The Ketogenic Diet Increases In Vivo Glutathione Levels in Patients with Epilepsy


 , , and
, , and
Abstract
1. Introduction
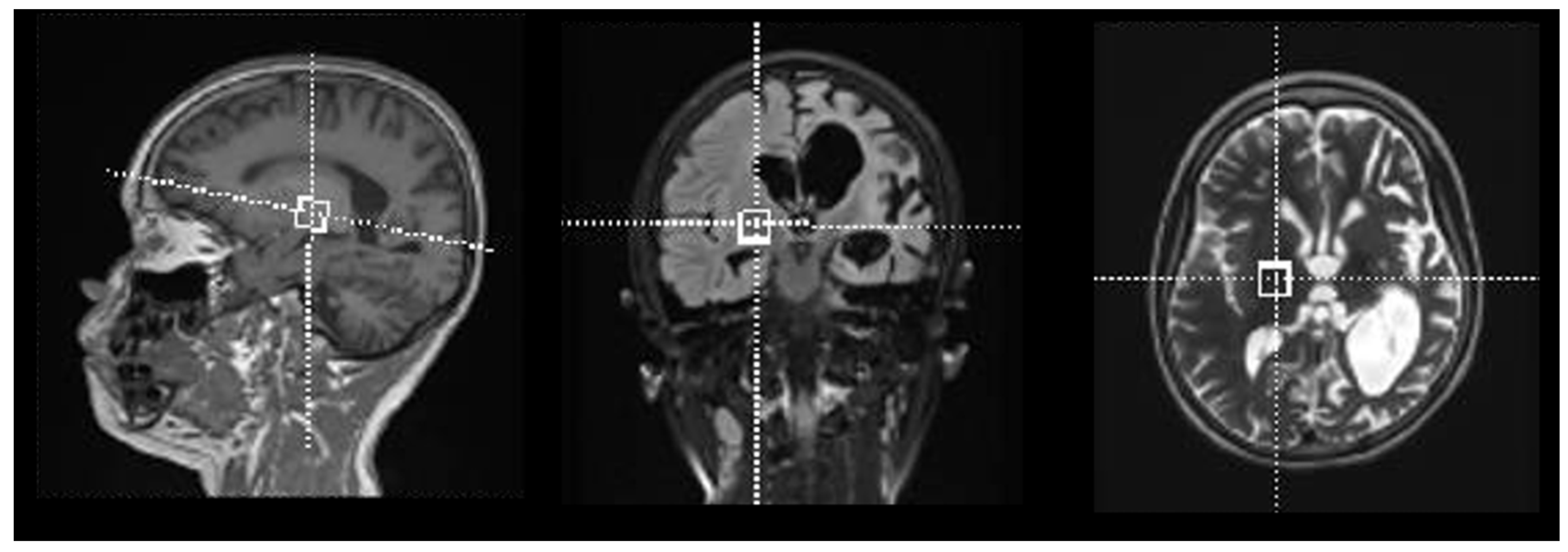
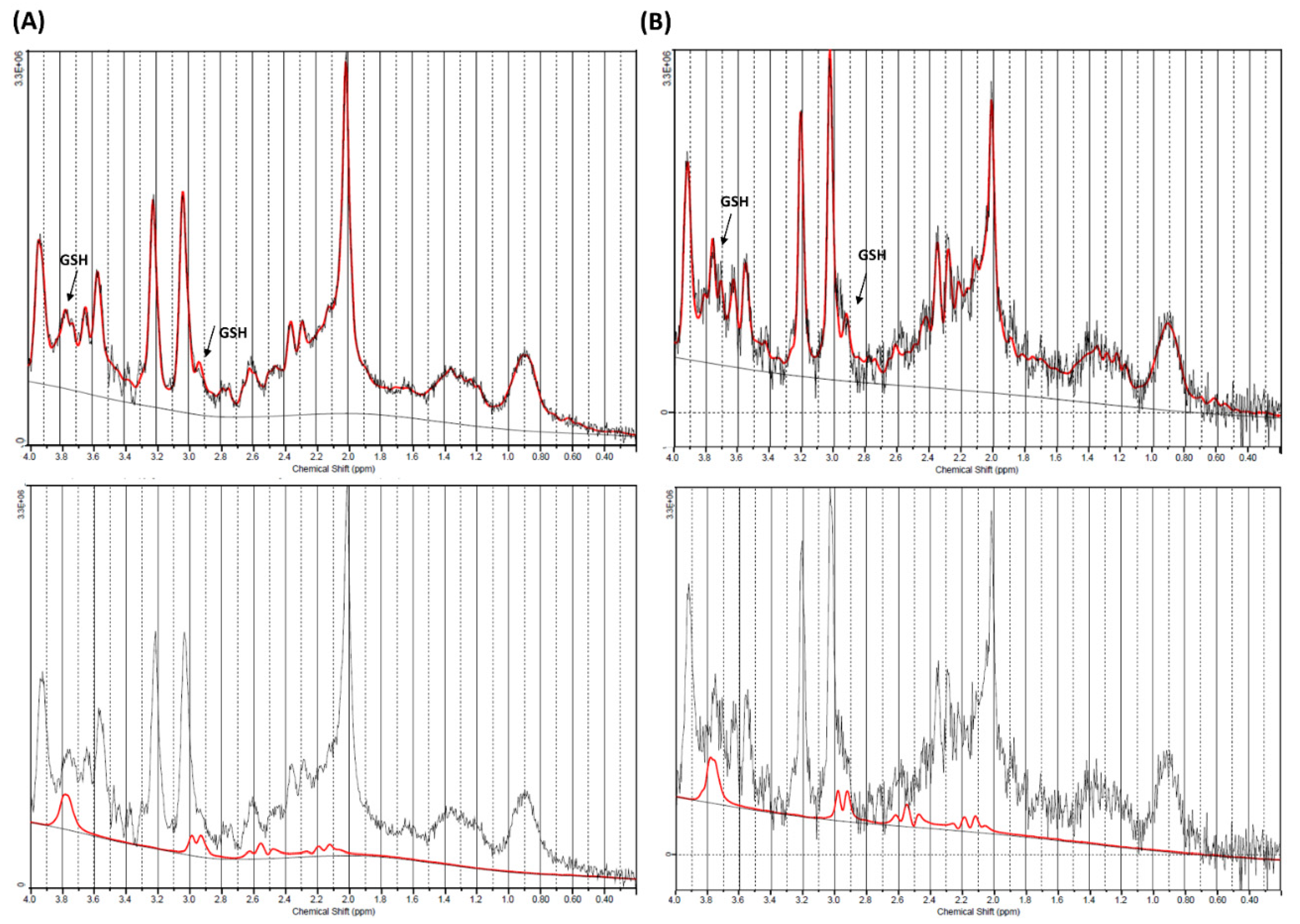
2. Results
3. Discussion
4. Materials and Methods
4.1. Standard Protocol Approvals, Registrations, and Patient Consents
4.2. Subjects
4.3. Data Acquisition
4.4. Data Processing
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Verrotti, A.; Iapadre, G.; Pisano, S.; Coppola, G. Ketogenic diet and childhood neurological disorders other than epilepsy: An overview. Expert Rev. Neurother. 2017, 17, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, D.R.; Ribeiro, L.C.; Hagenn, M.; Siqueira, I.R.; Araujo, E.; Torres, I.L.; Gottfried, C.; Netto, C.A.; Goncalves, C.A. Ketogenic diet increases glutathione peroxidase activity in rat hippocampus. Neurochem. Res. 2003, 28, 1793–1797. [Google Scholar] [CrossRef] [PubMed]
- Coppola, G.; Veggiotti, P.; Cusmai, R.; Bertoli, S.; Cardinali, S.; Dionisi-Vici, C.; Elia, M.; Lispi, M.L.; Sarnelli, C.; Tagliabue, A.; et al. The ketogenic diet in children, adolescents and young adults with refractory epilepsy: An Italian multicentric experience. Epilepsy Res. 2002, 48, 221–227. [Google Scholar] [CrossRef]
- Milder, J.B.; Liang, L.P.; Patel, M. Acute oxidative stress and systemic Nrf2 activation by the ketogenic diet. Neurobiol. Dis. 2010, 40, 238–244. [Google Scholar] [CrossRef]
- Longo, R.; Peri, C.; Cricri, D.; Coppi, L.; Caruso, D.; Mitro, N.; De Fabiani, E.; Crestani, M. Ketogenic Diet: A New Light Shining on Old but Gold Biochemistry. Nutrients 2019, 11, 2497. [Google Scholar] [CrossRef]
- Suzuki, M.; Kitamura, Y.; Mori, S.; Sato, K.; Dohi, S.; Sato, T.; Matsuura, A.; Hiraide, A. Beta-hydroxybutyrate, a cerebral function improving agent, protects rat brain against ischemic damage caused by permanent and transient focal cerebral ischemia. Jpn. J. Pharmacol. 2002, 89, 36–43. [Google Scholar] [CrossRef]
- Suzuki, M.; Sato, K.; Dohi, S.; Sato, T.; Matsuura, A.; Hiraide, A. Effect of beta-hydroxybutyrate, a cerebral function improving agent, on cerebral hypoxia, anoxia and ischemia in mice and rats. Jpn. J. Pharmacol. 2001, 87, 143–150. [Google Scholar] [CrossRef]
- Maalouf, M.; Sullivan, P.G.; Davis, L.; Kim, D.Y.; Rho, J.M. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 2007, 145, 256–264. [Google Scholar] [CrossRef]
