Deriving Lipid Classification Based on Molecular Formulas



Abstract
1. Introduction
2. Results
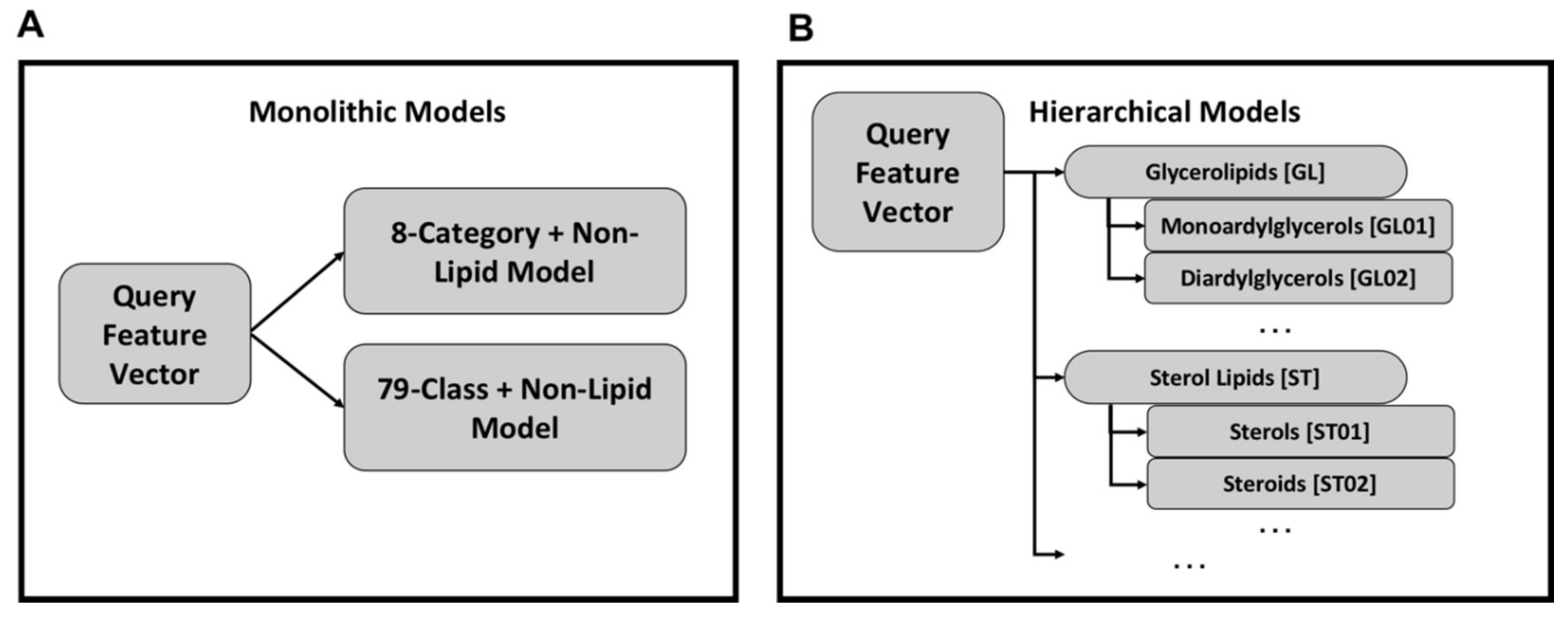
2.1. Monolithic Classifier Performance on Training Datasets
2.2. Multi-Classifier Performance on Training Datasets
2.3. Multi-Classifier Performance on Theoretical Molecular Formulas
2.4. Multi-Classifier Performance on Experimentally-Observed Molecular Formulas
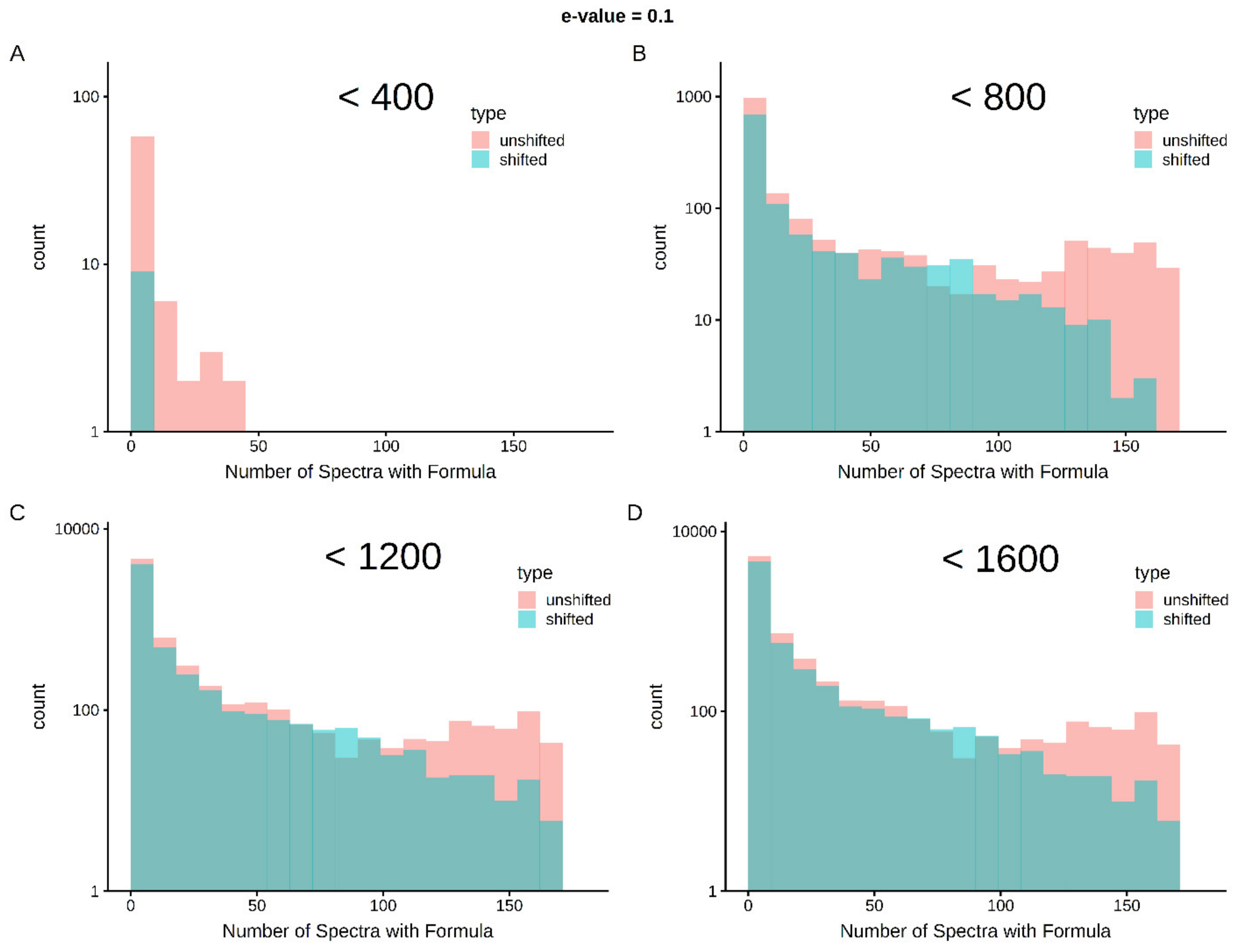
2.5. Cross-Sample Assignment Correspondence along with Lipid Classification Improves Assignment Quality
3. Discussion
3.1. Classifier Organization and Performance
3.2. LMSD vs LMISSD Trained Models
3.3. Classifier Generalization
3.4. Mass Error and Classification Results
3.5. Implications for Experimental Design
4. Materials and Methods
4.1. Structure of Chemically-Descriptive Feature Vectors
4.2. Derivation and Organization of Training Datasets
4.3. HMDB-Derived Molecular Formula Convex Hull Construction
4.4. Experimentally-Derived Molecular Formulas from Human Lung Cancer Samples
4.5. Classifier Construction
4.6. Evaluation of Lipid Classification Performance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
A.1. Paired Human NSCLC Cancer and Non-Cancer Tissue Samples
A.2. Mass Spectrometry Analysis of Tissue Samples
References
- Singer, S.J.; Nicolson, G.L. The fluid mosaic model of the structure of cell membranes. Science 1972, 175, 720–731. [Google Scholar] [CrossRef]
- Clamp, A.; Ladha, S.; Clark, D.; Grimble, R.; Lund, E. The influence of dietary lipids on the composition and membrane fluidity of rat hepatocyte plasma membrane. Lipids 1997, 32, 179–184. [Google Scholar] [CrossRef]
- Chen, J.J.; Yu, B.P. Alterations in mitochondrial membrane fluidity by lipid peroxidation products. Free Radic. Biol. Med. 1994, 17, 411–418. [Google Scholar] [CrossRef]
- Zechner, R.; Zimmermann, R.; Eichmann, T.O.; Kohlwein, S.D.; Haemmerle, G.; Lass, A.; Madeo, F. FAT SIGNALS-lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012, 15, 279–291. [Google Scholar] [CrossRef]
- Morrison, R.F.; Farmer, S.R. Hormonal signaling and transcriptional control of adipocyte differentiation. J. Nutr. 2000, 130, 3116S–3121S. [Google Scholar] [CrossRef]
