Enzyme Assembly for Compartmentalized Metabolic Flux Control



Abstract
1. Introduction
2. Scaffoldless Engineered Enzyme Assembly
2.1. Interaction Pair or Affinity Peptide Guided Enzyme Assembly
2.2. Enzyme Aggregation Guided by Active Inclusion Bodies
3. Enhancing Multi-Enzyme Biosynthesis Using Synthetic Scaffolds
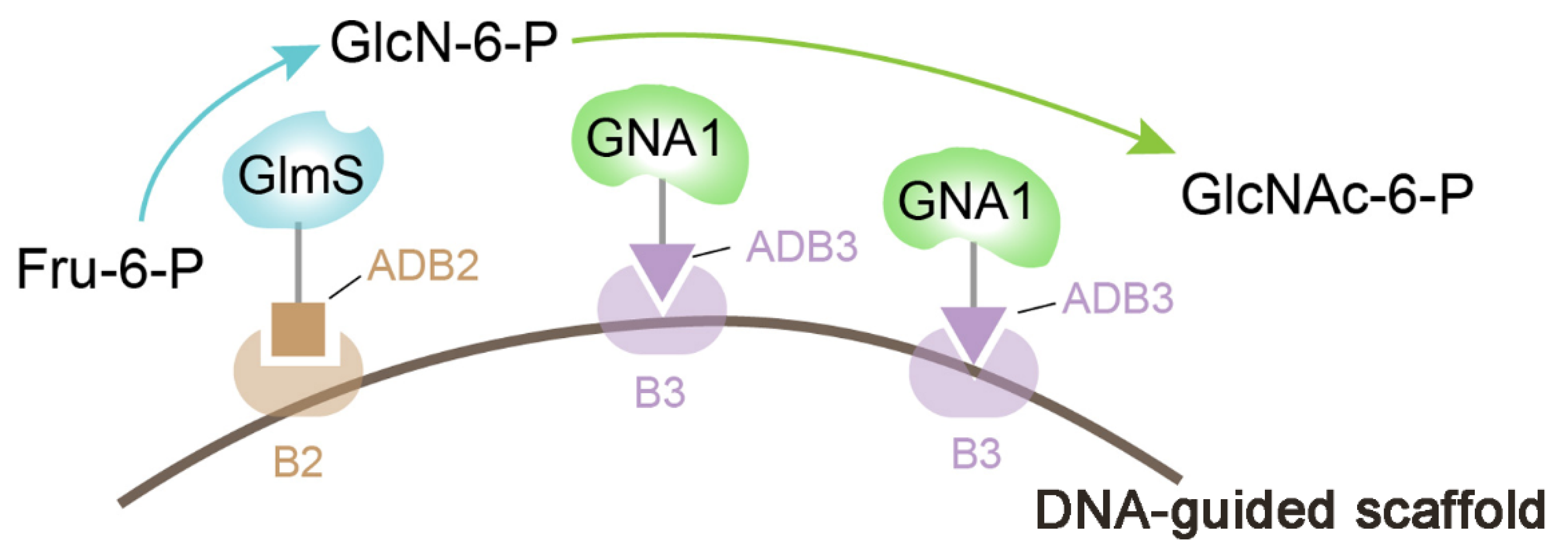
3.1. Nucleic Acid Scaffold
3.2. Protein Scaffolds
3.3. Lipid-Containing Scaffolds
4. Physical Compartments for Pathway Sequestration
4.1. Eukaryotic Physical Compartments
4.2. Protein-Based Compartments in Prokaryotic Cells
5. Perspectives and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nielsen, J.; Keasling, J.D. Engineering cellular metabolism. Cell 2016, 164, 1185–1197. [Google Scholar] [CrossRef]
- Keasling, J.D. Manufacturing molecules through metabolic engineering. Science 2010, 330, 1355–1358. [Google Scholar] [CrossRef]
- Na, D.; Yoo, S.M.; Chung, H.; Park, H.; Park, J.H.; Lee, S.Y. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat. Biotechnol. 2013, 31, 170–174. [Google Scholar] [CrossRef]
- Xu, P.; Gu, Q.; Wang, W.Y.; Wong, L.; Bower, A.G.; Collins, C.H.; Koffas, M.A. Modular optimization of multigene pathways for fatty acids production in E. coli. Nat. Commun. 2013, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Biggs, B.W.; de Paepe, B.; Santos, C.N.S.; De Mey, M.; Ajikumar, P.K. Multivariate modular metabolic engineering for pathway and strain optimization. Curr. Opin. Biotechnol. 2014, 29, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Anesiadis, N.; Cluett, W.R.; Mahadevan, R. Dynamic metabolic engineering for increasing bioprocess productivity. Metab. Eng. 2008, 10, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Venayak, N.; Anesiadis, N.; Cluett, W.R.; Mahadevan, R. Engineering metabolism through dynamic control. Curr. Opin. Biotechnol. 2015, 34, 142–152. [Google Scholar] [CrossRef]
- Lee, H.; DeLoache, W.C.; Dueber, J.E. Spatial organization of enzymes for metabolic engineering. Metab. Eng. 2012, 14, 242–251. [Google Scholar] [CrossRef]
- Jiang, H.; Wood, K.V.; Morgan, J.A. Metabolic engineering of the phenylpropanoid pathway in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2005, 71, 2962–2969. [Google Scholar] [CrossRef]
- Nicolaou, S.A.; Gaida, S.M.; Papoutsakis, E.T. A comparative view of metabolite and substrate stress and tolerance in microbial bioprocessing: From biofuels and chemicals, to biocatalysis and bioremediation. Metab. Eng. 2010, 12, 307–331. [Google Scholar] [CrossRef]
- Siu, K.H.; Chen, R.P.; Sun, Q.; Chen, L.; Tsai, S.L.; Chen, W. Synthetic scaffolds for pathway enhancement. Curr. Opin. Biotechnol. 2015, 36, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Baneyx, F.; Matthaei, J.F. Self-assembled two-dimensional protein arrays in bionanotechnology: From S-layers to designed lattices. Curr. Opin. Biotechnol. 2014, 28, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Brodin, J.D.; Carr, J.R.; Sontz, P.A.; Tezcan, F.A. Exceptionally stable, redox-active supramolecular protein assemblies with emergent properties. Proc. Natl. Acad. Sci. USA 2014, 111, 2897–2902. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.B.; Dinesh, S.D.; Deery, E.; Leech, H.K.; Brindley, A.A.; Heldt, D.; Frank, S.; Smales, C.M.; Lunsdorf, H.; Rambach, A.; et al. Biochemical and structural insights into bacterial organelle form and biogenesis. J. Biol. Chem. 2008, 283, 14366–14375. [Google Scholar] [CrossRef]
- Miles, E.W.; Rhee, S.; Davies, D.R. The molecular basis of substrate channeling. J. Biol. Chem. 1999, 274, 12193–12196. [Google Scholar] [CrossRef]
- Moon, T.S.; Dueber, J.E.; Shiue, E.; Prather, K.L. Use of modular, synthetic scaffolds for improved production of glucaric acid in engineered E. coli. Metab. Eng. 2010, 12, 298–305. [Google Scholar] [CrossRef]
- Qiu, X.; Xie, S.; Lu, M.; Wu, X.; Zhu, L.; Zhu, L. Spatial organization of enzymes to enhance synthetic pathways in microbial chassis: A systematic review. Microb. Cell Fact. 2018, 17, 120. [Google Scholar] [CrossRef]
- Agapakis, C.M.; Boyle, P.M.; Silver, P.A. Natural strategies for the spatial optimization of metabolism in synthetic biology. Nat. Chem. Biol. 2012, 8, 527–535. [Google Scholar] [CrossRef]
- Hinzpeter, F.; Gerland, U.; Tostevin, F. Optimal compartmentalization strategies for metabolic microcompartments. Biophys. J. 2017, 112, 767–779. [Google Scholar] [CrossRef]
- Good, M.C.; Zalatan, J.G.; Lim, W.A. Scaffold proteins: Hubs for controlling the flow of cellular information. Science 2011, 332, 680–686. [Google Scholar] [CrossRef]
- Bellapadrona, G.; Elbaum, M. Supramolecular protein assemblies in the nucleus of human cells. Angew. Chem. Int. Ed. 2014, 53, 1534–1537. [Google Scholar] [CrossRef] [PubMed]
