The Pentose Phosphate Pathway Dynamics in Cancer and Its Dependency on Intracellular pH

 , , ,
, , ,  ,
,  and
and
Abstract
1. Introduction
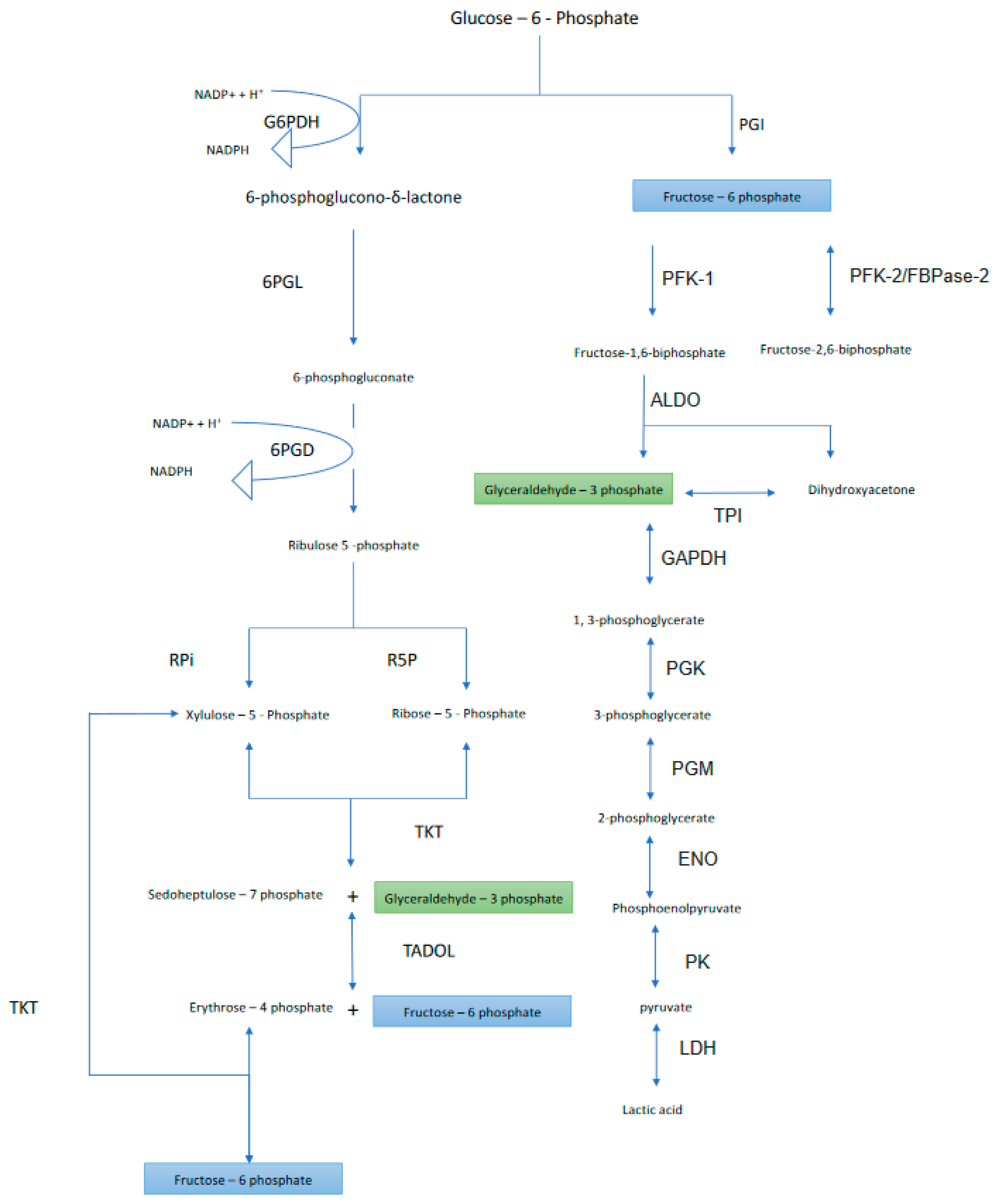
2. Oxidative Phase
2.1. First Step
Evolutionary Consequences of Population Selected G6PDH Deficiency
- G6PDH deficiency confers natural resistance against malaria [26].
- Free radical scavengers such as NADPH are cytoplasmic reducing agents (see below). In other words, NADPH is a cytoplasmic free radical scavenger. Overproduction of free radicals, reactive oxygen species (ROS) and reactive nitrogen species (RNS) by NAD(P)H oxidase isoforms and NO synthase respectively, induces cellular senescence and apoptosis [30], as well as necrosis [31].
- Cancer cells induce anti-apoptotic proteins [32] and are devoid of pro-apoptotic protein expression [33]. Hence, the ratio of the anti-apoptotic/apoptotic proteins is indicative of increased malignancy [34]. NADPH increases this ratio and, therefore, decreases apoptosis [35,36]. Also, the pro-apoptotic protein, BCL-2, has anti-oxidant properties [37], which increase the perturbations of the cellular redox status. So, two questions arise: is it preventive or prompting? And does it depend on the stage of the tumor?