- Neely, J.R.; Morgan, H.E. Relationship between carbohydrate and lipid metabolism and the energy balance of heart muscle. Annu. Rev. Physiol. 1974, 36, 413–459. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; Carneiro-Freire, N.; Seco-Filgueira, M.; Fernández-Fernández, C.; Mouriño-Bayolo, D. Mitochondrial β-oxidation of saturated fatty acids in humans. Mitochondrion 2018, 46, 73–90. [Google Scholar] [CrossRef]
- De Pablo, M.A.; De Cienfuegos, G.Á. Modulatory effects of dietary lipids on immune system functions. Immunol. Cell Biol. 2000, 78, 31–39. [Google Scholar] [CrossRef]
- Zhang, F.; Du, G. Dysregulated lipid metabolism in cancer. World J. Biol. Chem. 2012, 3, 167. [Google Scholar] [CrossRef]
- Ray, U.; Roy, S.S. Aberrant lipid metabolism in cancer cells–the role of oncolipid-activated signaling. FEBS J. 2018, 285, 432–443. [Google Scholar] [CrossRef]
- Ahadi, S.; Zhou, W.; Rose, S.M.S.-F.; Sailani, M.R.; Contrepois, K.; Avina, M.; Ashland, M.; Brunet, A.; Snyder, M. Personal aging markers and ageotypes revealed by deep longitudinal profiling. Nat. Med. 2020, 26, 83–90. [Google Scholar] [CrossRef]
- Kolovou, G.; Katsiki, N.; Pavlidis, A.; Bilianou, H.; Goumas, G.; Mikhailidis, D.P. Ageing mechanisms and associated lipid changes. Curr. Vasc. Pharmacol. 2014, 12, 682–689. [Google Scholar] [CrossRef]
- Lydic, T.A.; Goo, Y.-H. Lipidomics unveils the complexity of the lipidome in metabolic diseases. Clin. Transl. Med. 2018, 7, 4. [Google Scholar] [CrossRef]
- Horvath, S.E.; Daum, G. Lipids of mitochondria. Prog. Lipid Res. 2013, 52, 590–614. [Google Scholar] [CrossRef]
- Aviram, R.; Manella, G.; Kopelman, N.; Neufeld-Cohen, A.; Zwighaft, Z.; Elimelech, M.; Adamovich, Y.; Golik, M.; Wang, C.; Han, X. Lipidomics analyses reveal temporal and spatial lipid organization and uncover daily oscillations in intracellular organelles. Mol. Cell 2016, 62, 636–648. [Google Scholar] [CrossRef]
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H., Jr.; Murphy, R.C.; Raetz, C.R.; Russell, D.W.; Seyama, Y.; Shaw, W. A comprehensive classification system for lipids. Eur. J. Lipid Sci. Technol. 2005, 107, 337–364. [Google Scholar] [CrossRef]
- Köfeler, H.C.; Fauland, A.; Rechberger, G.N.; Trötzmüller, M. Mass spectrometry based lipidomics: An overview of technological platforms. Metabolites 2012, 2, 19–38. [Google Scholar] [CrossRef]
