Data-Independent Acquisition for the Quantification and Identification of Metabolites in Plasma

 and
and
Abstract
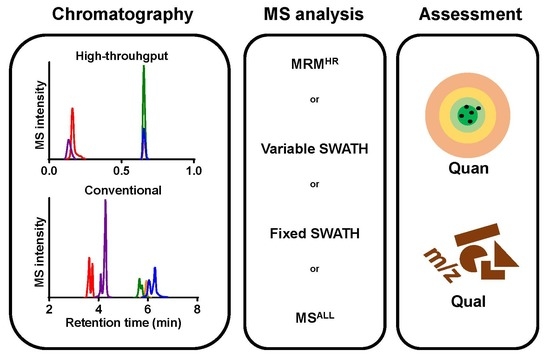
1. Introduction
2. Results and Discussion
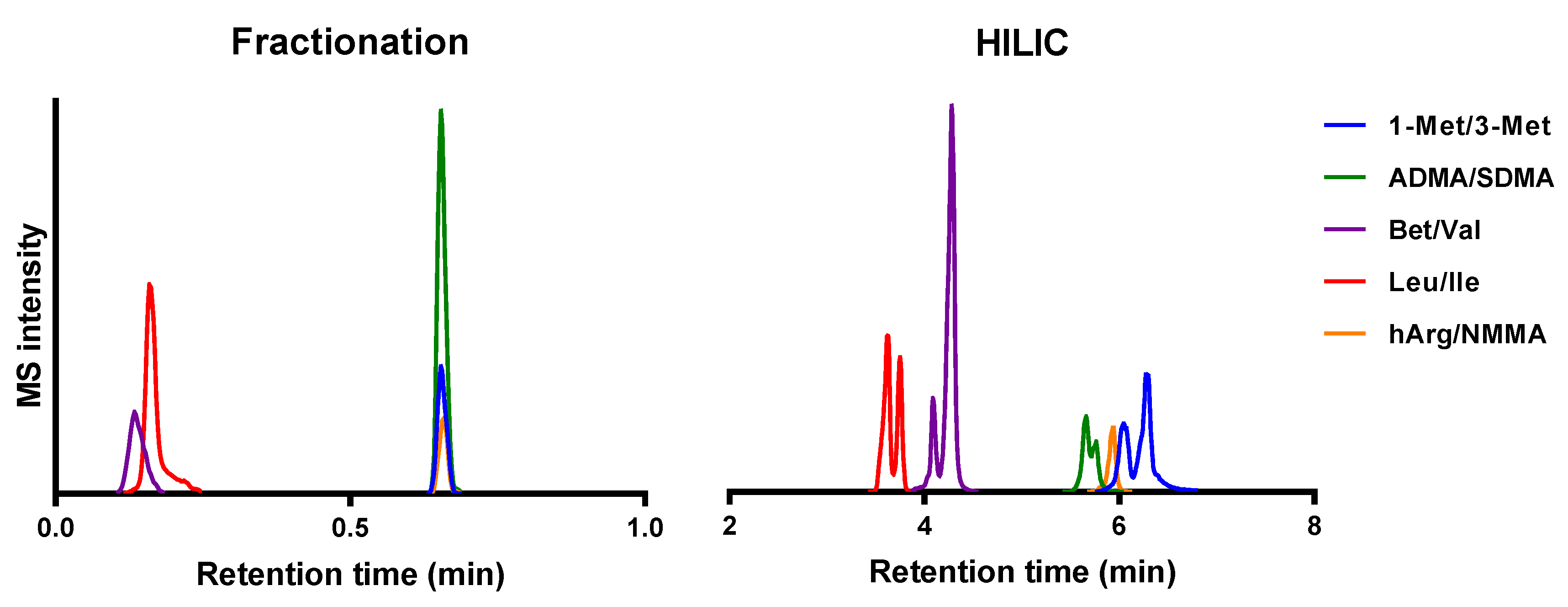
2.1. Method Development
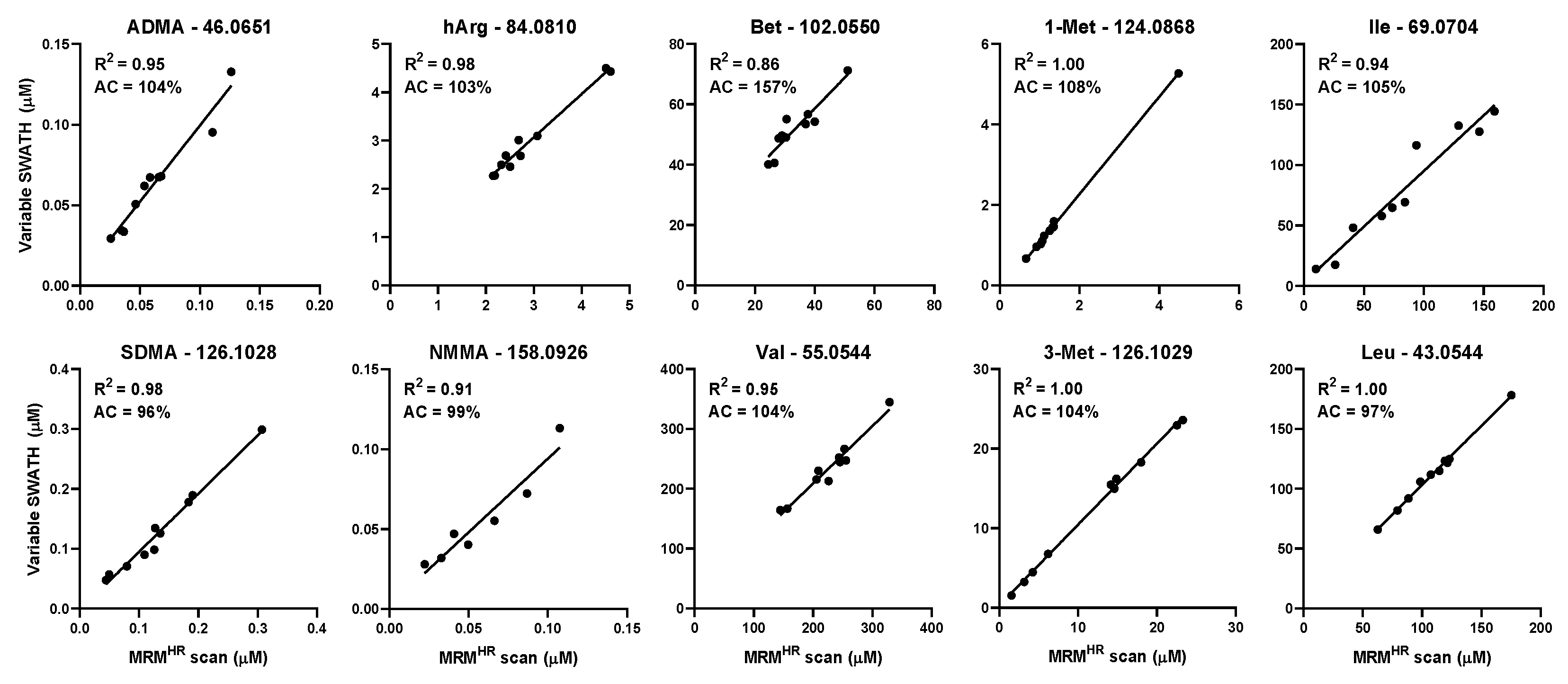
2.2. Quantitation of Structural Isomers
2.3. Identification of Metabolites
3. Conclusions
4. Materials and Methods
4.1. Chemicals
4.2. Standard and Internal Standards
4.3. Sample Preparation
4.4. LC-MS
4.5. Diagnostic Product Ions and Quantification of Structural Isomers
4.6. Identification of Metabolites
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, Z.; Klipfell, E.; Bennett, B.J.; A Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nat. Cell Biol. 2011, 472, 57–63. [Google Scholar] [CrossRef]
- Mayo Clinic. Rochester 2018 Interpretive Handbook. 2018. Available online: https://www.mayocliniclabs.com/test-catalog/pod/MayoTestCatalog-Rochester--SortedByTestName-duplex-interpretive.pdf (accessed on 2 November 2020).
