Metabolic Drug Response Phenotyping in Colorectal Cancer Organoids by LC-QTOF-MS

 ,
, 
Abstract
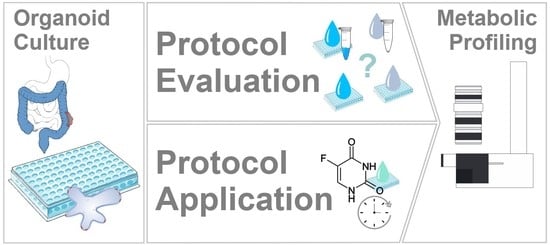
1. Introduction
2. Results and Discussion
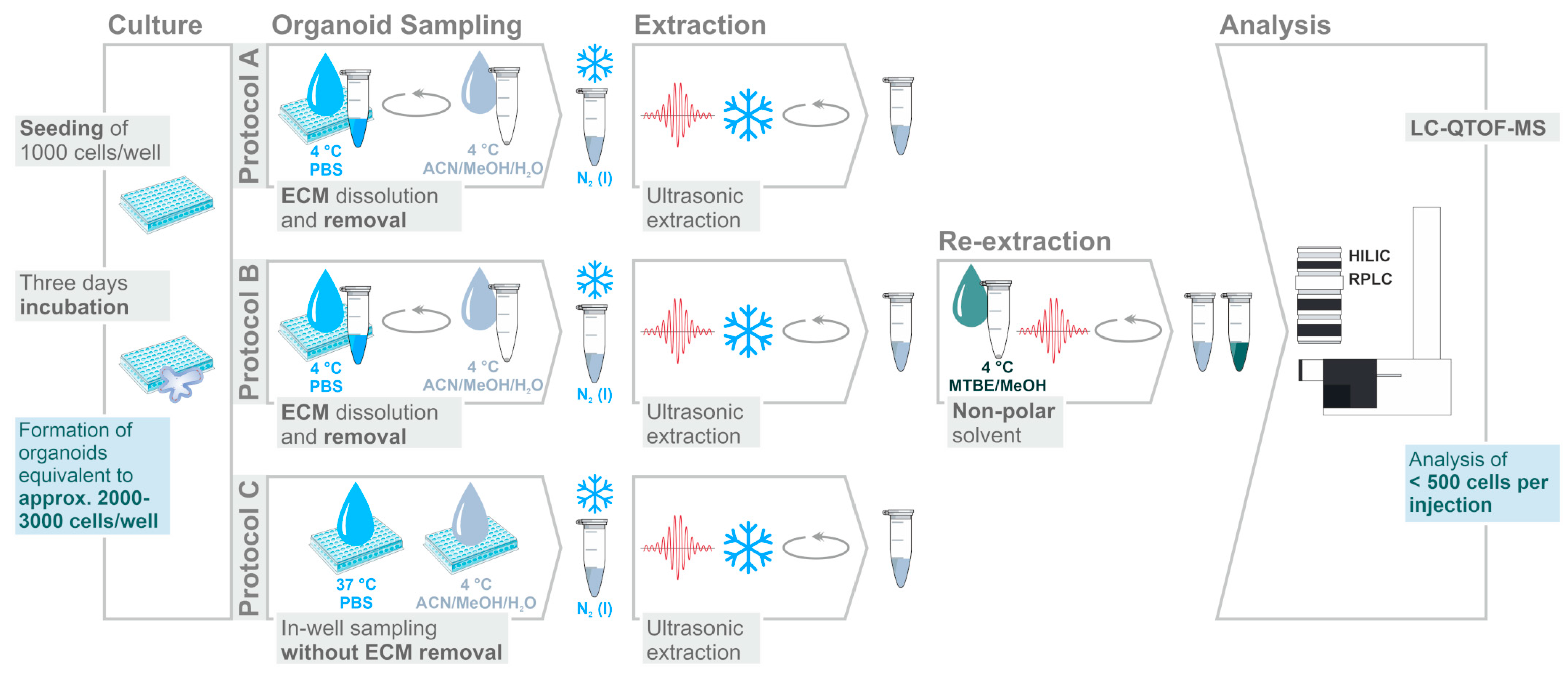
2.1. Assessment of Sample Preparation for Metabolomic and Lipidomic Profiling in CRC Organoids
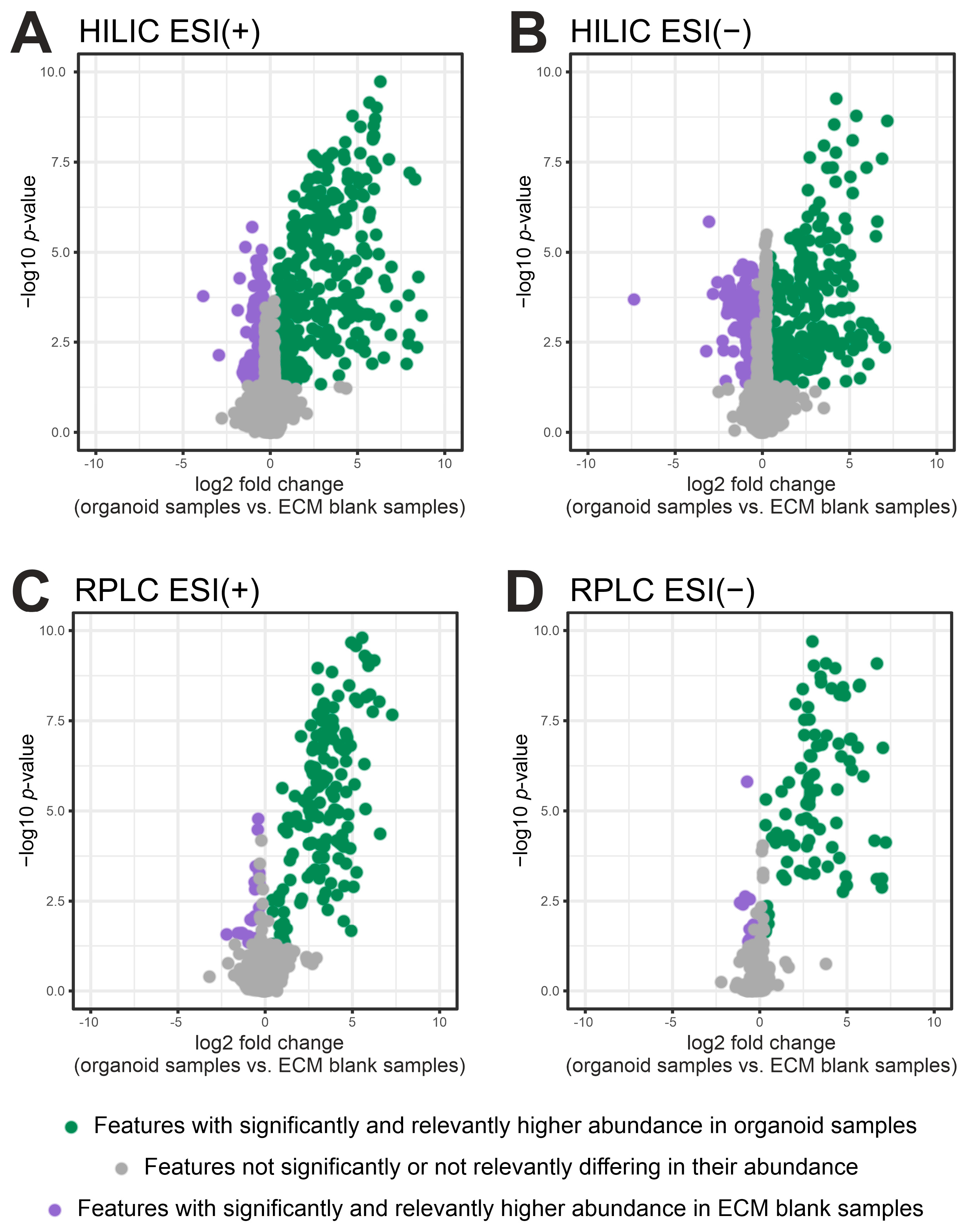
2.2. Filtering of ECM-Derived Background Features by Fold Change and p-Value
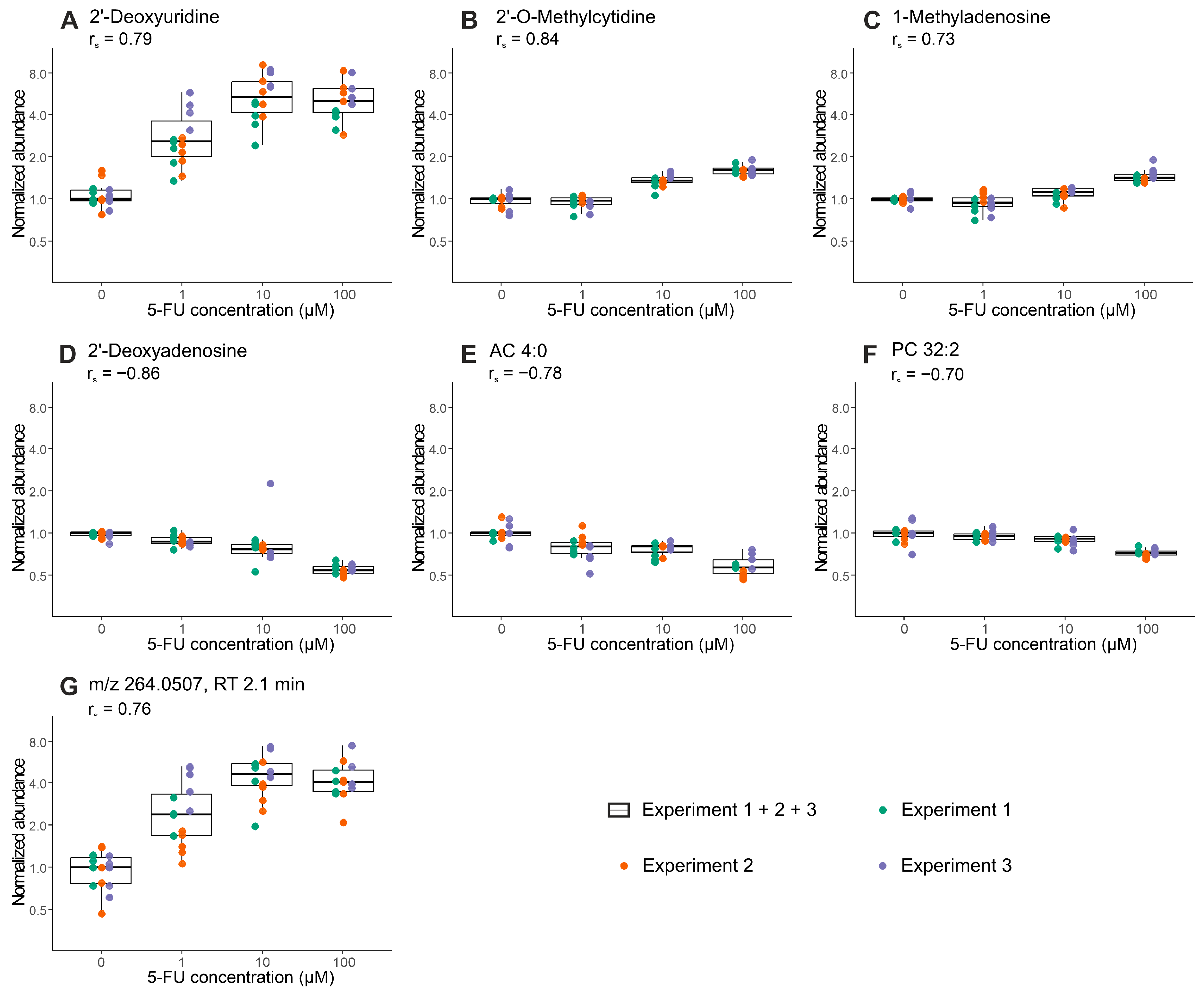
2.3. Proof-of-Concept: Early Metabolic Response of CRC Organoids to 5-Fluorouracil Treatment
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Patient Samples
3.3. Organoid Culture and Viability Assay
3.4. Sampling and Extraction Procedures
3.5. Sample Storage and Preparation
3.6. LC-QTOF-MS Analysis
3.7. Data Preprocessing and Statistical Analysis
3.7.1. Feature Extraction
3.7.2. Data Filtration, Normalization and Analysis
3.8. Metabolite Identification and Annotation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011, 472, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Huch, M.; Kujala, P.; van de Wetering, M.; Snippert, H.J.; van Es, J.H.; Sato, T.; Stange, D.E.; Begthel, H.; van den Born, M.; et al. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 2010, 6, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; van Es, J.H.; van den Brink, S.; van Houdt, W.J.; Pronk, A.; van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Sasai, Y. Cytosystems dynamics in self-organization of tissue architecture. Nature 2013, 493, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Manfrin, A.; Lutolf, M.P. Progress and potential in organoid research. Nat. Rev. Genet. 2018, 19, 671–687. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef]
- Artegiani, B.; Clevers, H. Use and application of 3D-organoid technology. Hum. Mol. Genet. 2018, 27, R99–R107. [Google Scholar] [CrossRef]
- Wild, C.P.; Weiderpass, E.; Stewart, B.W. World Cancer Report 2020; International Agency for Research on Cancer/World Health Organization: Lyon, France, 2020. [Google Scholar]
