16HBE Cell Lipid Mediator Responses to Mono and Co-Infections with Respiratory Pathogens

 ,
,  ,
, 
Abstract
1. Introduction
2. Results
2.1. Viability of 16HBE Post Infections
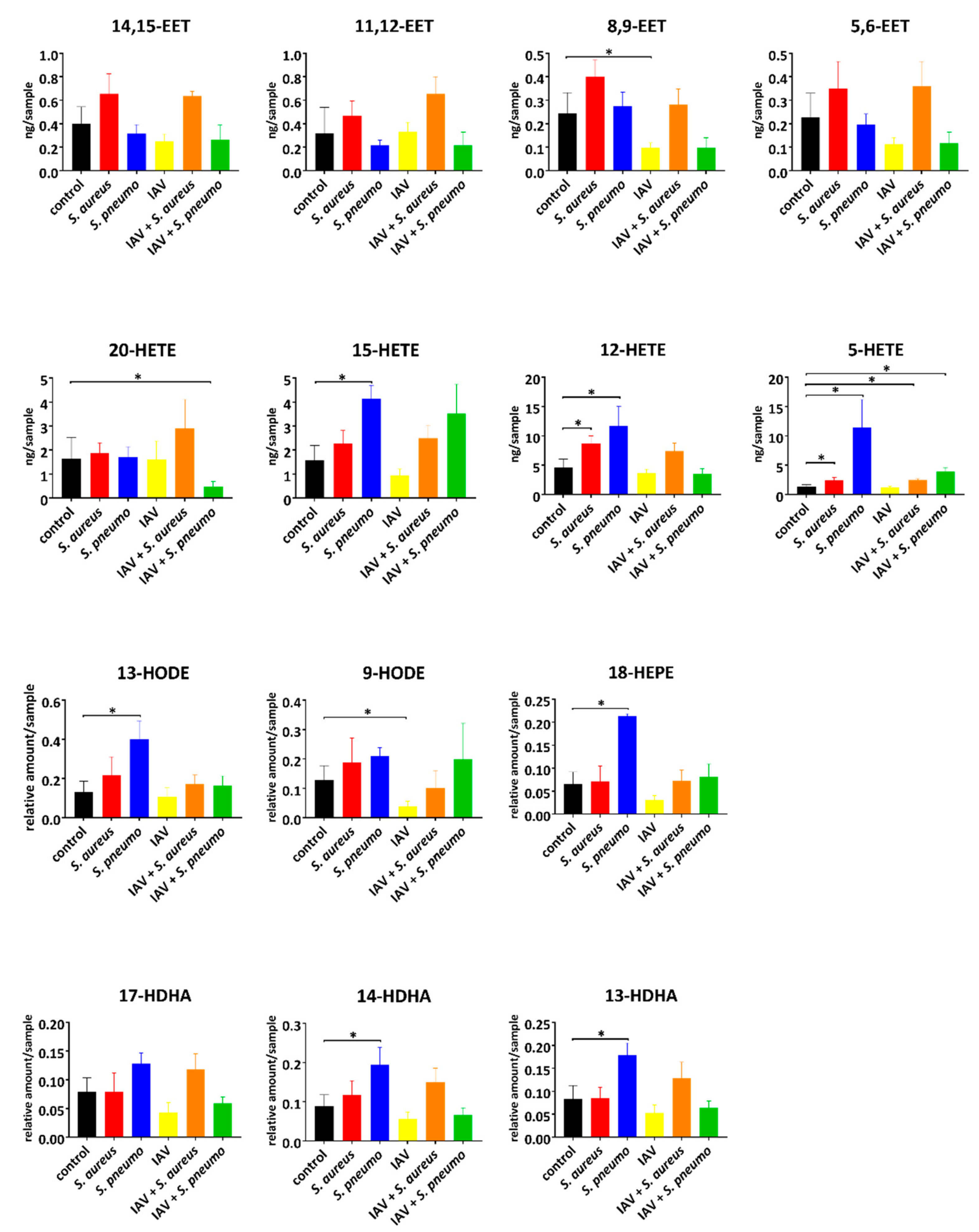
2.2. Intracellular Lipid Mediator Profile in Response to Bacterial and Viral Single and Co-Infections
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Bacterial and Virus Strains
4.4. Cell Culture Infections Experiments
4.5. Oxylipin Extraction
4.6. HPLC-MS/MS Measurement
4.7. Quantification and Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Roth, G.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef]
- Reiner, R.C.; Blacker, B.F.; Khalil, I.A.; Zimsen, S.R.M.; Albertson, S.B.; Abate, D.; Abdela, J.; Adhikari, T.B.; Aghayan, S.A.; Agrawal, S.; et al. Mortality, morbidity, and hospitalisations due to influenza lower respiratory tract infections, 2017: An analysis for the Global Burden of Disease Study 2017. Lancet Respir. Med. 2019, 7, 69–89. [Google Scholar]
- Aliberti, S.; Kaye, K.S. The Changing Microbiologic Epidemiology of Community-Acquired Pneumonia. Postgrad. Med. 2013, 125, 31–42. [Google Scholar] [CrossRef] [PubMed]
- McCullers, J.A. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat. Rev. Genet. 2014, 12, 252–262. [Google Scholar] [CrossRef]
- Tam, V.; Quehenberger, O.; Oshansky, C.M.; Suen, R.; Armando, A.M.; Treuting, P.M.; Thomas, P.G.; Dennis, E.A.; Aderem, A. Lipidomic profiling of influenza infection identifies mediators that induce and resolve inflammation. Cell 2013, 154, 213–227. [Google Scholar] [CrossRef]
- Duffney, P.F.; Falsetta, M.L.; Rackow, A.R.; Thatcher, T.; Phipps, R.; Sime, P.J. Key roles for lipid mediators in the adaptive immune response. J. Clin. Investig. 2018, 128, 2724–2731. [Google Scholar] [CrossRef]
- García-Sastre, A. Lessons from lipids in the fight against influenza. Cell 2013, 154, 22–23. [Google Scholar] [CrossRef]
- Tam, V. Lipidomic profiling of bioactive lipids by mass spectrometry during microbial infections. Semin. Immunol. 2013, 25, 240–248. [Google Scholar] [CrossRef]
- Arita, M. Mediator lipidomics in acute inflammation and resolution. J. Biochem. 2012, 152, 313–319. [Google Scholar] [CrossRef]
- Masoodi, M.; Eiden, M.; Koulman, A.; Spaner, D.; Volmer, D.A. Comprehensive Lipidomics Analysis of Bioactive Lipids in Complex Regulatory Networks. Anal. Chem. 2010, 82, 8176–8185. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, M.J. Arachidonic acid metabolism in airway epithelial cells. Annu. Rev. Physiol. 1992, 54, 303–329. [Google Scholar] [CrossRef] [PubMed]