- Quehenberger, O.; Armando, A.M.; Dennis, E.A. High sensitivity quantitative lipidomics analysis of fatty acids in biological samples by gas chromatography–mass spectrometry. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipid. 2011, 1811, 648–656. [Google Scholar] [CrossRef]
- Masoodi, M.; Nicolaou, A. Lipidomic analysis of twenty-seven prostanoids and isoprostanes by liquid chromatography/electrospray tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2006, 20, 3023–3029. [Google Scholar] [CrossRef]
- Sandra, K.; Dos Santos Pereira, A.; Vanhoenacker, G.; David, F.; Sandra, P. Comprehensive blood plasma lipidomics by liquid chromatography/quadrupole time-of-flight mass spectrometry. J. Chromatogr. A 2010, 1217, 4087–4099. [Google Scholar] [CrossRef]
- Valdes-Gonzalez, T.; Goto-Inoue, N.; Hirano, W.; Ishiyama, H.; Hayasaka, T.; Setou, M.; Taki, T. New approach for glyco-and lipidomics–Molecular scanning of human brain gangliosides by TLC-Blot and MALDI-QIT-TOF MS. J. Neurochem. 2011, 116, 678–683. [Google Scholar] [CrossRef]
- Eliuk, S.; Makarov, A. Evolution of orbitrap mass spectrometry instrumentation. Annu. Rev. Anal. Chem. 2015, 8, 61–80. [Google Scholar] [CrossRef]
- Carreer, W.; Flight, R.; Moseley, H. A computational framework for high-throughput isotopic natural abundance correction of omics-level ultra-high resolution FT-MS datasets. Metabolites 2013, 3, 853–866. [Google Scholar] [CrossRef]
- Jin, H.; Moseley, H.N.B. Moiety modeling framework for deriving moiety abundances from mass spectrometry measured isotopologues. BMC Bioinform. 2019, 20, 524. [Google Scholar] [CrossRef]
- Mitchell, J.M.; Flight, R.M.; Moseley, H.N. Small Molecule Isotope Resolved Formula Enumeration: A Methodology for Assigning Isotopologues and Metabolite Formulas in Fourier Transform Mass Spectra. Anal. Chem. 2019, 91, 8933–8940. [Google Scholar] [CrossRef]
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef]
- Li, J.; Hoene, M.; Zhao, X.; Chen, S.; Wei, H.; Häring, H.-U.; Lin, X.; Zeng, Z.; Weigert, C.; Lehmann, R. Stable isotope-assisted lipidomics combined with nontargeted isotopomer filtering, a tool to unravel the complex dynamics of lipid metabolism. Anal. Chem. 2013, 85, 4651–4657. [Google Scholar] [CrossRef]
- Postle, A.D.; Hunt, A.N. Dynamic lipidomics with stable isotope labelling. J. Chromatogr. B 2009, 877, 2716–2721. [Google Scholar] [CrossRef]