- Trivedi, D.K.; Hollywood, K.A.; Goodacre, R. Metabolomics for the masses: The future of metabolomics in a personalized world. Eur. J. Mol. Clin. Med. 2017, 3, 294–305. [Google Scholar] [CrossRef]
- Balashova, E.E.; Maslov, D.L.; Lokhov, P.G. A metabolomics approach to pharmacotherapy personalization. J. Pers. Med. 2018, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.F.; Zhou, B.; Ressom, H.W. Metabolite identification and quantitation in LC-MS/MS-based metabolomics. TrAC Trends Anal. Chem. 2012, 32, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Innovation: Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.D.; Souza, A.L.; Gerszten, R.E.; Clish, C.B. Targeted metabolomics. Curr. Protoc. Mol. Biol. 2012, 98, 3021–30224. [Google Scholar] [CrossRef] [PubMed]
- Fenaille, F.; Saint-Hilaire, P.B.; Rousseau, K.; Junot, C. Data acquisition workflows in liquid chromatography coupled to high resolution mass spectrometry-based metabolomics: Where do we stand? J. Chromatogr. A 2017, 1526, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yin, Y.; Zhu, Z.-J. Advancing untargeted metabolomics using data-independent acquisition mass spectrometry technology. Anal. Bioanal. Chem. 2019, 411, 4349–4357. [Google Scholar] [CrossRef]
- Stahl, D.C.; Swiderek, K.M.; Davis, M.T.; Lee, T.D. Data-controlled automation of liquid chromatography/tandem mass spectrometry analysis of peptide mixtures. J. Am. Soc. Mass Spectrom. 1996, 7, 532–540. [Google Scholar] [CrossRef]
- Johnson, D.; Boyes, B.; Fields, T.; Kopkin, R.; Orlando, R. Optimization of data-dependent acquisition parameters for coupling high-speed separations with LC-MS/MS for protein identifications. J. Biomol. Tech. 2013, 24, 62–72. [Google Scholar] [CrossRef]
- Zhang, Y.; Bilbao, A.; Bruderer, T.; Luban, J.; Strambio-De-Castillia, C.; Lisacek, F.; Hopfgartner, G.; Varesio, E. The use of variable Q1 isolation windows improves selectivity in LC–SWATH–MS acquisition. J. Proteome Res. 2015, 14, 4359–4371. [Google Scholar] [CrossRef] [PubMed]
- Bonner, R.; Hopfgartner, G. SWATH acquisition mode for drug metabolism and metabolomics investigations. Bioanalysis 2016, 8, 1735–1750. [Google Scholar] [CrossRef] [PubMed]
- Plumb, R.S.; Johnson, K.A.; Rainville, P.; Smith, B.W.; Wilson, I.D.; Castro-Perez, J.M.; Nicholson, J.K. UPLC/MSE; a new approach for generating molecular fragment information for biomarker structure elucidation Robert. Rapid Commun. Mass Spectrom. 2008, 20, 4129–4138. [Google Scholar]
- Navarro-Reig, M.; Jaumot, J.; Piña, B.; Moyano, E.; Galceran, M.T.; Tauler, R. Metabolomic analysis of the effects of cadmium and copper treatment in Oryza sativa L. using untargeted liquid chromatography coupled to high resolution mass spectrometry and all-ion fragmentation. Metallomics 2017, 9, 660–675. [Google Scholar] [CrossRef]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 2012, 11, 1–17. [Google Scholar] [CrossRef]
- Dubbelman, A.-C.; Cuyckens, F.; Dillen, L.; Gross, G.; Vreeken, R.J.; Hankemeier, T. Mass spectrometric recommendations for Quan/Qual analysis using liquid-chromatography coupled to quadrupole time-of-flight mass spectrometry. Anal. Chim. Acta 2018, 1020, 62–75. [Google Scholar] [CrossRef]
- Naz, S.; Gallart-Ayala, H.; Reinke, S.N.; Mathon, C.; Blankley, R.; Chaleckis, R.; Wheelock, C.E. Development of a liquid chromatography–high resolution mass spectrometry metabolomics method with high specificity for metabolite identification using all Ion fragmentation acquisition. Anal. Chem. 2017, 89, 7933–7942. [Google Scholar] [CrossRef]
- Peris-Díaz, M.D.; Sweeney, S.R.; Rodak, O.; Sentandreu, E.; Tiziani, S. R-metabolist 2: A flexible tool for metabolite annotation from high-resolution data-independent acquisition mass spectrometry analysis. Metabolites 2019, 9, 187. [Google Scholar] [CrossRef]
- The Human Serum Metabolome. Available online: https://serummetabolome.ca/ (accessed on 2 November 2020).