2.2. Second Step
2.3. Third Step
3. Nonoxidative Phase
3.1. Fourth Step
3.1.1. Ribose-5 Phosphate Formation
3.1.2. Xylulose 5-Phosphate Formation
3.2. Fifth Step
3.3. Sixth Step
3.4. Seventh Step
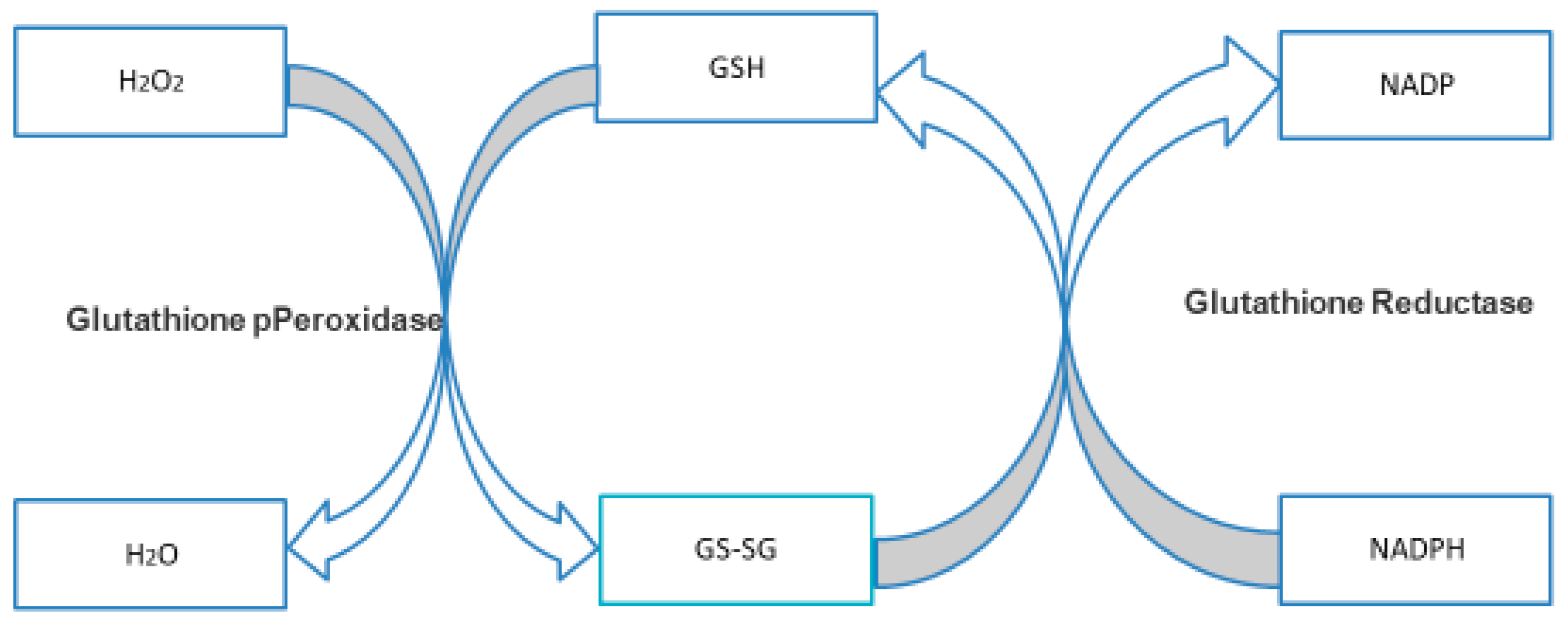
3.4.1. NADPH and GSH
3.4.2. The Overview of PPP and Cancer
3.4.3. The Possible Crosstalk between the Glycolysis and the Pentose Phosphate Pathway (NAPDH Is a DoublE-edged Sword)
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| G6PDH | Glucose-6-phosphate dehydrogenase |
| 6PGL | 6-phosphogluconolactonase |
| 6PGD | 6-phosphogluconate dehydrogenase |
| R5P | Ribose-5-phosphate isomerase |
| RPE | Ribulose 5-Phosphate 3-Epimerase |
| TKT | Transketolase |
| TADOL | Transaldolase |
| PGI | Phosphoglucose isomerase |
| PFK1 | Phosphofructokinase-1 |
| PFK2 | Phosphofructokinase-2 |
| ALDO | Fructose-bisphosphate aldolase |
| TPI | Triosephosphate isomerase |
| GAPDH | Glyceraldehyde phosphate dehydrogenase |
| PGK | Phosphoglycerate kinase |
| PGM | Phosphoglycerate mutase |
| ENO | Enolase |
| PK | Pyruvate Kinase |
| LDH | Lactate Dehydrogenase |
References
- Warburg, O.; Christian, W.; Griese, A. Wasserstoff{ü}bertragendes Co-Ferment, seine Zusammensetzung und Wirkungsweise. Biochem. Z. 1935, 282, 157–205. [Google Scholar]
- Warburg, O.; Christian, W. Optischer Nachweis der Hydrierung und Dehydrierung des Pyridins im Gärungs-Co-Ferment. Biochem. Z. 1936, 286, 81. [Google Scholar]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.-M.; Krüger, A.; Alam, M.T.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. 2015, 90, 927–963. [Google Scholar] [CrossRef] [PubMed]
- Alfarouk, K.O.; Verduzco, D.; Rauch, C.; Muddathir, A.K.; Bashir, A.H.H.; Elhassan, G.O.; Ibrahim, M.E.; Orozco, J.D.P.; Cardone, R.A.; Reshkin, S.J.; et al. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience 2014, 1, 777. [Google Scholar] [CrossRef] [PubMed]
- Olive, C.; Levy, H.R. The preparation and some properties of crystalline glucose 6-phosphate dehydrogenase from Leuconostoc mesenteroides. Biochemistry 1967, 6, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Anderson, W.B.; Nordlie, R.C. Glucose dehydrogenase activity of yeast glucose 6-phosphate dehydrogenase. I. Selective stimulation by bicarbonate, phosphate, and sulfate. Biochemistry 1968, 7, 1479–1485. [Google Scholar] [CrossRef]
- Türkoğlu, V.; Aldemir, S.; Çiftçi, M. Purification and Characterization of Glucose 6-Phosphate Dehydrogenase from Sheep Liver. Turk. J. Chem. 2003, 27, 395–402. [Google Scholar]
- Mahmoud, A.I.; Abdel-Hady, M.G.; Ahmed, M.H.S.; Mohamed, A.G.; Mohamed, M.A.-M. Purification and Characterization of glucose-6-phosphate Dehydrogenase From Camel Liver. Enzym. Res. 2014, 2014, 714054. [Google Scholar]
- Pessôa, B.S.; Peixoto, E.B.M.I.; Papadimitriou, A.; Lopes de Faria, J.M.; Lopes de Faria, J.B. Spironolactone improves nephropathy by enhancing glucose-6-phosphate dehydrogenase activity and reducing oxidative stress in diabetic hypertensive rat. J. Renin-Angiotensin-Aldosterone Syst. 2012, 13, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Manos, P.; Nakayama, R.; Holten, D. Regulation of glucose-6-phosphate dehydrogenase synthesis and mRNA abundance in cultured rat hepatocytes. Biochem. J. 1991, 276 Pt 1, 245–250. [Google Scholar] [CrossRef]
- Salati, L.M.; Adkins-Finke, B.; Clarke, S.D. Free fatty acid inhibition of the insulin induction of glucose-6-phosphate dehydrogenase in rat hepatocyte monolayers. Lipids 1988, 23, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Apse, K.; Pang, J.; Stanton, R.C. High glucose inhibits glucose-6-phosphate dehydrogenase via cAMP in aortic endothelial cells. J. Biol. Chem. 2000, 275, 40042–40047. [Google Scholar] [CrossRef] [PubMed]
- Catanzaro, D.; Gaude, E.; Orso, G.; Giordano, C.; Guzzo, G.; Rasola, A.; Ragazzi, E.; Caparrotta, L.; Frezza, C.; Montopoli, M. Inhibition of glucose-6-phosphate dehydrogenase sensitizes cisplatin-resistant cells to death. Oncotarget 2015, 6, 30102–30114. [Google Scholar] [CrossRef] [PubMed]
- Mele, L.; La Noce, M.; Paino, F.; Regad, T.; Wagner, S.; Liccardo, D.; Papaccio, G.; Lombardi, A.; Caraglia, M.; Tirino, V.; et al. Glucose-6-phosphate dehydrogenase blockade potentiates tyrosine kinase inhibitor effect on breast cancer cells through autophagy perturbation. J. Exp. Clin. Cancer Res. 2019, 38, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Cai, P.; Xu, C.; Cao, D.; Yu, W.; Zhao, Z.; Huang, M.; Jin, J. Inhibition of glucose-6-phosphate dehydrogenase reverses cisplatin resistance in lung cancer cells via the redox system. Front. Pharmacol. 2018, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef]