- Allen, D.K.; Bates, P.D.; Tjellstroem, H. Tracking the metabolic pulse of plant lipid production with isotopic labeling and flux analyses: Past, present and future. Prog. Lipid Res. 2015, 58, 97–120. [Google Scholar] [CrossRef]
- Schwudke, D.; Schuhmann, K.; Herzog, R.; Bornstein, S.R.; Shevchenko, A. Shotgun lipidomics on high resolution mass spectrometers. Cold Spring Harb. Perspect. Biol. 2011, 3, a004614. [Google Scholar] [CrossRef]
- Almeida, R.; Pauling, J.K.; Sokol, E.; Hannibal-Bach, H.K.; Ejsing, C.S. Comprehensive lipidome analysis by shotgun lipidomics on a hybrid quadrupole-orbitrap-linear ion trap mass spectrometer. J. Am. Soc. Mass Spectrom. 2014, 26, 133–148. [Google Scholar] [CrossRef]
- Peake, D.A.; Yokoi, Y.; Wang, J.; Huang, Y. A New Lipid Software Workflow for Processing Orbitrap-based Global Lipidomics Data in Translational and Systems Biology Research; THERMO SCIENTIFIC application note; ThermoFisher Scientific: Waltham, MA, USA, 2013. [Google Scholar]
- Lane, A.N.; Fan, T.W.-M.; Xie, Z.; Moseley, H.N.; Higashi, R.M. Isotopomer analysis of lipid biosynthesis by high resolution mass spectrometry and NMR. Anal. Chim. Acta 2009, 651, 201–208. [Google Scholar] [CrossRef]
- Mitchell, J.M.; Flight, R.M.; Wang, Q.J.; Higashi, R.M.; Fan, T.W.-M.; Lane, A.N.; Moseley, H.N. New methods to identify high peak density artifacts in Fourier transform mass spectra and to mitigate their effects on high-throughput metabolomic data analysis. Metabolomics 2018, 14, 125. [Google Scholar] [CrossRef]
- Moseley, H.N. Error analysis and propagation in metabolomics data analysis. Comput. Struct. Biotechnol. J. 2013, 4, e201301006. [Google Scholar] [CrossRef]
- Bielawski, J.; Pierce, J.S.; Snider, J.; Rembiesa, B.; Szulc, Z.M.; Bielawska, A. Sphingolipid analysis by high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS). In Sphingolipids as Signaling and Regulatory Molecules; Springer: Berlin, Germany, 2010; pp. 46–59. [Google Scholar]
- Chekmeneva, E.; Dos Santos Correia, G.a.; Chan, Q.; Wijeyesekera, A.; Tin, A.; Young, J.H.; Elliott, P.; Nicholson, J.K.; Holmes, E. Optimization and application of direct infusion nanoelectrospray HRMS method for large-scale urinary metabolic phenotyping in molecular epidemiology. J. Proteome Res. 2017, 16, 1646–1658. [Google Scholar] [CrossRef]