- Peters-Golden, M.; Gleason, M.M.; Togias, A. Cysteinyl leukotrienes: Multi-functional mediators in allergic rhinitis. Clin. Exp. Allergy 2006, 36, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Obinata, H.; Izumi, T. G2A as a receptor for oxidized free fatty acids. Prostaglandins Other Lipid Mediat. 2009, 89, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Schwab, J.M.; Chiang, N.; Arita, M.; Serhan, C.N. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 2007, 447, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Kronke, G.; Katzenbeisser, J.; Uderhardt, S.; Zaiss, M.M.; Scholtysek, C.; Schabbauer, G.; Zarbock, A.; Koenders, M.I.; Axmann, R.; Zwerina, J.; et al. 12/15-lipoxygenase counteracts inflammation and tissue damage in arthritis. J. Immunol. 2009, 183, 3383–3389. [Google Scholar] [CrossRef]
- Thomson, S.J.; Askari, A.; Bishop-Bailey, D. Anti-Inflammatory Effects of Epoxyeicosatrienoic Acids. Int. J. Vasc. Med. 2012, 2012, 605101. [Google Scholar] [CrossRef]
- Sanak, M. Eicosanoid Mediators in the Airway Inflammation of Asthmatic Patients: What is New? Allergy, Asthma Immunol. Res. 2016, 8, 481–490. [Google Scholar] [CrossRef]
- Barnig, C.; Bezema, T.; Calder, P.C.; Charloux, A.; Frossard, N.; Garssen, J.; Haworth, O.; Dilevskaya, K.; Levi-Schaffer, F.; Lonsdorfer, E.; et al. Activation of Resolution Pathways to Prevent and Fight Chronic Inflammation: Lessons From Asthma and Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 1699. [Google Scholar] [CrossRef]
- Chiang, N.; Fredman, G.; Bäckhed, F.; Oh, S.F.; Vickery, T.; Schmidt, B.A.; Serhan, C.N. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 2012, 484, 524–528. [Google Scholar] [CrossRef]
- Blaho, V.A.; Buczynski, M.W.; Brown, C.; A Dennis, E. Lipidomic Analysis of Dynamic Eicosanoid Responses during the Induction and Resolution of Lyme Arthritis*. J. Boil. Chem. 2009, 284, 21599–21612. [Google Scholar] [CrossRef] [PubMed]
- Morello, E.; Pérez-Berezo, T.; Boisseau, C.; Baranek, T.; Guillon, A.; Bréa, D.; Lanotte, P.; Carpena, X.; Pietrancosta, N.; Hervé, V.; et al. Pseudomonas aeruginosa Lipoxygenase LoxA Contributes to Lung Infection by Altering the Host Immune Lipid Signaling. Front. Microbiol. 2019, 10, 1826. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Kuba, K.; Ichikawa, A.; Nakayama, M.; Katahira, J.; Iwamoto, R.; Watanebe, T.; Sakabe, S.; Daidoji, T.; Nakamura, S.; et al. The Lipid Mediator Protectin D1 Inhibits Influenza Virus Replication and Improves Severe Influenza. Cell 2013, 153, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.; Methling, K.; Rothe, M.; Lalk, M.; KoInfekt Study Group. Eicosanoid Profile of Influenza A Virus Infected Pigs. Metabolites 2019, 9, 130. [Google Scholar] [CrossRef] [PubMed]