- Howorka, S. Rationally engineering natural protein assemblies in nanobiotechnology. Curr. Opin. Biotechnol. 2011, 22, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Conrado, R.J.; Varner, J.D.; DeLisa, M.P. Engineering the spatial organization of metabolic enzymes: Mimicking nature’s synergy. Curr. Opin. Biotechnol. 2008, 19, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Horn, A.; Sticht, H. Synthetic protein scaffolds based on peptide motifs and cognate adaptor domains for improving metabolic productivity. Front. Bioeng. Biotechnol. 2015, 3, 191. [Google Scholar] [CrossRef] [PubMed]
- Myhrvold, C.; Silver, P.A. Using synthetic RNAs as scaffolds and regulators. Nat. Struct. Mol. Biol. 2015, 22, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Giessen, T.W.; Silver, P.A. Encapsulation as a strategy for the design of biological compartmentalization. J. Mol. Biol. 2016, 428, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Mantell, J.; Hodgson, L.; Alibhai, D.; Fletcher, J.M.; Brown, I.R.; Frank, S.; Xue, W.F.; Verkade, P.; Woolfson, D.N.; et al. Engineered synthetic scaffolds for organizing proteins within the bacterial cytoplasm. Nat. Chem. Biol. 2018, 14, 142–147. [Google Scholar] [CrossRef]
- Kang, W.; Ma, T.; Liu, M.; Qu, J.; Liu, Z.; Zhang, H.; Shi, B.; Fu, S.; Ma, J.; Lai, L.T.F.; et al. Modular enzyme assembly for enhanced cascade biocatalysis and metabolic flux. Nat. Commun. 2019, 10, 4248. [Google Scholar] [CrossRef]
- Meynial Salles, I.; Forchhammer, N.; Croux, C.; Girbal, L.; Soucaille, P. Evolution of a Saccharomyces cerevisiae metabolic pathway in Escherichia coli. Metab. Eng. 2007, 9, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yoon, S.H.; Jang, H.J.; Chung, Y.R.; Kim, J.Y.; Choi, E.S.; Kim, S.W. Metabolic engineering of Escherichia coli for a-farnesene production. Metab. Eng. 2011, 13, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Bulow, L.; Ljungcrantz, P.; Mosbach, K. Preparation of a soluble bifunctionalenzyme by gene fusion. Nat. Biotechnol. 1985, 3, 821–823. [Google Scholar] [CrossRef]
- Young, E.J.; Burton, R.; Mahalik, J.P.; Sumpter, B.G.; Fuentes-Cabrera, M.; Kerfeld, C.A.; Ducat, D.C. Engineering the bacterial microcompartment domain for molecular scaffolding applications. Front. Microbiol. 2017, 8, 1441. [Google Scholar] [CrossRef] [PubMed]
- Pawson, T.; Nash, P. Protein-protein interactions define specificity in signal transduction. Genes Dev. 2000, 14, 1027–1047. [Google Scholar] [PubMed]
- Tonikian, R.; Zhang, Y.; Sazinsky, S.L.; Currell, B.; Yeh, J.-H.; Reva, B.; Held, H.A.; Appleton, B.A.; Evangelista, M.; Wu, Y.; et al. A specificity map for the PDZ domain family. PLoS Biol. 2008, 6, e239. [Google Scholar] [CrossRef] [PubMed]
- Dueber, J.E.; Yeh, B.J.; Bhattacharyya, R.P.; Lim, W.A. Rewiring cell signaling: The logic and plasticity of eukaryotic protein circuitry. Curr. Opin. Struct. Biol. 2004, 14, 690–699. [Google Scholar] [CrossRef]
- Peisajovich, S.G.; Garbarino, J.E.; Wei, P.; Lim, W.A. Rapid diversification of cell signaling phenotypes by modular domain recombination. Science 2010, 328, 368–372. [Google Scholar] [CrossRef]
- Remenyi, A.; Good, M.C.; Lim, W.A. Docking interactions in protein kinase and phosphatase networks. Curr. Opin. Struct. Biol. 2006, 16, 676–685. [Google Scholar] [CrossRef]
- Gao, X.; Yang, S.; Zhao, C.; Ren, Y.; Wei, D. Artificial multienzyme supramolecular device: Highly ordered self-assembly of oligomeric enzymes in vitro and in vivo. Angew. Chem. Int. Ed. Engl. 2014, 53, 14027–14030. [Google Scholar] [CrossRef]
- Kragl, U.; Vasic-Racki, D.; Wandrey, C. Continuous production of L-tert-leucine in series of two enzyme membrane reactors. Bioprocess Eng. 1996, 14, 291–297. [Google Scholar] [CrossRef]
- Fuh, G.; Pisabarro, M.T.; Li, Y.; Quan, C.; Lasky, L.A.; Sidhu, S.S. Analysis of PDZ domain–ligand interactions using carboxyl-terminal phage display. J. Biol. Chem. 2000, 275, 21486–21491. [Google Scholar] [CrossRef]
- Price, J.V.; Chen, L.; Whitaker, W.B.; Papoutsakis, E.; Chen, W. Scaffoldless engineered enzyme assembly for enhanced methanol utilization. Proc. Natl. Acad. Sci. USA 2016, 113, 12691–12696. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Wang, Y.; Zhao, S.; Ren, Y. Photocontrolled reversible self-assembly of dodecamer nitrilase. Bioresour. Bioprocess. 2017, 4, 36. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rossi, E.A.; Goldenberg, D.M.; Chang, C.H. The dock-and-lock method combines recombinant engineering with site-specific covalent conjugation to generate multifunctional structures. Bioconjug. Chem. 2012, 23, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.; Scott, J.D. AKAP signalling complexes: Focal points in space and time. Nat. Rev. Mol. Cell Biol. 2004, 5, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Sarma, G.N.; Kinderman, F.S.; Kim, C.; von Daake, S.; Chen, L.; Wang, B.C.; Taylor, S.S. Structure of D-AKAP2:PKA RI complex: Insights into AKAP specificity and selectivity. Structure 2010, 18, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.R.; Lygren, B.; Berge, T.; Hoshi, N.; Wong, W.; Taskén, K.; Scott, J.D. Delineation of type I protein kinase A-selective signaling events using an RI anchoring disruptor. J. Biol. Chem. 2006, 281, 21535–21545. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.G.; Lygren, B.; Dokurno, P.; Hoshi, N.; McConnachie, G.; Taskén, K.; Carlson, C.R.; Scott, J.D.; Barford, D. Molecular basis of AKAP specificity for PKA regulatory subunits. Mol. Cell 2006, 24, 383–395. [Google Scholar] [CrossRef]