- Sullivan, P.G.; Dube, C.; Dorenbos, K.; Steward, O.; Baram, T.Z. Mitochondrial uncoupling protein-2 protects the immature brain from excitotoxic neuronal death. Ann. Neurol. 2003, 53, 711–717. [Google Scholar] [CrossRef]
- Bough, K.J.; Wetherington, J.; Hassel, B.; Pare, J.F.; Gawryluk, J.W.; Greene, J.G.; Shaw, R.; Smith, Y.; Geiger, J.D.; Dingledine, R.J. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 2006, 60, 223–235. [Google Scholar] [CrossRef]
- Nazarewicz, R.R.; Ziolkowski, W.; Vaccaro, P.S.; Ghafourifar, P. Effect of short-term ketogenic diet on redox status of human blood. Rejuvenation Res. 2007, 10, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Rae, C.D.; Williams, S.R. Glutathione in the human brain: Review of its roles and measurement by magnetic resonance spectroscopy. Anal. Biochem. 2017, 529, 127–143. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.G.; Trabesinger, A.H.; Boesiger, P.; Wieser, H.G. Brain glutathione levels in patients with epilepsy measured by in vivo (1)H-MRS. Neurology 2001, 57, 1422–1427. [Google Scholar] [CrossRef]
- Jarrett, S.G.; Milder, J.B.; Liang, L.P.; Patel, M. The ketogenic diet increases mitochondrial glutathione levels. J. Neurochem. 2008, 106, 1044–1051. [Google Scholar] [CrossRef]
- Choi, I.Y.; Lee, S.P.; Denney, D.R.; Lynch, S.G. Lower levels of glutathione in the brains of secondary progressive multiple sclerosis patients measured by 1H magnetic resonance chemical shift imaging at 3 T. Mult. Scler. J. 2011, 17, 289–296. [Google Scholar] [CrossRef]
- Weiduschat, N.; Mao, X.; Hupf, J.; Armstrong, N.; Kang, G.; Lange, D.J.; Mitsumoto, H.; Shungu, D.C. Motor cortex glutathione deficit in ALS measured in vivo with the J-editing technique. Neurosci. Lett. 2014, 570, 102–107. [Google Scholar] [CrossRef]
- Srinivasan, R.; Ratiney, H.; Hammond-Rosenbluth, K.E.; Pelletier, D.; Nelson, S.J. MR spectroscopic imaging of glutathione in the white and gray matter at 7 T with an application to multiple sclerosis. Magn. Reson. Imaging 2010, 28, 163–170. [Google Scholar] [CrossRef]
- Goncalves, C.A.; Rodrigues, L.; Bobermin, L.D.; Zanotto, C.; Vizuete, A.; Quincozes-Santos, A.; Souza, D.O.; Leite, M.C. Glycolysis-Derived Compounds From Astrocytes That Modulate Synaptic Communication. Front. Neurosci. 2018, 12, 1035. [Google Scholar] [CrossRef]