- Mitchell, J.M.; Fan, T.W.-M.; Lane, A.N.; Moseley, H.N. Development and in silico evaluation of large-scale metabolite identification methods using functional group detection for metabolomics. Front. Genet. 2014, 5, 237. [Google Scholar] [CrossRef]
- Schrimpe-Rutledge, A.C.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A. Untargeted metabolomics strategies—challenges and emerging directions. J. Am. Soc. Mass Spectrom. 2016, 27, 1897–1905. [Google Scholar] [CrossRef]
- Moseley, H.N.; Carreer, W.J.; Mitchell, J.; Flight, R.M. Method and System for Identification of Metabolites. U.S. Patent 15/642,143, 11 January 2018. [Google Scholar]
- Moseley, H.N. Correcting for the effects of natural abundance in stable isotope resolved metabolomics experiments involving ultra-high resolution mass spectrometry. BMC Bioinform. 2010, 11, 139. [Google Scholar] [CrossRef]
- Günther, D.; Horn, I.; Hattendorf, B. Recent trends and developments in laser ablation-ICP-mass spectrometry. Fresen. J. Anal. Chem. 2000, 368, 4–14. [Google Scholar] [CrossRef]
- Ammann, A.A. Inductively coupled plasma mass spectrometry (ICP MS): A versatile tool. J. Mass Spectrom. 2007, 42, 419–427. [Google Scholar] [CrossRef]
- Kim, S.; Kramer, R.W.; Hatcher, P.G. Graphical method for analysis of ultrahigh-resolution broadband mass spectra of natural organic matter, the van Krevelen diagram. Anal. Chem. 2003, 75, 5336–5344. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Ubach, A.; Liu, Y.; Bianchi, T.S.; Tolić, N.; Jansson, C.; Pasa-Tolic, L. Moving beyond the van Krevelen diagram: A new stoichiometric approach for compound classification in organisms. Anal. Chem. 2018, 90, 6152–6160. [Google Scholar] [CrossRef] [PubMed]
- Feunang, Y.D.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E. ClassyFire: Automated chemical classification with a comprehensive, computable taxonomy. J. Cheminformatics 2016, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Brockman, S.A.; Roden, E.V.; Hegeman, A.D. Van Krevelen diagram visualization of high resolution-mass spectrometry metabolomics data with OpenVanKrevelen. Metabolomics 2018, 14, 48. [Google Scholar] [CrossRef] [PubMed]