- Moreau, L.-C.; Rajan, R.; Thirlwell, M.P.; Alcindor, T. Response to chemotherapy in metastatic colorectal cancer after exposure to oxaliplatin in the adjuvant setting. Anticancer Res. 2013, 33, 1765–1768. [Google Scholar]
- Schaeffeler, E.; Büttner, F.; Reustle, A.; Klumpp, V.; Winter, S.; Rausch, S.; Fisel, P.; Hennenlotter, J.; Kruck, S.; Stenzl, A.; et al. Metabolic and Lipidomic Reprogramming in Renal Cell Carcinoma Subtypes Reflects Regions of Tumor Origin. Eur. Urol. Focus 2018, 5, 608–618. [Google Scholar] [CrossRef]
- Njoku, K.; Sutton, C.J.; Whetton, A.D.; Crosbie, E.J. Metabolomic Biomarkers for Detection, Prognosis and Identifying Recurrence in Endometrial Cancer. Metabolites 2020, 10, 314. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.E.; Short, S.P.; Williams, C.S. Colorectal Cancer and Metabolism. Curr. Colorectal Cancer Rep. 2018, 14, 226–241. [Google Scholar] [CrossRef] [PubMed]
- Pakiet, A.; Kobiela, J.; Stepnowski, P.; Sledzinski, T.; Mika, A. Changes in lipids composition and metabolism in colorectal cancer: A review. Lipids Health Dis. 2019, 18, 29. [Google Scholar] [CrossRef] [PubMed]
- Perttula, K.; Edmands, W.M.B.; Grigoryan, H.; Cai, X.; Iavarone, A.T.; Gunter, M.J.; Naccarati, A.; Polidoro, S.; Hubbard, A.; Vineis, P.; et al. Evaluating Ultra-long-Chain Fatty Acids as Biomarkers of Colorectal Cancer Risk. Cancer Epidemiol. Biomarkers Prev. 2016, 25, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- La Vecchia, S.; Sebastián, C. Metabolic pathways regulating colorectal cancer initiation and progression. Semin. Cell Dev. Biol. 2020, 98, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Leuthold, P.; Schaeffeler, E.; Winter, S.; Buttner, F.; Hofmann, U.; Murdter, T.E.; Rausch, S.; Sonntag, D.; Wahrheit, J.; Fend, F.; et al. Comprehensive Metabolomic and Lipidomic Profiling of Human Kidney Tissue: A Platform Comparison. J. Proteome Res. 2017, 16, 933–944. [Google Scholar] [CrossRef]
- Ivanisevic, J.; Zhu, Z.-J.; Plate, L.; Tautenhahn, R.; Chen, S.; O’Brien, P.J.; Johnson, C.H.; Marletta, M.A.; Patti, G.J.; Siuzdak, G. Toward ‘omic scale metabolite profiling: A dual separation-mass spectrometry approach for coverage of lipid and central carbon metabolism. Anal. Chem. 2013, 85, 6876–6884. [Google Scholar] [CrossRef]