- Worrall, D.M.; Goss, N.H. The formation of biologically active beta-galactosidase inclusion bodies in Escherichia coli. Aust. J. Biotechnol. 1989, 3, 28–32. [Google Scholar]
- Diener, M.; Kopka, B.; Pohl, M.; Jaeger, K.; Krauss, U. Fusion of a coiled-coil domain facilitates the high-level production of catalytically active enzyme inclusion bodies. ChemCatChem 2016, 8, 142–152. [Google Scholar] [CrossRef]
- Zhou, B.; Xing, L.; Wu, W.; Zhang, X.E.; Lin, Z. Small surfactant-like peptides can drive soluble proteins into active aggregates. Microb. Cell Fact. 2012, 11, 10. [Google Scholar] [CrossRef]
- Jäger, V.; Lamm, R.; Kloß, R.; Kaganovitch, E.; Grünberger, A.; Pohl, M.; Büchs, J.; Jaeger, K.; Krauss, U. A synthetic reaction cascade implemented by co-localization of two proteins within catalytically-active inclusion bodies. ACS Synth. Biol. 2018, 7, 2282–2295. [Google Scholar] [CrossRef] [PubMed]
- Arié, J.P.; Miot, M.; Sassoon, N.; Betton, J.M. Formation of active inclusion bodies in the periplasm of Escherichia coli. Mol. Microbiol. 2006, 62, 427–437. [Google Scholar] [CrossRef]
- Nahalka, J.; Nidetzky, B. Fusion to a pull-down domain: A novel approach of producing Trigonopsis variabilis D-amino acid oxidase as insoluble enzyme aggregates. Biotechnol. Bioeng. 2007, 97, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Fruitos, E.; Gonzalez-Montalban, N.; Morell, M.; Vera, A.; Ferraz, R.M.; Aris, A.; Ventura, S.; Villaverde, A. Aggregation as bacterial inclusion bodies does not imply inactivation of enzymes and fluorescent proteins. Microb. Cell Fact. 2005, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Kloss, R.; Karmainski, T.; Jäger, V.D.; Hahn, D.; Grünberger, A.; Baumgart, M.; Krauss, U.; Jaeger, K.E.; Wiechert, W.; Pohl, M. Tailor-made catalytically active inclusion bodies for different applications in biocatalysis. Catal. Sci. Technol. 2018, 8, 5816. [Google Scholar] [CrossRef]
- Lv, X.; Jin, K.; Wu, Y.; Zhang, C.; Cui, S.; Zhu, X.; Li, J.; Du, G.; Liu, L. Enzyme assembly guided by SPFH-induced functional inclusion bodies for enhanced cascade biocatalysis. Biotechnol. Bioeng. 2020. [Google Scholar] [CrossRef]
- Pinheiro, A.V.; Han, D.R.; Shih, W.M.; Yan, H. Challenges and opportunities for structural DNA nanotechnology. Nat. Nanotechnol. 2011, 6, 763–772. [Google Scholar] [CrossRef]
- Linko, V.; Dietz, H. The enabled state of DNA nanotechnology. Curr. Opin. Biotechnol. 2013, 24, 555–561. [Google Scholar] [CrossRef]
- Zadeh, J.N.; Steenberg, C.D.; Bois, J.S.; Wolfe, B.R.; Pierce, M.B.; Khan, A.R.; Dirks, R.M.; Pierce, N.A. NUPACK: Analysis and design of nucleic acid systems. J. Comput. Chem. 2011, 32, 170–173. [Google Scholar] [CrossRef]
- Douglas, S.M.; Marblestone, A.H.; Teerapittayanon, S.; Vazquez, A.; Church, G.M.; Shih, W.M. Rapid prototyping of 3D DNA-origami shapes with caDNAno. Nucleic Acids Res. 2009, 37, 5001–5006. [Google Scholar] [CrossRef]
- Castro, C.E.; Kilchherr, F.; Kim, D.N.; Shiao, E.L.; Wauer, T.; Wortmann, P.; Bathe, M.; Dietz, H. A primer to scaffolded DNA origami. Nat. Methods 2011, 8, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Boileau, C.R.; d’Hauteville, H.M.; Sansonetti, P.J. DNA hybridization technique to detect Shigella species and enteroinvasive Escherichia coli. J. Clin. Microbiol. 1984, 20, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Barbu, V. Molecular hybridization techniques of nucleic acids. Innov. Rom. Food Biotechnol. 2007, 1, 1–12. [Google Scholar]
- Rothemund, P.W. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef]
- Douglas, S.M.; Dietz, H.; Liedl, T.; Högberg, B.; Graf, F.; Shih, W.M. Self-assembly of DNA into nanoscale three-dimensional shapes. Nature 2010, 459, 414–418. [Google Scholar] [CrossRef]
- Han, D.R.; Pal, S.; Nangreave, J.; Deng, Z.; Liu, Y.; Yan, H. DNA origami with complex curvatures in three-dimensional space. Science 2011, 332, 342–346. [Google Scholar] [CrossRef]
- Zhang, F.; Jiang, S.; Wu, S.; Li, Y.; Mao, C.; Liu, Y.; Yan, H. Complex wireframe DNA origami nanostructures with multi-arm junction vertices. Nat. Nanotechnol. 2015, 10, 779–784. [Google Scholar] [CrossRef]
- Wilner, O.I.; Weizmann, Y.; Gill, R.; Lioubashevski, O.; Freeman, R.; Willner, I. Enzyme cascades activated on topologically programmed DNA scaffolds. Nat. Nanotechnol. 2009, 4, 249–254. [Google Scholar] [CrossRef]
- Fu, J.; Liu, M.; Liu, Y.; Yan, H. Spatially-interactive biomolecular networks organized by nucleic acid nanostructures. Acc. Chem. Res. 2012, 45, 1215–1226. [Google Scholar] [CrossRef]
- Funke, J.J.; Dietz, H. Placing molecules with Bohr radius resolution using DNA origami. Nat. Nanotechnol. 2016, 11, 47–52. [Google Scholar] [CrossRef]