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; Dennis, E.A.; Glass, C.K.; Merrill Jr, A.H.; Murphy, R.C.; Raetz, C.R.; Russell, D.W. Lmsd: Lipid maps structure database. Nucleic Acids Res. 2006, 35, D527–D532. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E. HMDB 3.0—the human metabolome database in 2013. Nucleic Acids Res. 2012, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Chen, T.; Cao, Y.; Zhang, Y.; Liu, J.; Bao, Y.; Wang, C.; Jia, W.; Zhao, A. Random forest in clinical metabolomics for phenotypic discrimination and biomarker selection. Evid. Based Complement. Altern. Med. 2013, 2013, 298183. [Google Scholar] [CrossRef]
- Wang-Sattler, R.; Yu, Z.; Herder, C.; Messias, A.C.; Floegel, A.; He, Y.; Heim, K.; Campillos, M.; Holzapfel, C.; Thorand, B. Novel biomarkers for pre-diabetes identified by metabolomics. Mol. Syst. Biol. 2012, 8, 615. [Google Scholar] [CrossRef]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random Forest: A Classification and Regression Tool for Compound Classification and QSAR Modeling. J. Chem. Inf. Model. 2003, 43, 1947–1958. [Google Scholar] [CrossRef]
- Polikar, R. Ensemble learning. In Ensemble Machine Learning; Springer: Berlin, Germany, 2012; pp. 1–34. [Google Scholar]
- Foster, J.M.; Moreno, P.; Fabregat, A.; Hermjakob, H.; Steinbeck, C.; Apweiler, R.; Wakelam, M.J.; Vizcaino, J.A. LipidHome: A database of theoretical lipids optimized for high throughput mass spectrometry lipidomics. PLoS ONE 2013, 8, e61951. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Fiehn, O. Metabolomic database annotations via query of elemental compositions: Mass accuracy is insufficient even at less than 1 ppm. BMC Bioinform. 2006, 7, 234. [Google Scholar] [CrossRef] [PubMed]
- Wieser, M.E.; Holden, N.; Coplen, T.B.; Böhlke, J.K.; Berglund, M.; Brand, W.A.; De Bièvre, P.; Gröning, M.; Loss, R.D.; Meija, J. Atomic weights of the elements 2011 (IUPAC Technical Report). Pure Appl. Chem. 2013, 85, 1047–1078. [Google Scholar] [CrossRef]
- Berglund, M.; Wieser, M.E. Isotopic compositions of the elements 2009 (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 397–410. [Google Scholar] [CrossRef]
- Ihaka, R.; Gentleman, R. R: A language for data analysis and graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar]
- Harrison, J. RSelenium: R Bindings for Selenium WebDriver. R Package Version 1.4. 2016. Available online: https://CRAN.R-project.org/package=RSelenium (accessed on 3 February 2020).
