 (−1.4) | H2O C3H6O4 C3H6O4, HCl C6H12O4 C7H12O4 C7H12O4, Cl• | Plasma: Urine: H. urine: | -/6 h 72/24 h 72/72 h |
M6c 1,2 | Trihydroxylation + loss of 2H C16H18ClN2O4+ | 9.04 | T: 337.0950 E: 337.0955 −1.7 ppm | 319.0847 (−1.0) 301.0742 (−1.3) | H2O 2xH2O | Plasma: Urine: H. urine: | - 24/24 h 6/24 h |
| M7 | Glucuronidation C22H28ClN2O7+ | 8.95 | T: 467.1580 E: 467.1580 0.0 ppm | 291.1259 (0.0) 273.1155 (−0.8) 233.0841 (−0.5) 193.0529 (−1.1) 179.0371 (−0.3) 167.0372 (−0.9) 142.0527 (−1.4) 107.0856 (−0.7) | C6H8O6 H2O, C6H8O6 C3H6O, C6H8O6 C6H10O, C6H8O6 C7H12O, C6H8O6 C8H12O, C6H8O6 C7H14O, Cl•, C6H8O6 C8H5ClN2, 2xH2O, C6H8O6 | Plasma: Urine: H. urine: | -/6 h 24/24 h - |
 | |||||||
| M8a | Monohydroxylation + glucuronidation C22H28ClN2O8+ | 7.13 | T: 483.1529 E: 483.1531 −0.4 ppm | 307.1210 (−0.8) 289.1104 (−0.7) 231.0685 (−0.7) 195.0919 (−1.2) 189.0216 (−1.1) 177.0215 (−0.6) 142.0527 (−1.4) | C6H8O6 H2O, C6H8O6 C3H8O2, C6H8O6 C3H8O2, HCl, C6H8O6 C6H14O2, C6H8O6 C7H14O2, C6H8O6 C7H14O2, Cl•, C6H8O6 | Plasma: Urine: H. urine: | 36/36 h 48/48 h 6/7 h |
 | |||||||
| M8b | Monohydroxylation + glucuronidation C22H28ClN2O8+ | 7.61 | T: 483.1529 E: 483.1527 0.4 ppm | 465.1428 (−1.2) 289.1105 (−1.0) 271.0999 (−1.0) 217.0529 (−1.0) 179.0372 (−0.9) 123.0805 (−0.5) | H2O H2O, C6H8O6 2xH2O, C6H8O6 C4H10O2, C6H8O6 C7H12O2, C6H8O6 C8H7ClN2, H2O, C6H8O6 | Plasma: Urine: H. urine: | - 6/7 h - |
 | |||||||
| M8c | Monohydroxylation + glucuronidation C22H28ClN2O8+ | 7.85 | T: 483.1529 E: 483.1524 1.0 ppm | 307.1210 (−0.8) 289.1105 (−1.0) 271.1001 (−1.7) 233.0481 (−2.1) 193.0530 (−1.6) 179.0372 (−0.9) 142.0528 (−2.1) | C6H8O6 H2O, C6H8O6 2xH2O, C6H8O6 C4H10O, C6H8O6 C6H10O2, C6H8O6 C7H12O2, C6H8O6 C7H14O2, Cl•, C6H8O6 | Plasma: Urine: H. urine: | - 24/24 h - |
 | |||||||
| M8d | Monohydroxylation + glucuronidation C22H28ClN2O8+ | 8.00 | T: 483.1529 E: 483.1530 −0.2 ppm | 465.1425 (−0.5) 307.1209 (−0.5) 289.1104 (−0.7) 271.0999 (−1.0) 233.0841 (−0.5) 193.0528 (−0.6) 179.0371 (−0.3) 167.0372 (−0.9) 123.0805 (−0.5) | H2O C6H8O6 H2O, C6H8O6 2xH2O, C6H8O6 C3H6O2, C6H8O6 C6H10O2, C6H8O6 C7H12O2, C6H8O6 C8H12O2, C6H8O6 C8H7ClN2, H2O, C6H8O6 | Plasma: Urine: H. urine: | 36/36 h 96/48 h -/7 h |
 | |||||||
| M9a | Dihydroxylation + glucuronidation C22H28ClN2O9+ | 6.21 | T: 499.1478 E: 499.1470 1.6 ppm | 323.1155 (0.5) 305.1053 (−0.6) 287.0947 (−0.5) 231.0685 (−0.7) 195.0919 (−1.2) 142.0527 (−1.4) | C6H8O6 H2O, C6H8O6 2xH2O, C6H8O6 C3H8O3, C6H8O6 C3H8O3, HCl, C6H8O6 C7H14O3, Cl•, C6H8O6 | Plasma: Urine: H. urine: | - 48/24 h 6/7 h |
 | |||||||
| M9b | Dihydroxylation + glucuronidation C22H28ClN2O9+ | 6.52 | T: 499.1478 E: 499.1478 0.0 ppm | 481.1376 (−0.9) 323.1160 (−1.0) 305.1053 (−0.6) 287.0948 (−0.9) 247.0635 (−1.0) 231.0685 (−0.7) 195.0918 (−0.7) 142.0527 (−1.4) | H2O C6H8O6 H2O, C6H8O6 2xH2O, C6H8O6 C3H8O2, C6H8O6 C3H8O3, C6H8O6 C3H8O3, HCl, C6H8O6 C7H14O3, Cl•, C6H8O6 | Plasma: Urine: H. urine: | - 48/24 h - |
 | |||||||
References
- Gao, W.; Dalton, J.T. Expanding the therapeutic use of androgens via selective androgen receptor modulators (SARMs). Drug Discov. Today 2007, 12, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Negro-Vilar, A. Selective androgen receptor modulators (SARMs): A Novel Approach to Androgen Therapy for the New Millennium. J. Clin. Endocrinol. Metab. 1999, 84, 3459–3462. [Google Scholar] [CrossRef] [PubMed]