- Horne, R.N.; Anderson, W.B.; Nordlie, R.C. Glucose dehydrogenase activity of yeast glucose 6-phosphate dehydrogenase. Inhibition by adenosine 5’-triphosphate and other nucleoside 5’-triphosphates and diphosphates. Biochemistry 1970, 9, 610–616. [Google Scholar] [CrossRef]
- Levy, H.R.; Daouk, G.H.; Katopes, M.A. Regulation of coenzyme utilization by the dual nucleotide-specific glucose 6-phosphate dehydrogenase from Leuconostoc mesenteroids. Arch. Biochem. Biophys. 1979, 198, 406–413. [Google Scholar] [CrossRef]
- Kawaguchi, A.; Bloch, K. Inhibition of glucose 6-phosphate dehydrogenase by palmitoyl coenzyme A. J. Biol. Chem. 1974, 249, 5793–5800. [Google Scholar]
- Levy, H.R.; Christoff, M.; Ingulli, J.; Ho, E.M. Glucose-6-phosphate dehydrogenase from Leuconostoc mesenteroides: Revised kinetic mechanism and kinetics of ATP inhibition. Arch. Biochem. Biophys. 1983, 222, 473–488. [Google Scholar] [CrossRef]
- Comín-Anduix, B.; Boros, L.G.G.; Marin, S.; Boren, J.; Callol-Massot, C.; Centelles, J.J.J.; Torres, J.L.L.; Agell, N.; Bassilian, S.; Cascante, M.; et al. Fermented Wheat Germ Extract Inhibits Glycolysis/Pentose Cycle Enzymes and Induces Apoptosis through Poly(ADP-ribose) Polymerase Activation in Jurkat T-cell Leukemia Tumor Cells. J. Biol. Chem. 2002, 277, 46408–46414. [Google Scholar] [CrossRef] [PubMed]
- Zhurakivska, K.; Troiano, G.; Caponio, V.; Dioguardi, M.; Arena, C.; Lo Muzio, L.; Zhurakivska, K.; Troiano, G.; Caponio, V.C.A.; Dioguardi, M.; et al. The Effects of Adjuvant Fermented Wheat Germ Extract on Cancer Cell Lines: A Systematic Review. Nutrients 2018, 10, 1546. [Google Scholar] [CrossRef] [PubMed]
- Mele, L.; Paino, F.; Papaccio, F.; Regad, T.; Boocock, D.; Stiuso, P.; Lombardi, A.; Liccardo, D.; Aquino, G.; Barbieri, A.; et al. A new inhibitor of glucose-6-phosphate dehydrogenase blocks pentose phosphate pathway and suppresses malignant proliferation and metastasis in vivo. Cell Death Dis. 2018, 9, 572. [Google Scholar] [CrossRef]
- Guindo, A.; Fairhurst, R.M.; Doumbo, O.K.; Wellems, T.E.; Diallo, D.A. X-linked G6PD deficiency protects hemizygous males but not heterozygous females against severe malaria. PLoS Med. 2007, 4, e66. [Google Scholar] [CrossRef] [PubMed]
- Farhud, D.; Yazdanpanah, L. Glucose-6-phosphate dehydrogenase (G6PD) Deficiency. Iran. J. Public Health 2008, 37, 1–18. [Google Scholar]
- Greene, L.S. G6PD deficiency as protection againstfalciparum malaria: An epidemiologic critique of population and experimental studies. Am. J. Phys. Anthropol. 1993, 36, 153–178. [Google Scholar] [CrossRef]
- Cocco, P. Does G6PD deficiency protect against cancer? A critical review. J. Epidemiol. Community Health 1987, 41, 89–93. [Google Scholar] [CrossRef]
- Cocco, P.; Dessí, S.; Avataneo, G.; Picchiri, G.; Heinemann, E. Glucose-6-phosphate dehydrogenase deficiency and cancer in a Sardinian male population: A case-control study. Carcinogenesis 1989, 10, 813–816. [Google Scholar] [CrossRef]
- Pisano, M.; Cocco, P.; Cherchi, R.; Onnis, R.; Cherchi, P. Glucose-6-phosphate dehydrogenase deficiency and lung cancer: A hospital based case-control study. Tumori J. 1991, 77, 12–15. [Google Scholar] [CrossRef]
- Kern, J.C.; Kehrer, J.P. Free radicals and apoptosis: Relationships with glutathione, thioredoxin, and the BCL family of proteins. Front. Biosci. 2005, 10, 1727–1738. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Certo, M.; Moore, V.D.G.; Nishino, M.; Wei, G.; Korsmeyer, S.; Armstrong, S.A.; Letai, A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006, 9, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Lin, A.W. Apoptosis in cancer. Carcinogenesis 2000, 21, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Beerheide, W.; Tan, Y.J.; Teng, E.; Ting, A.E.; Jedpiyawongse, A.; Srivatanakul, P. Downregulation of proapoptotic proteins Bax and Bcl-X(S) in p53 overexpressing hepatocellular carcinomas. Biochem. Biophys. Res. Commun. 2000, 273, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Edderkaoui, M.; Truong, P.; Ohno, I.; Jang, K.; Berti, A.; Pandol, S.J.; Gukovskaya, A.S. NADPH Oxidase Promotes Pancreatic Cancer Cell Survival via Inhibiting JAK2 Dephosphorylation by Tyrosine Phosphatases. Gastroenterology 2007, 133, 1637–1648. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Feng, Z.; Li, Y.; Wang, Y.; Wertz, K.; Weber, P.; Fu, Y.; Liu, J. Stimulation of GSH synthesis to prevent oxidative stress-induced apoptosis by hydroxytyrosol in human retinal pigment epithelial cells: Activation of Nrf2 and JNK-p62/SQSTM1 pathways. J. Nutr. Biochem. 2012, 23, 994–1006. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Jones, D.P. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J. Biol. Chem. 1998, 273, 11401–11404. [Google Scholar] [CrossRef]