- Barber, C.; Huhdanpaa, H.; Qhull. The Geometry Center, University of Minnesota. 1995. Available online: http://www.geom.umn.edu/software/qhull (accessed on 5 March 2019).
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Janitza, S.; Hornung, R. On the overestimation of random forest’s out-of-bag error. PLoS ONE 2018, 13, e0201904. [Google Scholar] [CrossRef]
- Sellers, K.; Fox, M.P.; Michael Bousamra, I.I.; Slone, S.P.; Higashi, R.M.; Miller, D.M.; Wang, Y.; Yan, J.; Yuneva, M.O.; Deshpande, R. Pyruvate carboxylase is critical for non–small-cell lung cancer proliferation. J. Clin. Investig. 2015, 125, 687. [Google Scholar] [CrossRef]
- Ren, J.-G.; Seth, P.; Clish, C.B.; Lorkiewicz, P.K.; Higashi, R.M.; Lane, A.N.; Fan, T.W.M.; Sukhatme, V.P. Knockdown of malic enzyme 2 suppresses lung tumor growth, induces differentiation and impacts PI3K/AKT signaling. Sci. Rep. 2014, 4, 5414. [Google Scholar] [CrossRef]
- Yang, Y.; Fan, T.W.M.; Lane, A.N.; Higashi, R.M. Chloroformate derivatization for tracing the fate of Amino acids in cells and tissues by multiple stable isotope resolved metabolomics (mSIRM). Anal. Chim. Acta 2017, 976, 63–73. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| LMSD + HMDB_non_Lipid Model Performance (Category) | |||||
|---|---|---|---|---|---|
| Category | Precision | Out-of-Bag Accuracy | Number of Entries | True Positives | False Positives |
| Fatty Acyls [FA] | 0.838 | 0.901 | 2031 | 1681 | 324 |
| Glycerolipids [GL] | 0.996 | 0.995 | 532 | 520 | 2 |
| Glycerophospholipids [GP] | 0.995 | 0.996 | 1886 | 1886 | 10 |
| Polyketides [PK] | 0.780 | 0.884 | 1376 | 954 | 269 |
| Prenol Lipids [PR] | 0.989 | 0.970 | 473 | 263 | 3 |
| Saccharolipids [SL] | 1.0 | 0.998 | 102 | 99 | 0 |
| Sphingolipids [SP] | 0.996 | 0.993 | 1404 | 1386 | 6 |
| Sterol Lipids [ST] | 0.935 | 0.972 | 824 | 707 | 49 |
| not_lipid | 0.930 | 0.798 | 7587 | 6830 | 513 |
| LMSD + LMISSD + HMDB_non_Lipid Model Performance (Category) | |||||
|---|---|---|---|---|---|
| Category | Precision | Out-of-Bag Accuracy | Number of Entries | True Positives | False Positives |
| Fatty Acyls [FA] | 0.837 | 0.939 | 2031 | 1659 | 322 |
| Glycerolipids [GL] | 0.995 | 0.993 | 2715 | 2696 | 14 |
| Glycerophospholipids [GP] | 0.979 | 0.979 | 9766 | 9706 | 206 |
| Polyketides [PK] | 0.768 | 0.933 | 1376 | 979 | 295 |
| Prenol Lipids [PR] | 0.985 | 0.983 | 473 | 259 | 4 |
| Saccharolipids [SL] | 1.000 | 0.998 | 102 | 99 | 0 |
| Sphingolipids [SP] | 0.976 | 0.976 | 3089 | 2875 | 72 |
| Sterol Lipids [ST] | 0.935 | 0.983 | 824 | 702 | 49 |
| not_lipid | 0.928 | 0.882 | 7587 | 6845 | 532 |
| LMSD + HMDB_non_lipid Model Performance for Convex Hull (Category) | ||
|---|---|---|
| Category | Predictions | % of Hull Formulas |