- Thevis, M.; Schänzer, W. Detection of SARMs in doping control analysis. Mol. Cell. Endocrinol. 2018, 464, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Dalton, J.T.; Taylor, R.P.; Mohler, M.L.; Steiner, M.S. Selective androgen receptor modulators for the prevention and treatment of muscle wasting associated with cancer. Curr. Opin. Support. Palliat. Care 2013, 7, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Papanicolaou, D.A.; Ather, S.N.; Zhu, H.; Zhou, Y.; Lutkiewicz, J.; Scott, B.B.; Chandler, J. A phase IIA randomized, placebo-controlled clinical trial to study the efficacy and safety of the selective androgen receptor modulator (SARM), MK-0773 in female participants with sarcopenia. J. Nutr. Health Aging 2013, 17, 533–543. [Google Scholar] [CrossRef] [PubMed]
- World Anti-Doping Agency. The World Anti-Doping Code—The 2008 Prohibitied List International Standard. Available online: https://www.wada-ama.org/sites/default/files/resources/files/WADA_Prohibited_List_2008_EN.pdf (accessed on 3 June 2020).
- World Anti-Doping Agency. The World Anti-Doping Code—The 2020 Prohibited List International Standard. Available online: https://www.wada-ama.org/sites/default/files/wada_2020_english_prohibited_list_0.pdf (accessed on 3 June 2020).
- International Federation of Horseracing Authorities—The International Agreement on Breeding, Racing and Wagering. Available online: https://www.ifhaonline.org/resources/ifAgreement.pdf (accessed on 26 June 2020).
- Grata, E.; Perrenoud, L.; Saugy, M.; Baume, N. SARM-S4 and metabolites detection in sports drug testing: A case report. Forensic Sci. Int. 2011, 213, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Cawley, A.T.; Smart, C.; Greer, C.; Liu Lau, M.; Keledjian, J. Detection of the selective androgen receptor modulator andarine (S-4) in a routine equine blood doping control sample. Drug Test. Anal. 2016, 8, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Starcevic, B.; Ahrens, B.D.; Butch, A.W. Detection of the selective androgen receptor modulator S-4 (Andarine) in a doping control sample. Drug Test. Anal. 2013, 5, 377–379. [Google Scholar] [CrossRef] [PubMed]
- Cox, H.D.; Eichner, D. Detection of LGD-4033 and its metabolites in athlete urine samples. Drug Test. Anal. 2017, 9, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Thevis, M.; Lagojda, A.; Kuehne, D.; Thomas, A.; Dib, J.; Hansson, A.; Hedeland, M.; Bondesson, U.; Wigger, T.; Karst, U.; et al. Characterization of a non-approved selective androgen receptor modulator drug candidate sold via the Internet and identification of in vitro generated phase-I metabolites for human sports drug testing. Rapid Commun. Mass Spectrom. 2015, 29, 991–999. [Google Scholar] [CrossRef]
- Thevis, M.; Thomas, A.A.; Piper, T.S.; Krug, O.R.; Delahaut, P.P.; Schänzer, W.L. Liquid chromatography-high resolution/high accuracy (tandem) mass spectrometrybased identification of in vivo generated metabolites of the selective androgen receptor modulator ACP-105 for doping control purposes. Eur. J. Mass Spectrom. 2014, 20, 73–83. [Google Scholar] [CrossRef]
- Guddat, S.; Fußhöller, G.; Beuck, S.; Thomas, A.; Geyer, H.; Rydevik, A.; Bondesson, U.; Hedeland, M.; Lagojda, A.; Schänzer, W.; et al. Synthesis, characterization, and detection of new oxandrolone metabolites as long-term markers in sports drug testing. Anal. Bioanal. Chem. 2013, 405, 8285–8294. [Google Scholar] [CrossRef]
- ILAC G7:02/2016 Accreditation Requirements and Operating Criteria for Horseracing Laboratories. Available online: https://ilac.org/publications-and-resources/ilac-guidance-series/ (accessed on 3 July 2020).