- Bauer, H.P.; Srihari, T.; Jochims, J.C.; Hofer, H.W. 6-Phosphogluconolactonase. Purification, Properties and Activities in Various Tissues. Eur. J. Biochem. 1983, 133, 163–168. [Google Scholar] [CrossRef]
- Sukhatme, V.P.; Chan, B. Glycolytic cancer cells lacking 6-phosphogluconate dehydrogenase metabolize glucose to induce senescence. FEBS Lett. 2012, 586, 2389–2395. [Google Scholar] [CrossRef]
- Mori-Iwamoto, S.; Kuramitsu, Y.; Ryozawa, S.; Mikuria, K.; Fujimoto, M.; Maehara, S.-I.; Maehara, Y.; Okita, K.; Nakamura, K.; Sakaida, I. Proteomics finding heat shock protein 27 as a biomarker for resistance of pancreatic cancer cells to gemcitabine. Int. J. Oncol. 2007, 31, 1345–1350. [Google Scholar] [CrossRef][Green Version]
- Tran, A.T.; Sadet, A.; Calligari, P.; Lopes, P.; Ouazzani, J.; Sollogoub, M.; Miclet, E.; Abergel, D. Targeting the Pentose Phosphate Pathway: Characterization of a New 6PGL Inhibitor. Biophys. J. 2018, 115, 2114–2126. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Kim, E.S.; Koo, J.S. Expression of pentose phosphate pathway-related proteins in breast cancer. Dis. Markers 2018, 2018, 9369358. [Google Scholar] [CrossRef] [PubMed]
- Ceyhan, D.; Danişan, A.; Öğüş, I.H.; Özer, N. Purification and Kinetic Properties of 6-Phosphogluconate Dehydrogenase from Rat Small Intestine. Protein J. 2005, 24, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Adem, S.; Ciftci, M. Purification and biochemical characterization of glucose 6-phosphate dehydrogenase, 6-phosphogluconate dehydrogenase and glutathione reductase from rat lung and inhibition effects of some antibiotics. J. Enzym. Inhib. Med. Chem. 2016, 31, 1342–1348. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lin, R.; Elf, S.; Shan, C.; Kang, H.-B.; Ji, Q.; Zhou, L.; Hitosugi, T.; Zhang, L.; Zhang, S.; Seo, J.H.; et al. 6-Phosphogluconate dehydrogenase links oxidative PPP, lipogenesis and tumour growth by inhibiting LKB1–AMPK signalling. Nat. Cell Biol. 2015, 17, 1484–1496. [Google Scholar] [CrossRef]
- Chan, B.; VanderLaan, P.A.; Sukhatme, V.P. 6-Phosphogluconate dehydrogenase regulates tumor cell migration in vitro by regulating receptor tyrosine kinase c-Met. Biochem. Biophys. Res. Commun. 2013, 439, 247–251. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Zhang, X.; Fan, R.; Gu, H.; Shi, Y.; Liu, H. Glucose-6-phosphate dehydrogenase expression is correlated with poor clinical prognosis in esophageal squamous cell carcinoma. Eur. J. Surg. Oncol. 2015, 41, 1293–1299. [Google Scholar] [CrossRef]
- Zheng, W.; Feng, Q.; Liu, J.; Guo, Y.; Gao, L.; Li, R.; Xu, M.; Yan, G.; Yin, Z.; Zhang, S.; et al. Inhibition of 6-phosphogluconate Dehydrogenase Reverses Cisplatin Resistance in Ovarian and Lung Cancer. Front. Pharmacol. 2017, 8, 421. [Google Scholar] [CrossRef]
- Guo, H.; Xiang, Z.; Zhang, Y.; Sun, D. Inhibiting 6-phosphogluconate dehydrogenase enhances chemotherapy efficacy in cervical cancer via AMPK-independent inhibition of RhoA and Rac1. Clin. Transl. Oncol. 2019, 21, 404–411. [Google Scholar] [CrossRef]
- Yang, X.; Peng, X.; Huang, J. Inhibiting 6-phosphogluconate dehydrogenase selectively targets breast cancer through AMPK activation. Clin. Transl. Oncol. 2018, 20, 1145–1152. [Google Scholar] [CrossRef]
- Basile, A.; Rigano, D.; Loppi, S.; Di Santi, A.; Nebbioso, A.; Sorbo, S.; Conte, B.; Paoli, L.; De Ruberto, F.; Molinari, A.M.; et al. Antiproliferative, Antibacterial and Antifungal Activity of the Lichen Xanthoria parietina and Its Secondary Metabolite Parietin. Int. J. Mol. Med. 2015, 16, 7861–7875. [Google Scholar] [CrossRef] [PubMed]
- Elf, S.; Lin, R.; Xia, S.; Pan, Y.; Shan, C.; Wu, S.; Lonial, S.; Gaddh, M.; Arellano, M.L.; Khoury, H.J.; et al. Targeting 6-phosphogluconate dehydrogenase in the oxidative PPP sensitizes leukemia cells to antimalarial agent dihydroartemisinin. Oncogene 2017, 36, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Zugic, A.; Jeremic, I.; Isakovic, A.; Arsic, I.; Savic, S.; Tadic, V. Evaluation of Anticancer and Antioxidant Activity of a Commercially Available CO2 Supercritical Extract of Old Man’s Beard (Usnea barbata). PLoS ONE 2016, 11, e0146342. [Google Scholar] [CrossRef] [PubMed]
- Pilz, R.B.; Willis, R.C.; Boss, G.R. The influence of ribose 5-phosphate availability on purine synthesis of cultured human lymphoblasts and mitogen-stimulated lymphocytes. J. Biol. Chem. 1984, 259, 2927–2935. [Google Scholar]
- Garcia, J.; Han, D.; Sancheti, H.; Yap, L.-P.; Kaplowitz, N.; Cadenas, E. Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J. Biol. Chem. 2010, 285, 39646–39654. [Google Scholar] [CrossRef] [PubMed]