- Cheng, B.; Yang, X.; An, L.; Gao, B.; Liu, X.; Liu, S. Ketogenic diet protects dopaminergic neurons against 6-OHDA neurotoxicity via up-regulating glutathione in a rat model of Parkinson’s disease. Brain Res. 2009, 1286, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Achanta, L.B.; Rae, C.D. 𝛽-Hydroxybutyrate in the Brain: One Molecule, Multiple Mechanisms. Neurochem. Res. 2017, 42, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Kashiwaya, Y.; Keon, C.A.; Tsuchiya, N.; King, M.T.; Radda, G.K.; Chance, B.; Clarke, K.; Veech, R.L. Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J. 1995, 9, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Agledal, L.; Niere, M.; Ziegler, M. The phosphate makes a difference: Cellular functions of NADP. Redox Rep. Commun. Free Radic. Res. 2010, 15, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Veech, R.L.; Bradshaw, P.C.; Clarke, K.; Curtis, W.; Pawlosky, R.; King, M.T. Ketone bodies mimic the life span extending properties of caloric restriction. IUBMB Life 2017, 69, 305–314. [Google Scholar] [CrossRef]
- Blacker, T.S.; Duchen, M.R. Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free Radic. Biol. Med. 2016, 100, 53–65. [Google Scholar] [CrossRef]
- Norwitz, N.G.; Hu, M.T.; Clarke, K. The Mechanisms by Which the Ketone Body D-beta-Hydroxybutyrate May Improve the Multiple Cellular Pathologies of Parkinson’s Disease. Front. Nutr. 2019, 6, 63. [Google Scholar] [CrossRef]
- Kops, G.J.; Dansen, T.B.; Polderman, P.E.; Saarloos, I.; Wirtz, K.W.; Coffer, P.J.; Huang, T.T.; Bos, J.L.; Medema, R.H.; Burgering, B.M. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 2002, 419, 316–321. [Google Scholar] [CrossRef]
- Ruttkay-Nedecky, B.; Kudr, J.; Nejdl, L.; Maskova, D.; Kizek, R.; Adam, V. G-quadruplexes as sensing probes. Molecules 2013, 18, 14760–14779. [Google Scholar] [CrossRef]
- Schoeler, N.E.; Leu, C.; Balestrini, S.; Mudge, J.M.; Steward, C.A.; Frankish, A.; Leung, M.A.; Mackay, M.; Scheffer, I.; Williams, R.; et al. Genome-wide association study: Exploring the genetic basis for responsiveness to ketogenic dietary therapies for drug-resistant epilepsy. Epilepsia 2018, 59, 1557–1566. [Google Scholar] [CrossRef]
- Cinnella, G.; Vendemiale, G.; Dambrosio, M.; Serviddio, G.; Pugliese, P.L.; Aspromonte, G.; Altomare, E. Effect of Propofol, Sevoflurane and Desflurane on systemic redox balance. Int. J. Immunopathol. Pharmacol. 2007, 20, 585–593. [Google Scholar] [CrossRef]