- Lin, C.; Liu, Y.; Yan, H. Designer DNA nanoarchitectures. Biochemistry 2009, 48, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Winfree, E.; Liu, F.; Wenzler, L.A.; Seeman, N.C. Design and self-assembly of two-dimensional DNA crystals. Nature 1998, 394, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Rinker, S.; Wang, X.; Liu, Y.; Seeman, N.C.; Yan, H. In vivo cloning of artificial DNA nanostructures. Proc. Natl. Acad. Sci. USA 2008, 105, 17626–17631. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Niemeyer, C.M. DNA-directed assembly of artificial multienzyme complexes. Biochem. Biophys. Res. Commun. 2008, 377, 67. [Google Scholar] [CrossRef]
- Fu, J.L.; Liu, M.H.; Liu, Y.; Woodbury, N.W.; Yan, H. Interenzyme substrate diffusion for an enzyme cascade organized on spatially addressable DNA nanostructures. J. Am. Chem. Soc. 2012, 134, 5516–5519. [Google Scholar] [CrossRef]
- Numajiri, K.; Yamazaki, T.; Kimura, M.; Kuzuya, A.; Komiyama, M. Discrete and active enzyme nanoarrays on DNA origami scaffolds purified by affinity tag separation. J. Am. Chem. Soc. 2010, 132, 9937–9939. [Google Scholar] [CrossRef]
- Erkelenz, M.; Kuo, C.H.; Niemeyer, C.M. DNA-mediated assembly of cytochrome P450 BM3 subdomains. J. Am. Chem. Soc. 2011, 133, 16111–16118. [Google Scholar] [CrossRef]
- Sun, Q.; Chen, W. HaloTag mediated artificial cellulosome assembly on a rolling circle amplification DNA template for efficient cellulose hydrolysis. Chem. Commun. 2016, 52, 6701–6704. [Google Scholar] [CrossRef]
- Kou, B.B.; Chai, Y.Q.; Yuan, Y.L.; Yuan, R. PtNPs as scaffolds to regulate interenzyme distance for construction of efficient enzyme cascade amplification for ultrasensitive electrochemical detection of MMP-2. Anal. Chem. 2017, 89, 9383–9387. [Google Scholar] [CrossRef]
- Fu, J.L.; Yang, Y.R.; Dhakal, S.; Zhao, Z.; Liu, M.; Zhang, T.; Walter, N.G.; Yan, H. Assembly of multienzyme complexes on DNA nanostructures. Nat. Protoc. 2016, 11, 2243–2273. [Google Scholar] [CrossRef]
- Negi, S.; Imanishi, M.; Matsumoto, M.; Sugiura, Y. New redesigned zinc-finger proteins: Design strategy and its application. Chemistry 2008, 14, 3236–3249. [Google Scholar] [CrossRef] [PubMed]
- Conrado, R.J.; Wu, G.C.; Boock, J.T.; Xu, H.; Chen, S.Y.; Lebar, T.; Turnšek, J.; Tomšič, N.; Avbelj, M.; Gaber, R.; et al. DNA-guided assembly of biosynthetic pathways promotes improved catalytic efficiency. Nucleic Acids Res. 2012, 40, 1879–1889. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Jung, S.C.; Bui, L.M.; Kang, K.H.; Song, J.J.; Kim, S.C. Improved production of L-threonine in Escherichia coli by use of a DNA scaffold system. Appl. Environ. Microbiol. 2013, 79, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhu, Y.; Ma, W.; Shin, H.D.; Li, J.; Liu, L.; Du, G.; Chen, J. Spatial modulation of key pathway enzymes by DNA-guided scaffold system and respiration chain engineering for improved N-acetylglucosamine production by Bacillus subtilis. Metab. Eng. 2014, 24, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, J.; Yin, P.; Voigt, C.A. Genetic encoding of DNA nanostructures and their self-assembly in living bacteria. Nat. Commun. 2016, 7, 11179. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.Y.; Qiu, X.Y.; Zhu, L.Y.; Wu, X.M.; Zhang, Y.; Zhu, Q.H.; Fan, D.Y.; Zhu, C.S.; Zhang, D.Y. Spatial organization of heterologous metabolic system in vivo based on TALE. Sci. Rep. 2016, 6, 26065. [Google Scholar] [CrossRef]
- Sara, M.; Benjamin, B.; Christiane, E.; Jasper, J.; Koch, O.; Summerer, D. Overcoming conservation in TALE-DNA interactions: A minimal repeat scaffold enables selective recognition of an oxidized 5-methylcytosine. Chem. Sci. 2018, 9, 7247–7252. [Google Scholar]
- Myhrvold, C.; Polka, J.K.; Silver, P.A. Synthetic lipid-containing scaffolds enhance production by colocalizing enzymes. ACS Synth. Biol. 2016, 5, 1396–1403. [Google Scholar] [CrossRef]
- SantaLucia, J. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA 1998, 95, 1460–1465. [Google Scholar] [CrossRef]
- Delebecque, C.J.; Silver, P.A.; Lindner, A.B. Designing and using RNA scaffolds to assemble proteins in vivo. Nat. Protoc. 2012, 7, 1797–1807. [Google Scholar] [CrossRef]
- Chworos, A.; Severcan, I.; Koyfman, A.Y.; Weinkam, P.; Oroudjev, E.; Hansma, H.G.; Jaeger, L. Building programmable jigsaw puzzles with RNA. Science 2004, 306, 2068–2072. [Google Scholar] [CrossRef] [PubMed]
- Severcan, I.; Geary, C.; Chworos, A.; Voss, N.; Jacovetty, E.; Jaeger, L. A polyhedron made of tRNAs. Nat. Chem. 2010, 2, 772–779. [Google Scholar] [CrossRef]
- Geary, C.; Rothemund, P.W.; Andersen, E.S. A single-stranded architecture for cotranscriptional folding of RNA nanostructures. Science 2014, 345, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Delebecque, C.J.; Lindner, A.B.; Silver, P.A.; Aldaye, F.A. Organization of intracellular reactions with rationally designed RNA assemblies. Science 2011, 333, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, G.; Garg, A.; Godding, D.; Way, J.C.; Silver, P.A. In vivo co-localization of enzymes on RNA scaffolds increases metabolic production in a geometrically dependent manner. Nucleic Acids Res. 2014, 42, 9493–9503. [Google Scholar] [CrossRef] [PubMed]