- Foyer, C.H.; Halliwell, B. The presence of glutathione and glutathione reductase in chloroplasts: A proposed role in ascorbic acid metabolism. Planta 1976, 133, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Noltmann, E.A. Aldose-ketose isomerases. In The Enzymes; Boyer, P.D., Ed.; Academic Press: New York, NY, USA; London, UK, 1972; pp. 272–354. [Google Scholar]
- Rangarajan, E.S.; Sivaraman, J.; Matte, A.; Cygler, M. Crystal structure of D-ribose-5-phosphate isomerase (RpiA) fromEscherichia coli. Proteins Struct. Funct. Genet. 2002, 48, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Stern, A.L.; Burgos, E.; Salmon, L.; Cazzulo, J.J. Ribose 5-phosphate isomerase type B from Trypanosoma cruzi: Kinetic properties and site-directed mutagenesis reveal information about the reaction mechanism. Biochem. J. 2007, 401, 279–285. [Google Scholar] [CrossRef]
- Riganti, C.; Gazzano, E.; Polimeni, M.; Aldieri, E.; Ghigo, D. The pentose phosphate pathway: An antioxidant defense and a crossroad in tumor cell fate. Free Radic. Biol. Med. 2012, 53, 421–436. [Google Scholar] [CrossRef]
- Sørensen, K.I.; Hove-Jensen, B. Ribose catabolism of Escherichia coli: Characterization of the rpiB gene encoding ribose phosphate isomerase B and of the rpiR gene, which is involved in regulation of rpiB expression. J. Bacteriol. 1996, 178, 1003–1011. [Google Scholar] [CrossRef]
- Zhang, R.-G.; Andersson, C.E.; Skarina, T.; Evdokimova, E.; Edwards, A.M.; Joachimiak, A.; Savchenko, A.; Mowbray, S.L. The 2.2 A resolution structure of RpiB/AlsB from Escherichia coli illustrates a new approach to the ribose-5-phosphate isomerase reaction. J. Mol. Biol. 2003, 332, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.K.; Dinesh, N.; Soumya, N.; Babu, N.K.; Singh, S. Identification and characterization of a novel Ribose 5-phosphate isomerase B from Leishmania donovani. Biochem. Biophys. Res. Commun. 2012, 421, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.-T.; Chen, L.-Y.; Tsai, S.-L.; Tu, H.-C.; Lu, J.-W.; Ciou, S.-C.; Wang, H.-D.; Yuh, C.-H. Ribose-5-phosphate isomerase A overexpression promotes liver cancer development in transgenic zebrafish via activation of ERK and β-catenin pathways. Carcinogenesis 2018, 40, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.-T.; Jiang, J.-K.; Yang, M.-H.; Lu, J.-W.; Lin, H.-K.; Wang, H.-D.; Yuh, C.-H. Identification of a noncanonical function for ribose-5-phosphate isomerase A promotes colorectal cancer formation by stabilizing and activating β-catenin via a novel C-terminal domain. PLoS Biol. 2018, 16, e2003714. [Google Scholar] [CrossRef] [PubMed]
- Heintze, J.; Costa, J.R.; Weber, M.; Ketteler, R. Ribose 5-phosphate isomerase inhibits LC3 processing and basal autophagy. Cell. Signal. 2016, 28, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef]
- Gonzalez, S.N.; Valsecchi, W.M.; José, D.M.; Delfino, M.; Cazzulo, J.J.; Maugeri, D.; Delfino, J.M.; Cazzulo, J.J. Structure, kinetic characterization and subcellular localization of the two ribulose 5-phosphate epimerase isoenzymes from Trypanosoma cruzi. PLoS ONE 2017, 12, e0172405. [Google Scholar] [CrossRef]
- Sobota, J.M.; Imlay, J.A. Iron enzyme ribulose-5-phosphate 3-epimerase in Escherichia coli is rapidly damaged by hydrogen peroxide but can be protected by manganese. Proc. Natl. Acad. Sci. USA 2011, 108, 5402–5407. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Shayoub, M.E.A.; Muddathir, A.K.; Elhassan, G.O.; Bashir, A.H.H. Evolution of tumor metabolism might reflect carcinogenesis as a reverse evolution process (dismantling of multicellularity). Cancers 2011, 3, 3002–3017. [Google Scholar] [CrossRef]
- Kabashima, T.; Kawaguchi, T.; Wadzinski, B.E.; Uyeda, K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc. Natl. Acad. Sci. USA 2003, 100, 5107–5112. [Google Scholar] [CrossRef] [PubMed]
- Liangpunsakul, S.; Ross, R.A.; Crabb, D.W. Activation of carbohydrate response element binding protein (ChREBP) by ethanol. J. Investig. Med. 2013, 61, 270. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Lu, X.; Li, Z.; Green, E.D.; Massa, H.; Trask, B.J.; Morris, C.A.; Keating, M.T. Complete physical map of the common deletion region in Williams syndrome and identification and characterization of three novel genes. Hum. Genet. 1998, 103, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Zhao, F.; Mancuso, A.; Gruber, J.J.; Thompson, C.B. The glucose-responsive transcription factor ChREBP contributes to glucose-dependent anabolic synthesis and cell proliferation. Proc. Natl. Acad. Sci. USA 2009, 106, 21660–21665. [Google Scholar] [CrossRef] [PubMed]