- Terpstra, M.; Henry, P.G.; Gruetter, R. Measurement of reduced glutathione (GSH) in human brain using LCModel analysis of difference-edited spectra. Magn. Reson. Med. 2003, 50, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Sanaei Nezhad, F.; Anton, A.; Parkes, L.M.; Deakin, B.; Williams, S.R. Quantification of glutathione in the human brain by MR spectroscopy at 3 Tesla: Comparison of PRESS and MEGA-PRESS. Magn. Reson. Med. 2017, 78, 1257–1266. [Google Scholar] [CrossRef]
- Duffy, S.L.; Lagopoulos, J.; Hickie, I.B.; Diamond, K.; Graeber, M.B.; Lewis, S.J.G.; Naismith, S.L. Glutathione relates to neuropsychological functioning in mild cognitive impairment. Alzheimers Dement. 2014, 10, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Lagopoulos, J.; Hermens, D.F.; Tobias-Webb, J.; Duffy, S.; Naismith, S.L.; White, D.; Scott, E.; Hickie, I.B. In vivo glutathione levels in young persons with bipolar disorder: A magnetic resonance spectroscopy study. J. Psychiatr. Res. 2013, 47, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Barbagallo, G.; Morelli, M.; Quattrone, A.; Chiriaco, C.; Vaccaro, M.G.; Gulla, D.; Rocca, F.; Caracciolo, M.; Novellino, F.; Sarica, A.; et al. In vivo evidence for decreased scyllo-inositol levels in the supplementary motor area of patients with Progressive Supranuclear Palsy: A proton MR spectroscopy study. Parkinsonism Relat. Disord. 2019, 62, 185–191. [Google Scholar] [CrossRef]
- Moss, H.G.; Brown, T.R.; Wiest, D.B.; Jenkins, D.D. N-Acetylcysteine rapidly replenishes central nervous system glutathione measured via magnetic resonance spectroscopy in human neonates with hypoxic-ischemic encephalopathy. J. Cereb. Blood Flow Metab. 2018, 38, 950–958. [Google Scholar] [CrossRef]
- Morley, K.C.; Lagopoulos, J.; Logge, W.; Chitty, K.; Baillie, A.; Haber, P.S. Neurometabolite Levels in Alcohol Use Disorder Patients During Baclofen Treatment and Prediction of Relapse to Heavy Drinking. Front. Psychiatry 2018, 9, 412. [Google Scholar] [CrossRef]
- Coles, L.D.; Tuite, P.J.; Oz, G.; Mishra, U.R.; Kartha, R.V.; Sullivan, K.M.; Cloyd, J.C.; Terpstra, M. Repeated-Dose Oral N-Acetylcysteine in Parkinson’s Disease: Pharmacokinetics and Effect on Brain Glutathione and Oxidative Stress. J. Clin. Pharmacol. 2018, 58, 158–167. [Google Scholar] [CrossRef]