- Yang, X. New features of the 2017 NIST tandem mass spectral library. In Proceedings of the Biomolecular Measurement Division Seminar, Indianapolis, IN, USA, 27 June 2017. [Google Scholar]
- mzCloud–Statistics. Available online: https://www.mzcloud.org/Stats. (accessed on 2 November 2020).
- Yan, Z.; Maher, N.; Torres, R.; Cotto, C.; Hastings, B. Isobaric metabolite interferences and the requirement for close examination of raw data in addition to stringent chromatographic separations in liquid chromtaography/tandem mass spectrometric analysis of drugs in biological matrix. Rapid Commun. Mass Spectrom. 2008, 22, 2021–2028. [Google Scholar] [CrossRef]
- Martens-Lobenhoffer, J.; Surdacki, A.; Bode-Böger, S.M. Fast and precise quantification of l-Homoarginine in human plasma by HILIC-Isotope dilution-MS–MS. Chromatographia 2013, 76, 1755–1759. [Google Scholar] [CrossRef]
- Armirotti, A.; Millo, E.; Damonte, G. How to discriminate between leucine and isoleucine by low energy ESI-TRAP MSn. J. Am. Soc. Mass Spectrom. 2007, 18, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Martens-Lobenhoffer, J.; Bode-Böger, S.M. Chromatographic-mass spectrometric methods for the quantification of l-arginine and its methylated metabolites in biological fluids. J. Chromatogr. B 2007, 851, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Van Der Laan, T.; Dubbelman, A.-C.; Duisters, K.; Kindt, A.; Harms, A.C.; Hankemeier, T. High-throughput fractionation coupled to mass spectrometry for improved quantitation in metabolomics. Anal. Chem. 2020, 92, 14330–14338. [Google Scholar] [CrossRef] [PubMed]
- Poole, C.F. The Essence of Chromatography; Elsevier: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Bilbao, A.; Varesio, E.; Luban, J.; Strambio-De-Castillia, C.; Hopfgartner, G.; Müller, M.; Lisacek, F. Processing strategies and software solutions for data-independent acquisition in mass spectrometry. Proteomics 2015, 15, 964–980. [Google Scholar] [CrossRef] [PubMed]
- Venable, J.D.; Dong, M.Q.; Wohlschlegel, J.; Dillin, A.; Yates, J.R. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods 2004, 1, 39–45. [Google Scholar] [CrossRef]
- Collins, B.C.; Hunter, C.L.; Liu, Y.; Schilling, B.; Rosenberger, G.; Bader, S.L.; Chan, D.W.; Gibson, B.W.; Gingras, A.-C.; Held, J.M.; et al. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Roemmelt, A.T.; Steuer, A.E.; Poetzsch, M.; Kraemer, T. Liquid chromatography, in combination with a quadrupole time-of-flight instrument (LC QTOF), with sequential window acquisition of all theoretical fragment-ion spectra (SWATH) acquisition: Systematic studies on its use for screenings in clinical and foren. Anal. Chem. 2014, 86, 11742–11749. [Google Scholar] [CrossRef]
- Scheidweiler, K.B.; Jarvis, M.J.Y.; Huestis, M.A. Nontargeted SWATH acquisition for identifying 47 synthetic cannabinoid metabolites in human urine by liquid chromatography-high-resolution tandem mass spectrometry. Anal. Bioanal. Chem. 2015, 407, 883–897. [Google Scholar] [CrossRef]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.T.; Ikeda, K.; Kanazawa, M.; Vander Gheynst, J.S.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, Y.; Subramanian, R. Comparison of information-dependent acquisition, SWATH, and MS All techniques in metabolite identification study employing ultrahigh-performance liquid chromatography-quadrupole time-of-flight mass spectrometry. Anal. Chem. 2014, 86, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- HMDB. Available online: http://www.hmdb.ca/ (accessed on 2 November 2020).