- Diolaiti, D.; McFerrin, L.; Carroll, P.A.; Eisenman, R.N. Functional interactions among members of the MAX and MLX transcriptional network during oncogenesis. Biochim. Biophys. Acta 2015, 1849, 484–500. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K. The transcription factor carbohydrate-response element-binding protein (ChREBP): A possible link between metabolic disease and cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 474–485. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Takenoshita, M.; Kabashima, T.; Uyeda, K. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc. Natl. Acad. Sci. USA 2001, 98, 13710–13715. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.J.; Collier, J.J.; Scott, D.K. cAMP opposes the glucose-mediated induction of the L-PK gene by preventing the recruitment of a complex containing ChREBP, HNF4α, and CBP. J. Am. Fed. Biol. Exp. 2009, 23, 2855–2865. [Google Scholar] [CrossRef]
- Miyamoto, K.; Ohta, H. Enzymatic decarboxylation of synthetic compounds. In Future Directions in Biocatalysis; Elsevier: Amsterdam, The Netherlands, 2007; pp. 305–343. [Google Scholar]
- Jahromi, R.R.F.; Morris, P.; Martinez-Torres, R.J.; Dalby, P.A. Structural stability of E. coli transketolase to temperature and pH denaturation. J. Biotechnol. 2011, 155, 209–216. [Google Scholar] [CrossRef]
- Datta, A.G.; Racker, E. Mechanism of action of transketolase. I. Properties of the crystalline yeast enzyme. J. Biol. Chem. 1961, 236, 617–623. [Google Scholar]
- Combs, G.F. Thiamin. In The Vitamins; Elsevier: Amsterdam, The Netherlands, 2012; pp. 261–276. [Google Scholar]
- Tseng, C.-W.; Kuo, W.-H.; Chan, S.-H.; Chan, H.-L.; Chang, K.-J.; Wang, L.-H. Transketolase Regulates the Metabolic Switch to Control Breast Cancer Cell Metastasis via the α-Ketoglutarate Signaling Pathway. Cancer Res. 2018, 78, 2799–2812. [Google Scholar] [CrossRef] [PubMed]
- Raïs, B.; Comin, B.; Puigjaner, J.; Brandes, J.L.; Creppy, E.; Saboureau, D.; Ennamany, R.; Paul Lee, W.-N.; Boros, L.G.; Cascante, M. Oxythiamine and dehydroepiandrosterone induce a G1 phase cycle arrest in Ehrlich’s tumor cells through inhibition of the pentose cycle. FEBS Lett. 1999, 456, 113–118. [Google Scholar] [CrossRef]
- Sica, D.A. Diuretics. In Side Effects of Drugs Annual; Elsevier: Amsterdam, The Netherlands, 2005; pp. 233–243. [Google Scholar]
- Venkataraman, R.; Racker, E. Mechanism of action of transaldolase. I. Crystalization and properties of yeast enzyme. J. Biol. Chem. 1961, 236, 1876–1882. [Google Scholar] [PubMed]
- Huang, S.-Y.; Zhang, Y.-H.P.; Zhong, J.-J. A thermostable recombinant transaldolase with high activity over a broad pH range. Appl. Microbiol. Biotechnol. 2012, 93, 2403–2410. [Google Scholar] [CrossRef] [PubMed]
- Banki, K.; Hutter, E.; Colombo, E.; Gonchoroff, N.J.; Perl, A. Glutathione levels and sensitivity to apoptosis are regulated by changes in transaldolase expression. J. Biol. Chem. 1996, 271, 32994–33001. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Morris, H.P.; Weber, G. Behavior of transaldolase (EC 2.2.1.2) and transketolase (EC 2.2.1.1) Activities in normal, neoplastic, differentiating, and regenerating liver. Cancer Res. 1976, 36, 3189–3197. [Google Scholar] [PubMed]
- Perl, A. The pathogenesis of transaldolase deficiency. IUBMB Life 2007, 59, 365–373. [Google Scholar] [CrossRef]
- Ding, Y.; Gong, C.; Huang, D.; Chen, R.; Sui, P.; Lin, K.H.; Liang, G.; Yuan, L.; Xiang, H.; Chen, J.; et al. Synthetic lethality between HER2 and transaldolase in intrinsically resistant HER2-positive breast cancers. Nat. Commun. 2018, 9, 4274. [Google Scholar] [CrossRef]
- Bennett, C.F.; Kwon, J.J.; Chen, C.; Russell, J.; Acosta, K.; Burnaevskiy, N.; Crane, M.M.; Bitto, A.; Vander Wende, H.; Simko, M.; et al. Transaldolase inhibition impairs mitochondrial respiration and induces a starvation-like longevity response in Caenorhabditis elegans. PLoS Genet. 2017, 13, e1006695. [Google Scholar] [CrossRef]
- Ogawa, T.; Murakami, K.; Yoshino, M. Inhibition by fructose 1,6-bisphosphate of transaldolase from Escherichia coli. FEMS Microbiol. Lett. 2016, 363, fnw183. [Google Scholar] [CrossRef]
- Light, S.H.; Anderson, W.F. Arabinose 5-phosphate covalently inhibits transaldolase. J. Struct. Funct. Genom. 2014, 15, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Egan, R.M.; Sable, H.Z. Transketolase Kinetics. The Slow Reconstitution of the Holoenzyme Is Due to Rate-Limiting Dimerization of the Subunits. J. Biol. Chem. 1981, 256, 4877–4883. [Google Scholar] [PubMed]
- Ballatori, N.; Krance, S.M.; Notenboom, S.; Shi, S.; Tieu, K.; Hammond, C.L. Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 2009, 390, 191–214. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Seefeldt, T.; Chen, W.; Wang, X.; Matthees, D.; Hu, Y.; Guan, X. Effects of glutathione reductase inhibition on cellular thiol redox state and related systems. Arch. Biochem. Biophys. 2009, 485, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P.; Sies, H. The Redox Code. Antioxid. Redox Signal. 2015, 23, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Salbitani, G.; Bottone, C.; Carfagna, S. Determination of Reduced and Total Glutathione Content in Extremophilic Microalga Galdieria phlegrea. Bio-Protocol 2017, 7, e2372. [Google Scholar] [CrossRef]