- Dueber, J.E.; Wu, G.C.; Malmirchegini, G.R.; Moon, T.S.; Petzold, C.J.; Ullal, A.V.; Prather, K.L.; Keasling, J.D. Synthetic protein scaffolds provide modular control over metabolic flux. Nat. Biotechnol. 2009, 27, 753–759. [Google Scholar] [CrossRef]
- Whitaker, W.R.; Dueber, J.E. Metabolic pathway flux enhancement by synthetic protein scaffolding. Methods Enzymol. 2011, 497, 447–468. [Google Scholar]
- Zarrinpar, A.; Park, S.; Lim, W.A. Optimization of specificity in a cellular protein interaction network by negative selection. Nature 2003, 426, 676–680. [Google Scholar] [CrossRef]
- Harris, B.Z.; Hillier, B.J.; Lim, W.A. Energetic determinants of internal motif recognition by PDZ domains. Biochemistry 2001, 40, 5921–5930. [Google Scholar] [CrossRef]
- Kim, A.S.; Kakalis, L.T.; Abdul-Manan, N.; Liu, G.A.; Rosen, M.K. Autoinhibition and activation mechanisms of the Wiskott–Aldrich syndrome protein. Nature 2000, 404, 151–158. [Google Scholar] [CrossRef]
- Reinke, A.W.; Grant, R.A.; Keating, A.E. A synthetic coiled-coil interactome provides heterospecific modules for molecular engineering. J. Am. Chem. Soc. 2010, 132, 6025–6031. [Google Scholar] [CrossRef]
- Levskaya, A.; Weiner, O.D.; Lim, W.A.; Voigt, C.A. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature 2009, 461, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, B.; Moks, T.; Jansson, B.; Abrahmsen, L.; Elmblad, A.; Holmgren, E.; Henrichson, C.; Jones, T.A.; Uhlen, M. A synthetic IgG-binding domain based on staphylococcal protein A. Protein Eng. 1987, 1, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Agapakis, C.M.; Ducat, D.C.; Boyle, P.M.; Wintermute, E.H.; Way, J.C.; Silver, P.A. Insulation of a synthetic hydrogen metabolism circuit in bacteria. J. Biol. Eng. 2010, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.M.; Mazumdar, S.; Lee, S.W.; Jung, M.Y.; Lim, J.H.; Seo, S.W.; Jung, G.Y.; Oh, M.K. Butyrate production in engineered Escherichia coli with synthetic scaffolds. Biotechnol. Bioeng. 2013, 110, 2790–2794. [Google Scholar] [CrossRef]
- Wang, Y.C.; Yu, O. Synthetic scaffolds increased resveratrol biosynthesis in engineered yeast cells. J. Biotechnol. 2012, 157, 258–260. [Google Scholar] [CrossRef]
- Liu, F.; Banta, S.; Chen, W. Functional assembly of a multi-enzyme methanol oxidation cascade on a surface-displayed trifunctional scaffold for enhanced NADH production. Chem. Commun. 2013, 49, 3766–3768. [Google Scholar] [CrossRef]
- Tsai, S.L.; Oh, J.; Singh, S.; Chen, R.; Chen, W. Functional assembly of minicellulosomes on the Saccharomyces cerevisiae cell surface for cellulose hydrolysis and ethanol production. Appl. Environ. Microbiol. 2009, 75, 6087–6093. [Google Scholar] [CrossRef]
- Hirakawa, H.; Kakitani, A.; Nagamune, T. Introduction of selective intersubunit disulfide bonds into self-assembly protein scaffold to enhance an artificial multienzyme complex’s activity. Biotechnol. Bioeng. 2013, 110, 1858–1864. [Google Scholar] [CrossRef]
- Hirakawa, H.; Nagamune, T. Molecular assembly of P450 with ferredoxin and ferredoxin reductase by fusion to PCNA. Chembiochem 2010, 11, 1517–1520. [Google Scholar] [CrossRef]
- Bayer, E.A.; Belaich, J.P.; Shoham, Y.; Lamed, R. The cellulosomes: Multienzyme machines for degradation of plant cell wall polysaccharides. Ann. Rev. Microbiol. 2004, 58, 521–554. [Google Scholar] [CrossRef] [PubMed]
- Doi, R.H.; Kosugi, A. Cellulosomes: Plant-cell-wall-degrading enzyme complexes. Nat. Rev. Microbiol. 2004, 2, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.L.; Zhu, J.; Wheeldon, I. Synthetic protein scaffolds for biosynthetic pathway co-localization on lipid droplet membranes. ACS Synth. Biol. 2017, 6, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Sarin, L.P.; Hirvonen, J.J.; Laurinmaki, P.; Butcher, S.J.; Bamford, D.H.; Poranen, M.M. Bacteriophage φ6 nucleocapsid surface protein 8 interacts with virus-specific membrane vesicles containing major envelope protein 9. J. Virol. 2012, 86, 5376–5379. [Google Scholar] [CrossRef]
- Chen, A.H.; Silver, P.A. Designing biological compartmentalization. Trends Cell Biol. 2012, 22, 662–670. [Google Scholar] [CrossRef]
- DeLoache, W.C.; Dueber, J.E. Compartmentalizing metabolic pathways in organelles. Nat. Biotechnol. 2013, 31, 320–321. [Google Scholar] [CrossRef]
- Ivessa, A.S.; Schneiter, R.; Kohlwein, S.D. Yeast acetyl-CoA carboxylase is associated with the cytoplasmic surface of the endoplasmic reticulum. Eur. J. Cell Biol. 1997, 74, 399–406. [Google Scholar]
- Markgraf, D.F.; Klemm, R.W.; Junker, M.; Hannibal-Bach, H.K.; Ejsing, C.S.; Rapoport, T.A. An ER protein functionally couples neutral lipid metabolism on lipid droplets to membrane lipid synthesis in the ER. Cell Rep. 2014, 6, 44–55. [Google Scholar] [CrossRef]