- Nelson, S.R.; Zhang, C.; Roche, S.; O’Neill, F.; Swan, N.; Luo, Y.; Larkin, A.; Crown, J.; Walsh, N. Modelling of pancreatic cancer biology: Transcriptomic signature for 3D PDX-derived organoids and primary cell line organoid development. Sci. Rep. 2020, 10, 3529. [Google Scholar] [CrossRef]
- Gonneaud, A.; Asselin, C.; Boudreau, F.; Boisvert, F.-M. Phenotypic Analysis of Organoids by Proteomics. Proteomics 2017, 17, 1700023. [Google Scholar] [CrossRef]
- Artati, A.; Prehn, C.; Adamski, J. LC-MS/MS-Based Metabolomics for Cell Cultures. Methods Mol. Biol. 2019, 1994, 119–130. [Google Scholar]
- Weygand, J.; Carter, S.E.; Salzillo, T.C.; Moussalli, M.; Dai, B.; Dutta, P.; Zuo, X.; Fleming, J.B.; Shureiqi, I.; Bhattacharya, P. Can an Organoid Recapitulate the Metabolome of its Parent Tissue?: A Pilot NMR Spectroscopy Study. JCPCR 2017, 8, 307. [Google Scholar]
- Yoshizaki, H.; Ogiso, H.; Okazaki, T.; Kiyokawa, E. Comparative lipid analysis in the normal and cancerous organoids of MDCK cells. J. Biochem. 2016, 159, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, O.D.K.; Athineos, D.; Cheung, E.C.; Lee, P.; Zhang, T.; van den Broek, N.J.F.; Mackay, G.M.; Labuschagne, C.F.; Gay, D.; Kruiswijk, F.; et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 2017, 544, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Lindeboom, R.G.; van Voorthuijsen, L.; Oost, K.C.; Rodríguez-Colman, M.J.; Luna-Velez, M.V.; Furlan, C.; Baraille, F.; Jansen, P.W.; Ribeiro, A.; Burgering, B.M.; et al. Integrative multi-omics analysis of intestinal organoid differentiation. Mol. Syst. Biol. 2018, 14, e8227. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.; Mukha, D.; Maor, I.I.; Sedov, E.; Koren, E.; Yosefzon, Y.; Shlomi, T.; Fuchs, Y. Blimp1+ cells generate functional mouse sebaceous gland organoids in vitro. Nat. Commun. 2019, 10, 2348. [Google Scholar] [CrossRef]
- Ser, Z.; Liu, X.; Tang, N.N.; Locasale, J.W. Extraction parameters for metabolomics from cultured cells. Anal. Biochem. 2015, 475, 22–28. [Google Scholar] [CrossRef]