- Baird, A.-M.; O’Byrne, K.J.; Gray, S.G. Reactive Oxygen Species and Reactive Nitrogen Species in Epigenetic Modifications. In Systems Biology of Free Radicals and Antioxidants; Springer: Berlin/Heidelberg, Germany, 2014; pp. 437–455. [Google Scholar]
- Valko, M.; Izakovic, M.; Mazur, M.; Rhodes, C.J.; Telser, J. Role of oxygen radicals in DNA damage and cancer incidence. Mol. Cell. Biochem. 2004, 266, 37–56. [Google Scholar] [CrossRef]
- Ríos-Arrabal, S.; Artacho-Cordón, F.; León, J.; Román-Marinetto, E.; Del Mar Salinas-Asensio, M.; Calvente, I.; Núñez, M.I. Involvement of free radicals in breast cancer. Springerplus 2013, 2, 404. [Google Scholar] [CrossRef]
- Dreher, D.; Junod, A.F. Role of oxygen free radicals in cancer development. Eur. J. Cancer 1996, 32A, 30–38. [Google Scholar] [CrossRef]
- Liu-Smith, F.; Dellinger, R.; Meyskens, F.L., Jr. Updates of reactive oxygen species in melanoma etiology and progression. Arch. Biochem. Biophys. 2014, 563, 51–55. [Google Scholar] [CrossRef]
- Augsten, M.; Sjoberg, E.; Frings, O.; Vorrink, S.U.; Frijhoff, J.; Olsson, E.; Borg, A.; Ostman, A. Cancer-Associated Fibroblasts Expressing CXCL14 Rely upon NOS1-Derived Nitric Oxide Signaling for Their Tumor-Supporting Properties. Cancer Res. 2014, 74, 2999–3010. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zou, L.; Huang, C.; Lei, Y. Redox Regulation of Cancer Metastasis: Molecular Signaling and Therapeutic Opportunities. Drug Dev. Res. 2014, 75, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Badana, A.K.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Simon, H.-U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Fubini, B.; Hubbard, A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic. Biol. Med. 2003, 34, 1507–1516. [Google Scholar] [CrossRef]
- Ahn, H.J.; Kim, K.I.; Hoan, N.N.; Kim, C.H.; Moon, E.; Choi, K.S.; Yang, S.S.; Lee, J.-S. Targeting Cancer Cells with Reactive Oxygen and Nitrogen Species Generated by Atmospheric-Pressure Air Plasma. PLoS ONE 2014, 9, e86173. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1–44. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Kowalik, M.A.; Columbano, A.; Perra, A. Emerging Role of the Pentose Phosphate Pathway in Hepatocellular Carcinoma. Front. Oncol. 2017, 7, 87. [Google Scholar] [CrossRef]
- James, E.L.; Michalek, R.D.; Pitiyage, G.N.; de Castro, A.M.; Vignola, K.S.; Jones, J.; Mohney, R.P.; Karoly, E.D.; Prime, S.S.; Parkinson, E.K. Senescent Human Fibroblasts Show Increased Glycolysis and Redox Homeostasis with Extracellular Metabolomes That Overlap with Those of Irreparable DNA Damage, Aging, and Disease. J. Proteome Res. 2015, 14, 1854–1871. [Google Scholar] [CrossRef]
- Sousa, B.; Pereira, J.; Paredes, J.; Sousa, B.; Pereira, J.; Paredes, J. The Crosstalk Between Cell Adhesion and Cancer Metabolism. Int. J. Mol. Sci. 2019, 20, 1933. [Google Scholar] [CrossRef] [PubMed]
- Vizán, P.; Alcarraz-Vizán, G.; Díaz-Moralli, S.; Solovjeva, O.N.; Frederiks, W.M.; Cascante, M. Modulation of pentose phosphate pathway during cell cycle progression in human colon adenocarcinoma cell line HT29. Int. J. Cancer 2009, 124, 2789–2796. [Google Scholar] [CrossRef]
- Uyeda, K.; Yamashita, H.; Kawaguchi, T. Carbohydrate responsive element-binding protein (ChREBP): A key regulator of glucose metabolism and fat storage. Biochem. Pharmacol. 2002, 63, 2075–2080. [Google Scholar] [CrossRef]
- Iizuka, K.; Wu, W.; Horikawa, Y.; Takeda, J. Role of glucose-6-phosphate and xylulose-5-phosphate in the regulation of glucose-stimulated gene expression in the pancreatic β cell line, INS-1E. Endocr. J. 2013, 60, 473–482. [Google Scholar] [CrossRef] [PubMed]
- De Santis, M.C.; Porporato, P.E.; Martini, M.; Morandi, A. Signaling Pathways Regulating Redox Balance in Cancer Metabolism. Front. Oncol. 2018, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Koceva-Chyła, A.; Jedrzejczak, M.; Skierski, J.; Kania, K.; Jóźwiak, Z. Mechanisms of induction of apoptosis by anthraquinone anticancer drugs aclarubicin and mitoxantrone in comparison with doxorubicin: Relation to drug cytotoxicity and caspase-3 activation. Apoptosis 2005, 10, 1497–1514. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M.S.; Zong, W.-X. Chemotherapeutic approaches for targeting cell death pathways. Oncologist 2006, 11, 342–357. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Vacchelli, E.; Kroemer, G. Cell Death Signaling and Anticancer Therapy. Front. Oncol. 2011, 1, 5. [Google Scholar] [CrossRef]