- Salina, A.C.; Souza, T.P.; Serezani, C.; Medeiros, A.I. Efferocytosis-induced prostaglandin E2 production impairs alveolar macrophage effector functions during Streptococcus pneumoniae infection. Innate Immun. 2016, 23, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Krause, J.; Geginat, G.; Tammer, I. Prostaglandin E2 from Candida albicans Stimulates the Growth of Staphylococcus aureus in Mixed Biofilms. PLoS ONE 2015, 10, e0135404. [Google Scholar] [CrossRef]
- Sheppe, A.E.F.; Kummari, E.; Walker, A.; Richards, A.; Hui, W.W.; Lee, J.H.; Mangum, L.; Borazjani, A.; Ross, M.K.; Edelmann, M.J. PGE2 Augments Inflammasome Activation and M1 Polarization in Macrophages Infected With Salmonella Typhimurium and Yersinia enterocolitica. Front. Microbiol. 2018, 9, 2447. [Google Scholar] [CrossRef]
- Ikeh, M.A.C.; Fidel, P.L.; Noverr, M.C. Identification of Specific Components of the Eicosanoid Biosynthetic and Signaling Pathway Involved in Pathological Inflammation during Intra-abdominal Infection with Candida albicans and Staphylococcus aureus. Infect. Immun. 2018, 86, e00144-18. [Google Scholar] [CrossRef]
- Shambat, S.M.; Chen, P.; Hoang, A.T.N.; Bergsten, H.; Vandenesch, F.; Siemens, N.; Lina, G.; Monk, I.R.; Foster, T.J.; Arakere, G.; et al. Modelling staphylococcal pneumonia in a human 3D lung tissue model system delineates toxin-mediated pathology. Dis. Model. Mech. 2015, 8, 1413–1425. [Google Scholar] [CrossRef]
- Marks, L.; Parameswaran, G.I.; Hakansson, A.P. Pneumococcal Interactions with Epithelial Cells Are Crucial for Optimal Biofilm Formation and Colonization In Vitro and In Vivo. Infect. Immun. 2012, 80, 2744–2760. [Google Scholar] [CrossRef]
- Blom, R.A.M.; Erni, S.T.; Krempaská, K.; Schaerer, O.; Van Dijk, R.M.; Amacker, M.; Moser, C.; Hall, S.R.R.; Von Garnier, C.; Blank, F. A Triple Co-Culture Model of the Human Respiratory Tract to Study Immune-Modulatory Effects of Liposomes and Virosomes. PLoS ONE 2016, 11, e0163539. [Google Scholar] [CrossRef] [PubMed]
- Shirey, K.A.; Perkins, D.J.; Lai, W.; Zhang, W.; Fernando, L.R.; Gusovsky, F.; Blanco, J.C.G.; Vogel, S.; Arditi, M.; Kagan, J. Influenza "Trains" the Host for Enhanced Susceptibility to Secondary Bacterial Infection. mBio 2019, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, M.; Standiford, T.J. Postinfluenza Bacterial Pneumonia: Host Defenses Gone Awry. J. Interf. Cytokine Res. 2010, 30, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Obinata, H.; Hattori, T.; Nakane, S.; Tatei, K.; Izumi, T. Identification of 9-Hydroxyoctadecadienoic Acid and Other Oxidized Free Fatty Acids as Ligands of the G Protein-coupled Receptor G2A. J. Boil. Chem. 2005, 280, 40676–40683. [Google Scholar] [CrossRef] [PubMed]
- Bittleman, D.B.; Casale, T.B. 5-Hydroxyeicosatetraenoic acid (HETE)-induced neutrophil transcellular migration is dependent upon enantiomeric structure. Am. J. Respir. Cell Mol. Boil. 1995, 12, 260–267. [Google Scholar] [CrossRef]