- Villaret-Cazadamont, J.; Poupin, N.; Tournadre, A.; Batut, A.; Gales, L.; Zalko, D.; Cabaton, N.J.; Bellvert, F. Bertrand-Michel, J. An Optimized Dual Extraction Method for the Simultaneous and Accurate Analysis of Polar Metabolites and Lipids Carried out on Single Biological Samples. Metabolites 2020, 10, 338. [Google Scholar] [CrossRef]
- Haag, M.; Schmidt, A.; Sachsenheimer, T.; Brügger, B. Quantification of Signaling Lipids by Nano-Electrospray Ionization Tandem Mass Spectrometry (Nano-ESI MS/MS). Metabolites 2012, 2, 57–76. [Google Scholar] [CrossRef]
- Tirinato, L.; Pagliari, F.; Di Franco, S.; Sogne, E.; Marafioti, M.G.; Jansen, J.; Falqui, A.; Todaro, M.; Candeloro, P.; Liberale, C.; et al. ROS and Lipid Droplet accumulation induced by high glucose exposure in healthy colon and Colorectal Cancer Stem Cells. Genes Dis. 2019. [Google Scholar] [CrossRef]
- Rusz, M.; Rampler, E.; Keppler, B.K.; Jakupec, M.A.; Koellensperger, G. Single Spheroid Metabolomics: Optimizing Sample Preparation of Three-Dimensional Multicellular Tumor Spheroids. Metabolites 2019, 9, 304. [Google Scholar] [CrossRef]
- Broadhurst, D.; Goodacre, R.; Reinke, S.N.; Kuligowski, J.; Wilson, I.D.; Lewis, M.R.; Dunn, W.B. Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies. Metabolomics 2018, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Verkh, Y.; Rozman, M.; Petrovic, M. Extraction and cleansing of data for a non-targeted analysis of high-resolution mass spectrometry data of wastewater. MethodsX 2018, 5, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Ivanisevic, J.; Want, E.J. From Samples to Insights into Metabolism: Uncovering Biologically Relevant Information in LC-HRMS Metabolomics Data. Metabolites 2019, 9, 308. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, C.; Petrick, L.; Perttula, K.; Yano, Y.; Carlsson, H.; Whitehead, T.; Metayer, C.; Hayes, J.; Rappaport, S.; Dudoit, S. Filtering procedures for untargeted LC-MS metabolomics data. BMC Bioinform. 2019, 20, 334. [Google Scholar] [CrossRef]