- Wijtenburg, S.A.; Near, J.; Korenic, S.A.; Gaston, F.E.; Chen, H.; Mikkelsen, M.; Chen, S.; Kochunov, P.; Hong, L.E.; Rowland, L.M. Comparing the reproducibility of commonly used magnetic resonance spectroscopy techniques to quantify cerebral glutathione. JMRI 2019, 49, 176–183. [Google Scholar] [CrossRef]
- Durieux, A.M.S.; Horder, J.; Mendez, M.A.; Egerton, A.; Williams, S.C.R.; Wilson, C.E.; Spain, D.; Murphy, C.; Robertson, D.; Barker, G.J.; et al. Cortical and subcortical glutathione levels in adults with autism spectrum disorder. Autism Res. Off. J. Int. Soc. Autism Res. 2016, 9, 429–435. [Google Scholar] [CrossRef]
- Shukla, D.; Mandal, P.K.; Ersland, L.; Gruner, E.R.; Tripathi, M.; Raghunathan, P.; Sharma, A.; Chaithya, G.R.; Punjabi, K.; Splaine, C. A Multi-Center Study on Human Brain Glutathione Conformation using Magnetic Resonance Spectroscopy. J. Alzheimers Dis. 2018, 66, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Provencher, S.W. Automatic quantitation of localized in vivo 1H spectra with LCModel. NMR Biomed. 2001, 14, 260–264. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| KD | Age (y) | Diagnosis | Seizures (#/week) | Diet Duration (Months) | Drugs | Ketonemia (mM/L) | GSH | NAA | FWHM (ppm) | S/N | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Concentration (mM) | %SD | GSH/Cr + PCr | NAA/Cr + PCr | |||||||||
| 1 | 8.9 | DNM1L ME | 3 | 39.4 | topiramate, perampanel | 2.0 ± 0.01 | 2.6 | 13% | 0.44 | 0.63 | 0.064 | 5 |
| 2 | 3.4 | PDH | 28 | 35.7 | levetiracetam, nitrazepam, allopurinol, thiamine | 2.5 ± 0.01 | 2.8 | 11% | 0.48 | 0.72 | 0.038 | 9 |
| 3 | 10.6 | PDH | 0 | 109.0 | thiamine, carnitine, α-lipoic acid, coenzime Q, riboflavin, allopurinol | 2.2 ± 0.15 | 2.3 | 12% | 0.46 | 1.53 | 0.124 | 10 |
| 4 | 12.2 | GLUT-1 | 0 | 49.8 | allopurinol | 1.8 ± 0.1 | 1.4 | 11% | 0.28 | 1.36 | 0.067 | 21 |
| 5 | 16.0 | GCK-HI | 0 | 56.2 | allopurinol, vitamin D | 7.2 ± 0.09 | 2.8 | 9% | 0.47 | 1.26 | 0.086 | 10 |
| 6 | 2.6 | GLUT-1 | 0 | 21.5 | multivitamin | 4.4 ± 0.16 | 2.9 | 7% | 0.42 | 0.89 | 0.038 | 12 |
| 7 | 4.2 | NKH | 28 | 49.0 | levetiracetam, vigabatrin, levofolene, Na benzoate, dextromethorphan | 2.4 ± 0.18 | 3.1 | 7% | 0.38 | 0.53 | 0.093 | 11 |
| 8 | 6.7 | GCK-HI | 0 | 3.4 | ethosuximide, multivitamin | 3.7 ± 0.02 | 1.7 | 13% | 0.44 | 1.32 | 0.029 | 12 |