- O’Flaherty, J.T.; Thomas, M.J.; Lees, C.J.; McCall, C.E. Neutrophil-aggregating activity of monohydroxyeicosatetraenoic acids. Am. J. Pathol. 1981, 104, 55–62. [Google Scholar]
- Luo, M.; Lee, S.; Brock, T.G. Leukotriene synthesis by epithelial cells. Histol. Histopathol. 2003, 18, 587–595. [Google Scholar]
- Neels, J. A role for 5-lipoxygenase products in obesity-associated inflammation and insulin resistance. Adipocyte 2013, 2, 262–265. [Google Scholar] [CrossRef]
- Joshi, Y.B.; Praticò, D. The 5-lipoxygenase pathway: Oxidative and inflammatory contributions to the Alzheimer’s disease phenotype. Front. Cell. Neurosci. 2015, 8, 8. [Google Scholar] [CrossRef]
- Khan, R.; Spagnoli, V.; Tardif, J.-C.; L’Allier, P.L. Novel anti-inflammatory therapies for the treatment of atherosclerosis. Atherosclerosis 2015, 240, 497–509. [Google Scholar] [CrossRef]
- Clementsen, P.F.; Bisgaard, H.; Pedersen, M.; Permin, H.; Struve-Christensen, E.; Milman, N.; Nüchel-Petersen, B.; Norn, S. Staphylococcus aureus and influenza A virus stimulate human bronchoalveolar cells to release histamine and leukotrienes. Inflamm. Res. 1989, 27, 107–109. [Google Scholar] [CrossRef] [PubMed]
- De Vries, H. Eicosanoid production by rat cerebral endothelial cells: Stimulation by lipopolysaccharide, interleukin-1 and interleukin-6. J. Neuroimmunol. 1995, 59, 1–8. [Google Scholar] [CrossRef]
- Cauley, L.S.; Vella, A.T. Why is coinfection with influenza virus and bacteria so difficult to control? Discov. Med. 2015, 19, 33–40. [Google Scholar] [PubMed]
- Peltola, V.T.; Murti, K.G.; McCullers, J.A. Influenza virus neuraminidase contributes to secondary bacterial pneumonia. J. Infect. Dis. 2005, 192, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Han, S.W.; Inoue, H.; Flowers, L.C.; Sidell, N. Control of COX-2 gene expression through peroxisome proliferator-activated receptor gamma in human cervical cancer cells. Clin. Cancer Res. 2003, 9, 4627–4635. [Google Scholar] [PubMed]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef]
- Villapol, S. Roles of Peroxisome Proliferator-Activated Receptor Gamma on Brain and Peripheral Inflammation. Cell. Mol. Neurobiol. 2017, 38, 121–132. [Google Scholar] [CrossRef]
- Solleti, S.K.; Simon, D.M.; Srisuma, S.; Arikan, M.C.; Bhattacharya, S.; Rangasamy, T.; Bijli, K.M.; Rahman, A.; Crossno, J.T., Jr.; Shapiro, S.D.; et al. Airway epithelial cell ppargamma modulates cigarette smoke-induced chemokine expression and emphysema susceptibility in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L293–L304. [Google Scholar] [CrossRef]