| Fatty Acyls [FA] | 475,516 | 0.429 |
| Glycerolipids [GL] | 8205 | 0.007 |
| Glycerophospholipids [GP] | 1,145,418 | 1.033 |
| Polyketides [PK] | 84,333 | 0.076 |
| Prenol Lipids [PR] | 18,684 | 0.016 |
| Saccharolipids [SL] | 6708 | 0.006 |
| Sphingolipids [SP] | 7,494,579 | 6.761 |
| Sterol Lipids [ST] | 18,643 | 0.017 |
| not_lipid | 74,621,680 | 67.31 |
| no category | 29,202,459 | 26.34 |
| LMSD + LMISSD +HMDB_non_lipid Model Performance for Convex Hull (Category) | ||
|---|---|---|
| Category | Predictions | % of Hull Formulas |
| Fatty Acyls [FA] | 393,314 | 0.354 |
| Glycerolipids [GL] | 56,116 | 0.051 |
| Glycerophospholipids [GP] | 1,735,925 | 1.566 |
| Polyketides [PK] | 118,968 | 0.107 |
| Prenol Lipids [PR] | 15,881 | 0.014 |
| Saccharolipids [SL] | 2795 | 0.002 |
| Sphingolipids [SP] | 12,568,226 | 11.34 |
| Sterol Lipids [ST] | 15,670 | 0.014 |
| not_lipid | 73,562,707 | 66.36 |
| no category | 27,808,607 | 25.08 |
| LMSD + HMDB_non_lipid Model Performance for Unshifted Assignments | ||
|---|---|---|
| Category | Predictions | % of Assigned Formulas |
| Fatty Acyls [FA] | 639 | 0.502 |
| Glycerolipids [GL] | 795 | 0.624 |
| Glycerophospholipids [GP] | 8062 | 6.331 |
| Polyketides [PK] | 28 | 0.022 |
| Prenol Lipids [PR] | 1054 | 0.827 |
| Saccharolipids [SL] | 166 | 0.130 |
| Sphingolipids [SP] | 21,586 | 16.952 |
| Sterol Lipids [ST] | 358 | 0.281 |
| not_lipid | 54,389 | 42.71 |
| no category | 40,683 | 31.95 |
| LMSD + HMDB_non_lipid Model Performance for Shifted Assignments | ||
|---|---|---|
| Category | Predictions | % of Assigned Formulas |
| Fatty Acyls [FA] | 258 | 0.1951 |
| Glycerolipids [GL] | 923 | 0.7001 |
| Glycerophospholipids [GP] | 9517 | 7.227 |
| Polyketides [PK] | 37 | 0.0281 |
| Prenol Lipids [PR] | 1160 | 0.8808 |
| Saccharolipids [SL] | 233 | 0.1769 |
| Sphingolipids [SP] | 22,370 | 16.99 |
| Sterol Lipids [ST] | 257 | 0.1952 |
| not_lipid | 51,863 | 39.38 |
| no category | 45,663 | 34.67 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitchell, J.M.; Flight, R.M.; Moseley, H.N.B. Deriving Lipid Classification Based on Molecular Formulas. Metabolites 2020, 10, 122. https://doi.org/10.3390/metabo10030122
Mitchell JM, Flight RM, Moseley HNB. Deriving Lipid Classification Based on Molecular Formulas. Metabolites. 2020; 10(3):122. https://doi.org/10.3390/metabo10030122
Chicago/Turabian StyleMitchell, Joshua M., Robert M. Flight, and Hunter N.B. Moseley. 2020. "Deriving Lipid Classification Based on Molecular Formulas" Metabolites 10, no. 3: 122. https://doi.org/10.3390/metabo10030122
APA StyleMitchell, J. M., Flight, R. M., & Moseley, H. N. B. (2020). Deriving Lipid Classification Based on Molecular Formulas. Metabolites, 10(3), 122. https://doi.org/10.3390/metabo10030122