- Abe, Y.; Tada, A.; Isoyama, J.; Nagayama, S.; Yao, R.; Adachi, J.; Tomonaga, T. Improved phosphoproteomic analysis for phosphosignaling and active-kinome profiling in Matrigel-embedded spheroids and patient-derived organoids. Sci. Rep. 2018, 8, 29. [Google Scholar] [CrossRef]
- Zhang, Y.; Lukacova, V.; Reindl, K.; Balaz, S. Quantitative characterization of binding of small molecules to extracellular matrix. J. Biochem. Biophys. Methods 2006, 67, 107–122. [Google Scholar] [CrossRef][Green Version]
- Giobbe, G.G.; Crowley, C.; Luni, C.; Campinoti, S.; Khedr, M.; Kretzschmar, K.; de Santis, M.M.; Zambaiti, E.; Michielin, F.; Meran, L.; et al. Extracellular matrix hydrogel derived from decellularized tissues enables endodermal organoid culture. Nat. Commun. 2019, 10, 246. [Google Scholar] [CrossRef]
- de la Fuente, A.G.; Godzien, J.; López, M.F.; Rupérez, F.J.; Barbas, C.; Otero, A. Knowledge-based metabolite annotation tool: CEU Mass Mediator. J. Pharm. Biomed. Anal. 2018, 154, 138–149. [Google Scholar] [CrossRef]
- Gil-de-la-Fuente, A.; Godzien, J.; Saugar, S.; Garcia-Carmona, R.; Badran, H.; Wishart, D.S.; Barbas, C.; Otero, A. CEU Mass Mediator 3.0: A Metabolite Annotation Tool. J. Proteome Res. 2019, 18, 797–802. [Google Scholar] [CrossRef]
- Hughes, C.S.; Postovit, L.M.; Lajoie, G.A. Matrigel: A complex protein mixture required for optimal growth of cell culture. Proteomics 2010, 10, 1886–1890. [Google Scholar] [CrossRef]
- Li, L.; Andrén, P.E.; Sweedler, J.V. Editorial and Review: 29th ASMS Sanibel Conference on Mass Spectrometry—Peptidomics: Bridging the Gap between Proteomics and Metabolomics by MS. J. Am. Soc. Mass Spectrom. 2018, 29, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, N.K.; Montag, B.J.; Heidelberger, C. Studies on fluorinated pyrimidines. III. The metabolism of 5-fluorouracil-2-C14 and 5-fluoroorotic-2-C14 acid in vivo. Cancer Res. 1958, 18, 318–328. [Google Scholar] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Heidelberger, C.; Chaudhuri, N.K.; Danneberg, P.; Mooren, D.; Griesbach, L.; Duschinsky, R.; Schnitzer, R.J.; Pleven, E.; Scheiner, J. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature 1957, 179, 663–666. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.-M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef]