- Ziegler, J.; Facchini, P.J. Alkaloid biosynthesis: Metabolism and trafficking. Annu. Rev. Plant Biol. 2008, 59, 735–769. [Google Scholar] [CrossRef]
- Huttanus, H.M.; Feng, X. Compartmentalized metabolic engineering for biochemical and biofuel production. Biotechnol. J. 2017, 12, 1700052. [Google Scholar] [CrossRef]
- Abernathy, M.H.; He, L.; Tang, Y.J. Channeling in native microbial pathways: Implications and challenges for metabolic engineering. Biotechnol. Adv. 2017, 35, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Gnanasekaran, T.; Karcher, D.; Nielsen, A.Z.; Martens, H.J.; Ruf, S.; Kroop, X.; Olsen, C.E.; Motawie, M.S.; Pribil, M.; Møller, B.L.; et al. Transfer of the cytochrome P450-dependent dhurrin pathway from Sorghum bicolor into Nicotiana tabacum chloroplasts for light-driven synthesis. J. Exp. Bot. 2016, 67, 2495–2506. [Google Scholar] [CrossRef] [PubMed]
- Lassen, L.M.; Nielsen, A.Z.; Ziersen, B.; Gnanasekaran, T.; Møller, B.L.; Jensen, P.E. Redirecting photosynthetic electron flow into light-driven synthesis of alternative products including high-value bioactive natural compounds. ACS Synth. Biol. 2014, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, A.Z.; Mellor, S.B.; Vavitsas, K.; Wlodarczyk, A.J.; Gnanasekaran, T.; Perestrello Ramos, H.; de Jesus, M.; King, B.C.; Bakowski, K.; Jensen, P.E. Extending the biosynthetic repertoires of cyanobacteria and chloroplasts. Plant J. 2016, 87, 87–102. [Google Scholar] [CrossRef]
- Nielsen, A.Z.; Ziersen, B.; Jensen, K.; Lassen, L.M.; Olsen, C.E.; Møller, B.L.; Jensen, P.E. Redirecting photosynthetic reducing power toward bioactive natural product synthesis. ACS Synth. Biol. 2013, 2, 308–315. [Google Scholar] [CrossRef]
- Henriques de Jesus, M.P.R.; Zygadlo Nielsen, A.; Busck Mellor, S.; Matthes, A.; Burow, M.; Robinson, C.; Erik Jensen, P. Tat proteins as novel thylakoid membrane anchors organize a biosynthetic pathway in chloroplasts and increase product yield 5-fold. Metab. Eng. 2017, 44, 108–116. [Google Scholar] [CrossRef]
- Avalos, J.L.; Fink, G.R.; Stephanopoulos, G. Compartmentalization of metabolic pathways in yeast mitochondria improves the production of branched-chain alcohols. Nat. Biotechnol. 2013, 31, 335–341. [Google Scholar] [CrossRef]
- Chowdhury, C.; Sinha, S.; Chun, S.; Yeates, T.O.; Bobik, T.A. Diverse acterial microcompartment organelles. Microbiol. Mol. Biol. Rev. 2014, 78, 438–468. [Google Scholar] [CrossRef]
- Frank, S.; Lawrence, A.D.; Prentice, M.B.; Warren, M.J. Bacterial microcompartments moving into a synthetic biological world. J. Biotechnol. 2013, 163, 273–279. [Google Scholar] [CrossRef]
- Kerfeld, C.A.; Erbilgin, O. Bacterial microcompartments and the modular construction of microbial metabolism. Trends Microbiol. 2015, 23, 22–34. [Google Scholar] [CrossRef]
- Bonacci, W.; Teng, P.K.; Afonso, B.; Niederholtmeyer, H.; Grob, P.; Silver, P.A.; Savage, D.F. Modularity of a carbon-fixing protein organelle. Proc. Natl. Acad. Sci. USA 2012, 109, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Quin, M.B.; Sanders, M.A.; Johnson, E.T.; SchmidtDannert, C. Engineered protein nano-compartments for targeted enzyme localization. PLoS ONE 2012, 7, e33342. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, A.D.; Frank, S.; Newnham, S.; Lee, M.J.; Brown, I.R.; Xue, W.F.; Rowe, M.L.; Mulvihill, D.P.; Prentice, M.B.; Howard, M.J.; et al. Solution structure of a bacterial microcompartment targeting peptide and its application in the construction of an ethanol bioreactor. ACS Synth. Biol. 2014, 3, 454–465. [Google Scholar] [CrossRef]
- Cai, F.; Sutter, M.; Bernstein, S.L.; Kinney, J.N.; Kerfeld, C.A. Engineering bacterial microcompartment shells: Chimeric shell proteins and chimeric carboxysome shells. ACS Synth. Biol. 2015, 4, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Esquer, C.R.; Shubitowski, T.B.; Kerfeld, C.A. Streamlined construction of the cyanobacterial CO2-fixing organelle via protein domain fusions for use in plant synthetic biology. Plant Cell 2015, 27, 2637–2644. [Google Scholar] [CrossRef]
- Quin, M.B.; Perdue, S.A.; Hsu, S.Y.; Schmidt-Dannert, C. Encapsulation of multiple cargo proteins within recombinant Eut nanocompartments. Appl. Microbiol. Biotechnol. 2016, 100, 9187–9200. [Google Scholar] [CrossRef]
- Lee, M.; DeLoache, W.C.; Dueber, J.E. Employing bacterial microcompartment technology to engineer a shell-free enzyme aggregate for enhance 1-2 propanediol production in Escherichia coli. Metab. Eng. 2016, 36, 48–56. [Google Scholar] [CrossRef]
- Baumgart, M.; Huber, I.; Abdollahzadeh, I.; Gensch, T.; Frunzke, J. Heterologous expression of the Halothiobacillus neapolitanus carboxysomal gene cluster in Corynebacterium glutamicum. J. Biotechnol. 2017, 258, 126–135. [Google Scholar] [CrossRef]