| 9 | 8.5 | NKH | 1 | 98.4 | levofolene, clonazepam, lansoprazole, levetiracetam, trihexyphenidyl | 3.0 ± 0.01 | 2.6 | 14% | 0.33 | 0.59 | 0.043 | 8 |
| 10 | 7.3 | Unknown EE | 21 | 41.9 | levetiracetam, clobazam, phenobarbital | 2.9 ± 0.15 | 3.0 | 9% | 0.48 | 0.79 | 0.064 | 11 |
| 11 | 5.7 | ATP1A3 | 1 | 46.6 | valproic acid, levetiracetam | 2.0 ± 0.01 | 2.5 | 10% | 0.40 | 1.04 | 0.064 | 10 |
| 12 | 8.6 | Chromosome 21q and 15q deletions | 0 | 7.5 | vigabatrin, valproic acid | 1.7 ± 0.01 | 3.1 | 4% | 0.55 | 1.62 | 0.063 | 47 |
| 13 | 6.8 | Lissencephaly | 14 | 38.3 | vigabatrin, phenobarbital | 2.3 ± 0.32 | 2.2 | 8% | 0.46 | 1.31 | 0.024 | 23 |
| 14 | 12.4 | Unknown EE | 14 | 82.9 | felbamate, clobazam | 3.2 ± 0.01 | 2.5 | 8% | 0.46 | 1.17 | 0.071 | 12 |
| 15 | 0.4 | KCNT1 | 4 | 1.6 | topiramate, levetiracetam, vigabatrin | 4.4 ± 0.01 | 2.8 | 11% | 0.43 | 0.65 | 0.029 | 10 |
| 16 | 2.9 | PRRT2 | 14 | 13.9 | vigabatrin, clobazam | 1.2 ± 0.01 | 2.0 | 10% | 0.29 | 0.83 | 0.057 | 12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Napolitano, A.; Longo, D.; Lucignani, M.; Pasquini, L.; Rossi-Espagnet, M.C.; Lucignani, G.; Maiorana, A.; Elia, D.; De Liso, P.; Dionisi-Vici, C.; et al. The Ketogenic Diet Increases In Vivo Glutathione Levels in Patients with Epilepsy. Metabolites 2020, 10, 504. https://doi.org/10.3390/metabo10120504
Napolitano A, Longo D, Lucignani M, Pasquini L, Rossi-Espagnet MC, Lucignani G, Maiorana A, Elia D, De Liso P, Dionisi-Vici C, et al. The Ketogenic Diet Increases In Vivo Glutathione Levels in Patients with Epilepsy. Metabolites. 2020; 10(12):504. https://doi.org/10.3390/metabo10120504
Chicago/Turabian StyleNapolitano, Antonio, Daniela Longo, Martina Lucignani, Luca Pasquini, Maria Camilla Rossi-Espagnet, Giulia Lucignani, Arianna Maiorana, Domenica Elia, Paola De Liso, Carlo Dionisi-Vici, and et al. 2020. "The Ketogenic Diet Increases In Vivo Glutathione Levels in Patients with Epilepsy" Metabolites 10, no. 12: 504. https://doi.org/10.3390/metabo10120504
APA StyleNapolitano, A., Longo, D., Lucignani, M., Pasquini, L., Rossi-Espagnet, M. C., Lucignani, G., Maiorana, A., Elia, D., De Liso, P., Dionisi-Vici, C., & Cusmai, R. (2020). The Ketogenic Diet Increases In Vivo Glutathione Levels in Patients with Epilepsy. Metabolites, 10(12), 504. https://doi.org/10.3390/metabo10120504