- Grem, J.L. 5-Fluorouracil: Forty-plus and still ticking. A review of its preclinical and clinical development. Invest. New Drugs 2000, 18, 299–313. [Google Scholar] [CrossRef]
- Ser, Z.; Gao, X.; Johnson, C.; Mehrmohamadi, M.; Liu, X.; Li, S.; Locasale, J.W. Targeting One Carbon Metabolism with an Antimetabolite Disrupts Pyrimidine Homeostasis and Induces Nucleotide Overflow. Cell Rep. 2016, 15, 2367–2376. [Google Scholar] [CrossRef]
- Chong, L.; Tattersall, M.H.N. 5,10-Dideazatetrahydrofolic acid reduces toxicity and deoxyadenosine triphosphate pool expansion in cultured L1210 cells treated with inhibitors of thymidylate synthase. Biochem. Pharmacol. 1995, 49, 819–827. [Google Scholar] [CrossRef]
- Houghton, J.A.; Tillman, D.M.; Harwood, F.G. Ratio of 2’-deoxyadenosine-5’-triphosphate/thymidine-5’-triphosphate influences the commitment of human colon carcinoma cells to thymineless death. Clin. Cancer Res. 1995, 1, 723–730. [Google Scholar]
- O’Dwyer, P.J.; Laub, P.B.; DeMaria, D.; Qian, M.; Reilly, D.; Giantonio, B.; Johnston, A.L.; Wu, E.Y.; Bauman, L.; Clendeninn, N.J.; et al. Phase I trial of the thymidylate synthase inhibitor AG331 as a 5-day continuous infusion. Clin. Cancer Res. 1996, 2, 1685–1692. [Google Scholar] [PubMed]
- Rafi, I.; Boddy, A.V.; Calvete, J.A.; Taylor, G.A.; Newell, D.R.; Bailey, N.P.; Lind, M.J.; Green, M.; Hines, J.; Johnstone, A.; et al. Preclinical and phase I clinical studies with the nonclassical antifolate thymidylate synthase inhibitor nolatrexed dihydrochloride given by prolonged administration in patients with solid tumors. J. Clin. Oncol. 1998, 16, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.J.; Laurensse, E.; Leyva, A.; Pinedo, H.M. Purine nucleosides as cell-specific modulators of 5-fluorouracil metabolism and cytotoxicity. Eur. J. Cancer Clin. Oncol. 1987, 23, 1869–1881. [Google Scholar] [CrossRef]
- Tanigawara, Y.; Nishimuta, A.; Otani, Y.; Matsuo, M. Method for Determining Sensitivity to Fluorouracil in a Subject Having Colorectal Cancer. US 10,309,957 B2, 19 September 2019. [Google Scholar]