- Liang, M.; Frank, S.; Lünsdorf, H.; Warren, M.J.; Prentice, M.B. Bacterial microcompartment-directed polyphaste kinase promotes stable polyphosphate accumulation E. coli. Biotechnol. J. 2017, 12, 3. [Google Scholar] [CrossRef]
- Slininger, L.; Jakobson, C.M.; Tullman-Ercek, D. Evidence for improved encapsulated pathway behavior in a bacterial microcompartment through shell protein engineering. ACS Synth. Biol. 2017, 6, 1880–1891. [Google Scholar] [CrossRef]
- Wagner, H.J.; Capitain, C.C.; Richter, K.; Nessling, M.; Mampel, J. Engineering bacterial microcompartments with heterologous enzyme cargos. Eng. Life Sci. 2017, 17, 36–46. [Google Scholar] [CrossRef]
- Yung, M.; Bourguet, F.A.; Carpenter, T.S.; Coleman, M.A. Re-directing bacterial microcompartment systems to enhance recombinant protein expression of lysis protein E from bacteriophage 0X174. Microb. Cell Fact. 2017, 16, 71. [Google Scholar] [CrossRef] [PubMed]
- Crowley, C.S.; Cascio, D.; Sawaya, M.R.; Kopstein, J.S.; Bobik, T.A.; Yeates, T.O. Structural insight into the mechanisms of transport across the Salmonella enterica Pdu microcompartment shell. J. Biol. Chem. 2010, 285, 37838–37846. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.N.; Axen, S.D.; Kerfeld, C.A. Comparative analysis of carboxysome shell proteins. Photosynth. Res. 2011, 109, 21–32. [Google Scholar] [CrossRef]
- Kerfeld, C.A.; Heinhorst, S.; Cannon, G.C. Bacterial microcompartments. Annu. Rev. Microbiol. 2010, 64, 391–408. [Google Scholar] [CrossRef]
- Yeates, T.O.; Crowley, C.S.; Tanaka, S. Bacterial microcompartment organelles: Protein shell structure and evolution. Annu. Rev. Biophys. 2010, 39, 185–205. [Google Scholar] [CrossRef]
- Kerfeld, C.A.; Sawaya, M.R.; Tanaka, S.; Nguyen, C.V.; Phillips, M.; Beeby, M.; Yeates, T.O. Protein structures forming the shell of primitive bacterial organelles. Science 2005, 309, 936–938. [Google Scholar] [CrossRef]
- Lassila, J.K.; Bernstein, S.L.; Kinney, J.N.; Axen, S.D.; Kerfeld, C.A. Assembly of robust bacterial microcompartment shells using building blocks from an organelle of unknown function. J. Mol. Biol. 2014, 426, 2217–2228. [Google Scholar] [CrossRef]
- Keeling, T.J.; Samborska, B.; Demers, R.W.; Kimber, M.S. Interactions and structural variability of b-carboxysomal shell protein CcmL. Photosynth. Res. 2014, 121, 125–133. [Google Scholar] [CrossRef]
- Noël, C.R.; Cai, F.; Kerfeld, C.A. Purification and characterization of protein nanotubes assembled from a single bacterial microcompartment shell subunit. Adv. Mater. Interfaces 2015, 3, 1500295. [Google Scholar] [CrossRef]
- Heldt, D.; Frank, S.; Seyedarabi, A.; Ladikis, D.; Parsons, J.B.; Warren, M.J.; Pickersgill, R.W. Structure of a trimeric bacterial microcompartment shell protein, EtuB, associated with ethanol utilization in Clostridium kluyveri. Biochem. J. 2009, 423, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.B.; Frank, S.; Bhella, D.; Liang, M.; Prentice, M.B.; Mulvihill, D.P.; Warren, M.J. Synthesis of empty bacterial microcompartments, directed organelle protein incorporation, and evidence of filament-associated organelle movement. Mol. Cell 2010, 38, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Pitts, A.C.; Tuck, L.R.; Faulds-Pain, A.; Lewis, R.J.; Marles-Wright, J. Structural insight into the Clostridium difficile ethanolamine utilization microcompartment. PLoS ONE 2013, 7, e48360. [Google Scholar]
- Pang, A.; Frank, S.; Brown, I.; Warren, M.J.; Pickersgill, R.W. Structural insights into higher order assembly and function of the bacterial microcompartment protein PduA. J. Biol. Chem. 2014, 289, 22377–22384. [Google Scholar] [CrossRef]
- Sutter, M.; Faulkner, M.; Aussignargues, C.; Paasch, B.C.; Barrett, S.; Kerfeld, C.A.; Liu, L.N. Visualization of bacterial microcompartment facet assembly using high-speed atomic force microscopy. Nano Lett. 2015, 16, 1590–1595. [Google Scholar] [CrossRef]
- Held, M.; Kolb, A.; Perdue, S.; Hsu, S.Y.; Bloch, S.E.; Quin, M.B.; Schmidt-Dannert, C. Engineering formation of multiple recombinant Eut protein nanocompartments in E. coli. Sci. Rep. 2016, 6, 24359. [Google Scholar] [CrossRef]
- Fan, C.; Cheng, S.; Sinha, S. Interactions between the termini of lumen enzymes and shell proteins mediate enzyme encapsulation into bacterial microcompartments. Proc. Natl. Acad. Sci. USA 2012, 109, 14995–15000. [Google Scholar] [CrossRef]
- Aussignargues, C.; Paasch, B.C.; Gonzalez-Esquer, R.; Erbilgin, O.; Kerfeld, C.A. Bacterial microcompartment assembly: The key role of encapsulation peptides. Commun. Integr. Biol. 2015, 8, e1039755. [Google Scholar] [CrossRef]
- Jakobson, C.M.; Kim, E.Y.; Slininger, M.F.; Chien, A.; TullmanErcek, D. Localization of proteins to the 1, 2-propanediol utilization microcompartment by non-native signal sequences is mediated by a common hydrophobic motif. J. Biol. 2015, 290, 24519–24533. [Google Scholar] [CrossRef]
- Dryden, K.A.; Crowley, C.S.; Tanaka, S.; Yeates, T.O.; Yeager, M. Two dimensional crystals of carboxysome shell proteins recapitulate the hexagonal packing of three-dimensional crystals. Protein Sci. 2009, 18, 2629–2635. [Google Scholar] [CrossRef]