- Mansilla, S.; Llovera, L.; Portugal, J. Chemotherapeutic targeting of cell death pathways. Anti-Cancer Agents Med. Chem. 2012, 12, 226–238. [Google Scholar] [CrossRef]
- Ocker, M.; Höpfner, M. Apoptosis-Modulating Drugs for Improved Cancer Therapy. Eur. Surg. Res. 2012, 48, 111–120. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Stock, C.-M.; Taylor, S.; Walsh, M.; Muddathir, A.K.; Verduzco, D.; Bashir, A.H.H.; Mohammed, O.Y.; Elhassan, G.O.; Harguindey, S.; et al. Resistance to cancer chemotherapy: Failure in drug response from ADME to P-gp. Cancer Cell Int. 2015, 15, 71. [Google Scholar] [CrossRef] [PubMed]
- Zaman, G.J.; Lankelma, J.; van Tellingen, O.; Beijnen, J.; Dekker, H.; Paulusma, C.; Oude Elferink, R.P.; Baas, F.; Borst, P. Role of glutathione in the export of compounds from cells by the multidrug-resistance-associated protein. Proc. Natl. Acad. Sci. USA 1995, 92, 7690–7694. [Google Scholar] [CrossRef] [PubMed]
- Bagrij, T.; Klokouzas, A.; Hladky, S.B.; Barrand, M.A. Influences of glutathione on anionic substrate efflux in tumour cells expressing the multidrug resistance-associated protein, MRP1. Biochem. Pharmacol. 2001, 62, 199–206. [Google Scholar] [CrossRef]
- Lai, L.; Tan, T.M.C. Role of glutathione in the multidrug resistance protein 4 (MRP4/ABCC4)-mediated efflux of cAMP and resistance to purine analogues. Biochem. J. 2002, 361, 497–503. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Muddathir, A.K.; Shayoub, M.E.A. Tumor acidity as evolutionary spite. Cancers 2011, 3, 408–414. [Google Scholar] [CrossRef]
- Boros, L.G.; Puigjaner, J.; Cascante, M.; Lee, W.N.P.; Brandes, J.L.; Bassilian, S.; Yusuf, F.I.; Williams, R.D.; Muscarella, P.; Melvin, W.S.; et al. Oxythiamine and dehydroepiandrosterone inhibit the nonoxidative synthesis of ribose and tumor cell proliferation. Cancer Res. 1997, 57, 4242–4248. [Google Scholar] [PubMed]
- Alfarouk, K.O.; Ahmed, S.B.M.; Ahmed, A.; Elliott, R.L.; Ibrahim, M.E.; Ali, H.S.; Wales, C.C.; Nourwali, I.; Aljarbou, A.N.; Bashir, A.H.H.; et al. The interplay of dysregulated ph and electrolyte imbalance in cancer. Cancers 2020, 12, 898. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Bashir, A.H.H. Diabetes mellitus type 2 through oncology lens. Med. Hypotheses 2011, 76, 761–762. [Google Scholar] [CrossRef]
- Domagk, G.F.; Doering, K.M.; Chilla, R. Purification and Properties of Ribose-Phosphate Isomerase from Candida utilis. Eur. J. Biochem. 1973, 38, 259–264. [Google Scholar] [CrossRef]
- Tsolas, O.; Horecker, B.L. Transaldolase. Enzymes 1972, 7, 259–280. [Google Scholar]



{kind=link}
{kind=link}
| Enzyme | Optimum pH |
|---|---|
| Glucose-6-phosphate dehydrogenase (G6PDH) | 7.8 [8] |
| 6-phosphogluconolactonase (6PGL) | 7.4 [38] |
| 6-phosphogluconate dehydrogenase (6PGD) | Range from (7–10) depending on several factors including the buffer used in the experiment [43] |
| Ribose-5-phosphate isomerase (RPi) | 8.4 [132] |
| Ribulose 5-Phosphate 3-Epimerase (RPE) | 7.25–7.5 [69] |
| Transketolase (TKT) | 7.5–7.6 [82,90,96] |
| Transaldolase (TADOL) | 8 [133] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfarouk, K.O.; Ahmed, S.B.M.; Elliott, R.L.; Benoit, A.; Alqahtani, S.S.; Ibrahim, M.E.; Bashir, A.H.H.; Alhoufie, S.T.S.; Elhassan, G.O.; Wales, C.C.; et al. The Pentose Phosphate Pathway Dynamics in Cancer and Its Dependency on Intracellular pH. Metabolites 2020, 10, 285. https://doi.org/10.3390/metabo10070285
Alfarouk KO, Ahmed SBM, Elliott RL, Benoit A, Alqahtani SS, Ibrahim ME, Bashir AHH, Alhoufie STS, Elhassan GO, Wales CC, et al. The Pentose Phosphate Pathway Dynamics in Cancer and Its Dependency on Intracellular pH. Metabolites. 2020; 10(7):285. https://doi.org/10.3390/metabo10070285
Chicago/Turabian StyleAlfarouk, Khalid O., Samrein B. M. Ahmed, Robert L. Elliott, Amanda Benoit, Saad S. Alqahtani, Muntaser E. Ibrahim, Adil H. H. Bashir, Sari T. S. Alhoufie, Gamal O. Elhassan, Christian C. Wales, and et al. 2020. "The Pentose Phosphate Pathway Dynamics in Cancer and Its Dependency on Intracellular pH" Metabolites 10, no. 7: 285. https://doi.org/10.3390/metabo10070285
APA StyleAlfarouk, K. O., Ahmed, S. B. M., Elliott, R. L., Benoit, A., Alqahtani, S. S., Ibrahim, M. E., Bashir, A. H. H., Alhoufie, S. T. S., Elhassan, G. O., Wales, C. C., Schwartz, L. H., Ali, H. S., Ahmed, A., Forde, P. F., Devesa, J., Cardone, R. A., Fais, S., Harguindey, S., & Reshkin, S. J. (2020). The Pentose Phosphate Pathway Dynamics in Cancer and Its Dependency on Intracellular pH. Metabolites, 10(7), 285. https://doi.org/10.3390/metabo10070285