- SCIEX. A Compendium of Related Research Applications: X500R QTOF. 2016. Available online: https://https://sciex.com/Documents/tech%20notes/X500R_QTOF_Compendium.pdf. (accessed on 2 November 2020).
- SCIEX. SCIEX OS Software User Guide. 2018. Available online: https://sciex.com/Documents/manuals/sciex-os-software-user-guide-en.pdf. (accessed on 2 November 2020).
- Stein, S.E.; Scott, D.R. Optimization and testing of mass spectral library search algorithms for compound identification. J. Am. Soc. Mass Spectrom. 1994, 5, 859–866. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | MRM | MRMHR/PRM | DDA | DIA | |
|---|---|---|---|---|---|
| SWATH | MSALL/MSE/AIF | ||||
| Q1 size | Narrow | Narrow | Narrow | Medium | Wide |
| High/low resolution | Low | High | High | High | High |
| Precursor selection | User-defined | User-defined | Based on parent ion intensity * | No selection | No selection |
| Targeted/untargeted | Targeted | Targeted | Untargeted | Untargeted | Untargeted |
| Analyte | Variable SWATH | Fixed SWATH (%) | MSALL (%) | |||
|---|---|---|---|---|---|---|
| Fractionation | HILIC | Fractionation | HILIC | Fractionation | HILIC | |
| ADMA | 88 | 96 | 0 | 102 | 0 | 0 |
| SDMA | 107 | 101 | 96 | 110 | 106 | 0 |
| hArg | 109 | 99 | 389 | 102 | 271 | 0 |
| NMMA | 739 | 111 | 6383 | 226 | 1944 | 0 |
| Bet | 106 | 102 | 103 | 134 | 136 | 124 |
| Val | 98 | 91 | 93 | 91 | 99 | 101 |
| 1-Met | 157 | 113 | 0 | 109 | 0 | 102 |
| 3-Met | 97 | 99 | 97 | 89 | 193 | 89 |
| Ile | 106 | 112 | 113 | 99 | 129 | 90 |
| Leu | 144 | 110 | 148 | 117 | 171 | 168 |
| Analyte | Variable SWATH | Fixed SWATH | MSALL | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Frac | HILIC | Frac | HILIC | Frac | HILIC | |||||||
| Score (%) | Hits (#) | Score (%) | Score (%) | Score (%) | Hits (#) | Score (%) | Hits (#) | Score (%) | Hits (#) | Score (%) | Hits (#) | |
| Proline | 100 | 5/5 | 100 | 5/5 | 100 | 5/5 | 100 | 5/5 | 100 | 5/5 | 100 | 5/5 |
| Arginine | 100 | 5/5 | 100 | 5/5 | 0 | 0/5 | 99 | 5/5 | 100 | 5/5 | 99 | 5/5 |
| Acetyl-carnitine | 99 | 5/5 | 99 | 5/5 | 98 | 5/5 | 99 | 5/5 | 96 | 5/5 | 98 | 5/5 |
| Creatinine | 100 | 5/5 | 99 | 5/5 | 100 | 5/5 | 99 | 5/5 | 95 | 5/5 | 99 | 5/5 |
| Ornithine | 0 | 0/5 | 99 | 5/5 | 0 | 0/5 | 99 | 5/5 | 40 | 2/5 | 96 | 5/5 |