- Seki, T.; Ishimoto, T.; Sakurai, T.; Yasuda, Y.; Taniguchi, K.; Doi, M.; Sato, M.; Roman, R.J.; Miyata, N. Increased excretion of urinary 20-HETE in rats with cyclosporine-induced nephrotoxicity. J. Pharmacol. Sci. 2005, 97, 132–137. [Google Scholar] [CrossRef]
- Shambat, S.M.; Haggar, A.; Vandenesch, F.; Lina, G.; Van Wamel, W.J.B.; Arakere, G.; Svensson, M.; Norrby-Teglund, A. Levels of Alpha-Toxin Correlate with Distinct Phenotypic Response Profiles of Blood Mononuclear Cells and with agr Background of Community-Associated Staphylococcus aureus Isolates. PLoS ONE 2014, 9, e106107. [Google Scholar]
- Mairpady Shambat, S.; Siemens, N.; Monk, I.R.; Mohan, D.B.; Mukundan, S.; Krishnan, K.C.; Prabhakara, S.; Snall, J.; Kearns, A.; Vandenesch, F.; et al. A point mutation in agrc determines cytotoxic or colonizing properties associated with phenotypic variants of st22 mrsa strains. Sci. Rep. 2016, 6, 31360. [Google Scholar] [CrossRef] [PubMed]
- Eisfeld, A.J.; Neumann, G.; Kawaoka, Y. Influenza a virus isolation, culture and identification. Nat. Protoc. 2014, 9, 2663–2681. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Precursor | Key Enzymes | Measured Lipid Mediators |
| arachidonic acid | 5-LOX, 12-LOX, 15-LOX | 5-HETE; 12-HETE; 15-HETE |
| CYP ω-hydroxylase | 20-HETE | |
| CYP epoxygenases | 5,6-EET; 8,9-EET; 11,12-EET; 14,15-EET | |
| linoleic acid | 15-LOX | 9-HODE, 13-HODE |
| docosahexaenoic acid | 12-LOX; 15-LOX | 13-HDHA, 14-HDHA, 17-HDHA |
| eicosapentaenoic acid | COX-2; CYP enzymes | 18-HEPE |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schultz, D.; Surabhi, S.; Stelling, N.; Rothe, M.; KoInfekt Study Group; Methling, K.; Hammerschmidt, S.; Siemens, N.; Lalk, M. 16HBE Cell Lipid Mediator Responses to Mono and Co-Infections with Respiratory Pathogens. Metabolites 2020, 10, 113. https://doi.org/10.3390/metabo10030113
Schultz D, Surabhi S, Stelling N, Rothe M, KoInfekt Study Group, Methling K, Hammerschmidt S, Siemens N, Lalk M. 16HBE Cell Lipid Mediator Responses to Mono and Co-Infections with Respiratory Pathogens. Metabolites. 2020; 10(3):113. https://doi.org/10.3390/metabo10030113
Chicago/Turabian StyleSchultz, Daniel, Surabhi Surabhi, Nicolas Stelling, Michael Rothe, KoInfekt Study Group, Karen Methling, Sven Hammerschmidt, Nikolai Siemens, and Michael Lalk. 2020. "16HBE Cell Lipid Mediator Responses to Mono and Co-Infections with Respiratory Pathogens" Metabolites 10, no. 3: 113. https://doi.org/10.3390/metabo10030113
APA StyleSchultz, D., Surabhi, S., Stelling, N., Rothe, M., KoInfekt Study Group, Methling, K., Hammerschmidt, S., Siemens, N., & Lalk, M. (2020). 16HBE Cell Lipid Mediator Responses to Mono and Co-Infections with Respiratory Pathogens. Metabolites, 10(3), 113. https://doi.org/10.3390/metabo10030113