- Savage, D.F.; Afonso, B.; Chen, A.H.; Silver, P.A. Spatially ordered dynamics of the bacterial carbon fixation machinery. Science 2010, 327, 1258–1261. [Google Scholar] [CrossRef] [PubMed]
- Samborska, B.; Kimber, M.S. A dodecameric CcmK2 structure suggests β-carboxysomal shell facets have a double-layered organization. Structure 2012, 20, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Cameron, J.C.; Wilson, S.C.; Bernstein, S.L.; Kerfeld, C.A. Biogenesis of a bacterial organelle: The carboxysome assembly pathway. Cell 2013, 155, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, C.; Chun, S.; Pang, A.; Sawaya, M.R.; Sinha, S.; Yeates, T.O.; Bobik, T.A. Selective molecular transport through the protein shell of a bacterial microcompartment organelle. Proc. Natl. Acad. Sci. USA 2015, 112, 2990–2995. [Google Scholar] [CrossRef]
- Mourão, M.A.; Hakim, J.B.; Schnell, S. Connecting the dots: The effects of macromolecular crowding on cell physiology. Biophys. J. 2014, 107, 2761–2766. [Google Scholar] [CrossRef]
- Spitzer, J. From water and ions to crowded biomacromolecules: In vivo structuring of a prokaryotic cell. Microbiol. Mol. Biol. Rev. 2011, 75, 491–506. [Google Scholar] [CrossRef]
- Parry, B.R.; Surovtsev, I.V.; Cabeen, M.T.; O’Hern, C.S.; Dufresne, E.R.; Jacobs-Wagner, C. The bacterial cytoplasm has glass-like properties and is fluidized by metabolic activity. Cell 2014, 156, 183–194. [Google Scholar] [CrossRef]
- Minton, A.P. The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J. Biol. Chem. 2001, 276, 10577–10580. [Google Scholar] [CrossRef]
- Castellana, M.; Wilson, M.Z.; Xu, Y.; Joshi, P.; Cristea, I.M.; Rabinowitz, J.D.; Gitai, Z.; Wingreen, N.S. Enzyme clustering accelerates processing of intermediates through metabolic channeling. Nat. Biotechnol. 2014, 32, 1011–1018. [Google Scholar] [CrossRef]
- Wheeldon, I.; Minteer, S.D.; Banta, S.; Barton, S.C.; Atanassov, P.; Sigman, M. Substrate channelling as an approach to cascade reactions. Nat. Chem. 2016, 8, 299–309. [Google Scholar] [CrossRef]
- Bar-Even, A.; Noor, E.; Savir, Y.; Liebermeister, W.; Davidi, D.; Tawfik, D.S.; Milo, R. The moderately efficient enzyme: Evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry (Mosc) 2011, 50, 4402–4410. [Google Scholar] [CrossRef] [PubMed]
- Kafri, M.; Metzl-Raz, E.; Jona, G.; Barkai, N. The cost of protein production. Cell Rep. 2016, 14, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Rugbjerg, R.; Sommer, M.O.A. Overcoming genetic heterogeneity in industrial fermentations. Nat. Biotechnol. 2019, 37, 869–876. [Google Scholar] [CrossRef]
- Miles, E.W. Tryptophan synthase: A multienzyme complex with an intramolecular tunnel. Chem. Rec. 2001, 1, 140–151. [Google Scholar] [CrossRef]
- An, S.; Kumar, R.; Sheets, E.D.; Benkovic, S.J. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science 2008, 320, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Narayanaswamy, R.; Levy, M.; Tsechansky, M.; Stovall, G.M.; O’Connell, J.D.; Mirrielees, J.; Ellington, A.D.; Marcotte, E.M. Widespread reorganization of metabolic enzymes into reversible assemblies upon nutrient starvation. Proc. Natl. Acad. Sci. USA 2009, 106, 10147–10152. [Google Scholar] [CrossRef]
- Noree, C.; Sato, B.K.; Broyer, R.M.; Wilhelm, J.E. Identification of novel filament-forming proteins in Saccharomyces cerevisiae and Drosophila melanogaster. J. Cell Biol. 2010, 190, 541–551. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme Assembly Strategy | Number of Immobilized Enzymes | Reference | |
|---|---|---|---|
| Scaffold-free enzyme assembly | Interaction pair or affinity peptide guided enzyme assembly | 2–3 | [28,38,41] |
| CatIBs | 1–2 | [56] | |
| Nucleic acid scaffold | DNA scaffold | 2 | [84] |
| RNA scaffold | 2–4 | [95] | |
| Protein scaffold | (GBD)x–(SH3)y–(PDZ)z protein scaffold | 3 | [96] |
| Protein scaffold outside the cell | 3 | [108] | |
| Lipid-containing scaffold | 2 | [88] | |
| Physical compartment | Eukaryotic physical compartments | 3 | [125,126] |
| Protein-based compartments in prokaryotic cells | 2 | [27] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, X.; Cui, S.; Gu, Y.; Li, J.; Du, G.; Liu, L. Enzyme Assembly for Compartmentalized Metabolic Flux Control. Metabolites 2020, 10, 125. https://doi.org/10.3390/metabo10040125
Lv X, Cui S, Gu Y, Li J, Du G, Liu L. Enzyme Assembly for Compartmentalized Metabolic Flux Control. Metabolites. 2020; 10(4):125. https://doi.org/10.3390/metabo10040125
Chicago/Turabian StyleLv, Xueqin, Shixiu Cui, Yang Gu, Jianghua Li, Guocheng Du, and Long Liu. 2020. "Enzyme Assembly for Compartmentalized Metabolic Flux Control" Metabolites 10, no. 4: 125. https://doi.org/10.3390/metabo10040125
APA StyleLv, X., Cui, S., Gu, Y., Li, J., Du, G., & Liu, L. (2020). Enzyme Assembly for Compartmentalized Metabolic Flux Control. Metabolites, 10(4), 125. https://doi.org/10.3390/metabo10040125