- World Anti-Doping Agency. The World Anti-Doping Code—International Standards for Laboratories 9.0. 2016. Available online: https://www.wada-ama.org/sites/default/files/resources/files/isl_june_2016.pdf (accessed on 3 November 2020).
- Zhang, D.; Yang, Y.; Leakey, J.E.A.; Cerniglia, C.E. Phase I and phase II enzymes produced by Cunninghamella elegans for the metabolism of xenobiotics. FEMS Microbiol. Lett. 1996, 138, 221–226. [Google Scholar] [CrossRef][Green Version]
- Åberg, A.T.; Löfgren, H.; Bondesson, U.; Hedeland, M. Structural elucidation of N-oxidized clemastine metabolites by liquid chromatography/tandem mass spectrometry and the use of Cunninghamella elegans to facilitate drug metabolite identification. Rapid Commun. Mass Spectrom. 2010, 24, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Hansson, A.; Knych, H.; Stanley, S.; Berndtson, E.; Jackson, L.; Bondesson, U.; Thevis, M.; Hedeland, M. Equine in vivo-derived metabolites of the SARM LGD-4033 and comparison with human and fungal metabolites. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1074–1075, 91–98. [Google Scholar] [CrossRef]
- Rydevik, A.; Lagojda, A.; Thevis, M.; Bondesson, U.; Hedeland, M. Isolation and characterization of a β-glucuronide of hydroxylated SARM S1 produced using a combination of biotransformation and chemical oxidation. J. Pharm. Biomed. Anal. 2014, 98, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Schlienger, N.; Lund, B.W.; Pawlas, J.; Badalassi, F.; Bertozzi, F.; Lewinsky, R.; Fejzic, A.; Thygesen, M.B.; Tabatabaei, A.; Bradley, S.R.; et al. Synthesis, structure-activity relationships, and characterization of novel nonsteroidal and selective androgen receptor modulators. J. Med. Chem. 2009, 52, 7186–7191. [Google Scholar] [CrossRef] [PubMed]
- Thevis, M.; Piper, T.; Beuck, S.; Geyer, H.; Schänzer, W. Expanding sports drug testing assays: Mass spectrometric characterization of the selective androgen receptor modulator drug candidates RAD140 and ACP-105. Rapid Commun. Mass Spectrom. 2013, 27, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Subhahar, M.B.; Karakka Kal, A.K.; Philip, M.; Karatt, T.K.; Ibrahimwaseem, N.; Vazhat, R.A.; Muhammed Ajeebsanu, M.P. Detection and identification of ACP-105 and its metabolites in equine urine using LC/MS/MS after oral administration. Drug Test. Anal. 2020. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Broberg, M.N.; Knych, H.; Bondesson, U.; Pettersson, C.; Stanley, S.; Thevis, M.; Hedeland, M. Investigation of Equine In Vivo and In Vitro Derived Metabolites of the Selective Androgen Receptor Modulator (SARM) ACP-105 for Improved Doping Control. Metabolites 2021, 11, 85. https://doi.org/10.3390/metabo11020085
Broberg MN, Knych H, Bondesson U, Pettersson C, Stanley S, Thevis M, Hedeland M. Investigation of Equine In Vivo and In Vitro Derived Metabolites of the Selective Androgen Receptor Modulator (SARM) ACP-105 for Improved Doping Control. Metabolites. 2021; 11(2):85. https://doi.org/10.3390/metabo11020085
Chicago/Turabian StyleBroberg, Malin Nilsson, Heather Knych, Ulf Bondesson, Curt Pettersson, Scott Stanley, Mario Thevis, and Mikael Hedeland. 2021. "Investigation of Equine In Vivo and In Vitro Derived Metabolites of the Selective Androgen Receptor Modulator (SARM) ACP-105 for Improved Doping Control" Metabolites 11, no. 2: 85. https://doi.org/10.3390/metabo11020085
APA StyleBroberg, M. N., Knych, H., Bondesson, U., Pettersson, C., Stanley, S., Thevis, M., & Hedeland, M. (2021). Investigation of Equine In Vivo and In Vitro Derived Metabolites of the Selective Androgen Receptor Modulator (SARM) ACP-105 for Improved Doping Control. Metabolites, 11(2), 85. https://doi.org/10.3390/metabo11020085