- Hills, D.C.; Cotten, M.L.; Horowitz, J. Isolation and characterization of two 5-fluorouracil-substituted Escherichia coli initiator methionine transfer ribonucleic acids. Biochemistry 1983, 22, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Dobrzyńska, I.; Szachowicz-Petelska, B.; Sulkowski, S.; Figaszewski, Z. Changes in electric charge and phospholipids composition in human colorectal cancer cells. Mol. Cell. Biochem. 2005, 276, 113–119. [Google Scholar] [CrossRef]
- Dueck, D.A.; Chan, M.; Tran, K.; Wong, J.T.; Jay, F.T.; Littman, C.; Stimpson, R.; Choy, P.C. The modulation of choline phosphoglyceride metabolism in human colon cancer. Mol. Cell. Biochem. 1996, 162, 97–103. [Google Scholar] [CrossRef]
- Kurabe, N.; Hayasaka, T.; Ogawa, M.; Masaki, N.; Ide, Y.; Waki, M.; Nakamura, T.; Kurachi, K.; Kahyo, T.; Shinmura, K.; et al. Accumulated phosphatidylcholine (16:0/16:1) in human colorectal cancer; possible involvement of LPCAT4. Cancer Sci. 2013, 104, 1295–1302. [Google Scholar] [CrossRef]
- Neef, S.K.; Winter, S.; Hofmann, U.; Mürdter, T.E.; Schaeffeler, E.; Horn, H.; Buck, A.; Walch, A.; Hennenlotter, J.; Ott, G.; et al. Optimized protocol for metabolomic and lipidomic profiling in formalin-fixed paraffin-embedded kidney tissue by LC-MS. Anal. Chim. Acta 2020, 1134, 125–135. [Google Scholar] [CrossRef]
- Leuthold, P.; Schwab, M.; Hofmann, U.; Winter, S.; Rausch, S.; Pollak, M.N.; Hennenlotter, J.; Bedke, J.; Schaeffeler, E.; Haag, M. Simultaneous Extraction of RNA and Metabolites from Single Kidney Tissue Specimens for Combined Transcriptomic and Metabolomic Profiling. J. Proteome Res. 2018, 17, 3039–3049. [Google Scholar] [CrossRef]
- Chetnik, K.; Petrick, L.; Pandey, G. MetaClean: A machine learning-based classifier for reduced false positive peak detection in untargeted LC-MS metabolomics data. Metabolomics 2020, 16, 117. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Slowikowski, K. ggrepel: Automatically Position Non-Overlapping Text Labels with ‘ggplot2’: R Package Version 0.8.0. 2018. Available online: https://CRAN.R-project.org/package=ggrepel (accessed on 11 September 2020).