| Methionine | 76 | 5/5 | 99 | 5/5 | 0 | 0/5 | 99 | 5/5 | 0 | 0/5 | 99 | 5/5 |
| Carnitine | 98 | 5/5 | 98 | 5/5 | 96 | 5/5 | 98 | 5/5 | 78 | 5/5 | 97 | 5/5 |
| Citrulline | 98 | 5/5 | 97 | 5/5 | 99 | 5/5 | 94 | 5/5 | 0 | 0/5 | 50 | 3/5 |
| Tyrosine | 94 | 5/5 | 97 | 5/5 | 94 | 5/5 | 96 | 5/5 | 92 | 5/5 | 98 | 5/5 |
| Phenylalanine | 76 | 5/5 | 96 | 5/5 | 57 | 4/5 | 96 | 5/5 | 41 | 3/5 | 97 | 5/5 |
| Histidine | 98 | 5/5 | 95 | 5/5 | 98 | 5/5 | 95 | 5/5 | 99 | 5/5 | 3 | 1/5 |
| Serine | 0 | 0/5 | 93 | 5/5 | 0 | 0/5 | 97 | 5/5 | 0 | 0/5 | 19 | 3/5 |
| Tryptophan | 63 | 5/5 | 93 | 5/5 | 77 | 5/5 | 91 | 5/5 | 0 | 0/5 | 94 | 5/5 |
| Glutamine | 93 | 5/5 | 93 | 5/5 | 92 | 5/5 | 94 | 5/5 | 97 | 5/5 | 93 | 5/5 |
| Cystine | 88 | 5/5 | 92 | 5/5 | 92 | 5/5 | 91 | 5/5 | 0 | 0/5 | 93 | 5/5 |
| Taurine | 0 | 0/5 | 92 | 5/5 | 0 | 0/5 | 89 | 5/5 | 0 | 0/5 | 0 | 0/5 |
| Lysine | 0 | 0/5 | 91 | 5/5 | 88 | 5/5 | 89 | 5/5 | 0 | 0/5 | 93 | 5/5 |
| Asparagine | 0 | 0/5 | 82 | 5/5 | 0 | 0/5 | 92 | 5/5 | 0 | 0/5 | 0 | 0/5 |
| Glutamic acid | 90 | 5/5 | 62 | 4/5 | 94 | 5/5 | 82 | 5/5 | 97 | 5/5 | 0 | 0/5 |
| γ-butyrobetaine | 54 | 5/5 | 44 | 5/5 | 73 | 5/5 | 60 | 5/5 | 0 | 0/5 | 0 | 0/5 |
| Average | 66 | 91 | 63 | 93 | 47 | 66 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Laan, T.; Boom, I.; Maliepaard, J.; Dubbelman, A.-C.; Harms, A.C.; Hankemeier, T. Data-Independent Acquisition for the Quantification and Identification of Metabolites in Plasma. Metabolites 2020, 10, 514. https://doi.org/10.3390/metabo10120514
van der Laan T, Boom I, Maliepaard J, Dubbelman A-C, Harms AC, Hankemeier T. Data-Independent Acquisition for the Quantification and Identification of Metabolites in Plasma. Metabolites. 2020; 10(12):514. https://doi.org/10.3390/metabo10120514
Chicago/Turabian Stylevan der Laan, Tom, Isabelle Boom, Joshua Maliepaard, Anne-Charlotte Dubbelman, Amy C. Harms, and Thomas Hankemeier. 2020. "Data-Independent Acquisition for the Quantification and Identification of Metabolites in Plasma" Metabolites 10, no. 12: 514. https://doi.org/10.3390/metabo10120514
APA Stylevan der Laan, T., Boom, I., Maliepaard, J., Dubbelman, A.-C., Harms, A. C., & Hankemeier, T. (2020). Data-Independent Acquisition for the Quantification and Identification of Metabolites in Plasma. Metabolites, 10(12), 514. https://doi.org/10.3390/metabo10120514