- Kassambara, A. ggpubr: ‘ggplot2’ Based Publication Ready Plots.: R Package Version 0.3.0. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 11 September 2020).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. JOSS 2019, 4, 1686. [Google Scholar] [CrossRef]
- Bengtsson, H. matrixStats: Functions that Apply to Rows and Columns of Matrices (and to Vectors): R Package Version 0.56.0. 2020. Available online: https://CRAN.R-project.org/package=matrixStats (accessed on 11 September 2020).
- Koncevicius, K. matrixTests: Fast Statistical Hypothesis Tests on Rows and Columns of Matrices: R Package Version 0.1.9. 2020. Available online: https://CRAN.R-project.org/package=matrixTests (accessed on 11 September 2020).
- Pounds, S.; Fofana, D. HybridMTest: Hybrid Multiple Testing: R Package Version 1.32.0. 2020. Available online: https://www.bioconductor.org/packages/release/bioc/html/HybridMTest.html (accessed on 11 September 2020).
- Gao, C.-H. ggVennDiagram: A ‘ggplot2’ Implement of Venn Diagram.: R Package Version 0.3. 2019. Available online: https://CRAN.R-project.org/package=ggVennDiagram (accessed on 3 November 2020).
- Gant, T.W.; Sauer, U.G.; Zhang, S.-D.; Chorley, B.N.; Hackermüller, J.; Perdichizzi, S.; Tollefsen, K.E.; van Ravenzwaay, B.; Yauk, C.; Tong, W.; et al. A generic Transcriptomics Reporting Framework (TRF) for ‘omics data processing and analysis. Regul. Toxicol. Pharmacol. 2017, 91, S36–S45. [Google Scholar] [CrossRef] [PubMed]
- Sangster, T.; Major, H.; Plumb, R.; Wilson, A.J.; Wilson, I.D. A pragmatic and readily implemented quality control strategy for HPLC-MS and GC-MS-based metabonomic analysis. Analyst 2006, 131, 1075–1078. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Weindl, D.; Wegner, A.; Hiller, K. Metabolome-Wide Analysis of Stable Isotope Labeling-Is It Worth the Effort? Front. Physiol. 2015, 6, 344. [Google Scholar] [CrossRef]
- Weindl, D.; Wegner, A.; Jäger, C.; Hiller, K. Isotopologue ratio normalization for non-targeted metabolomics. J. Chromatogr. A 2015, 1389, 112–119. [Google Scholar] [CrossRef]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V. O’Donovan, C. MetaboLights: A resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2020, 48, D440–D444. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protocol | Analytical Mode | No. of Significant and Relevant Metabolites (Organoids vs. ECM Controls) | Median CV of Significant and Relevant Metabolites (%) |
|---|---|---|---|
| A | RPLC ESI (−) | 17 | 21.7 |
| RPLC ESI (+) | 12 | 14.7 | |
| B | RPLC ESI (−) | 13 | 7.0 |
| RPLC ESI (+) | 13 | 8.9 | |
| A/B 1 | HILIC ESI (−) | 15 | 25.7 |
| HILIC ESI (+) | 19 | 33.5 | |
| C | RPLC ESI (−) | 44 | 13.6 |
| RPLC ESI (+) | 54 | 10.4 | |
| HILIC ESI (−) | 17 | 26.8 | |
| HILIC ESI (+) | 25 | 16.2 |
| Analytical Mode | No. of Experiments 1 | Mean Mass | Retention Time | Regulation | Annotation | MSI Level 4 |
|---|---|---|---|---|---|---|
| HILIC ESI (+) | 3 | 111.0436 | 3.21 | ↑ | Cytosine 2 | 2 |
| 251.1026 | 2.42 | ↓ | 2′-Deoxyadenosine | 1 | ||
| 257.1022 | 3.21 | ↑ | 2′-O-Methylcytidine | 1 | ||
| 2 | 231.1468 | 5.95 | ↓ | AC 4:0 | 2 | |
| 268.0828 | 4.89 | ↑ | Inosine | 2 | ||
| 281.1115 | 7.90 | ↑ | 1-Methyladenosine | 1 | ||
| 633.4739 | 3.78 | ↓ | LysoPC 26:1 | 2 | ||
| HILIC ESI (−) | 3 | 228.0731 | 2.12 | ↑ | 2′-Deoxyuridine | 2 |
| 264.0507 | 2.12 | ↑ | na 3 | - | ||
| 2 | 536.1892 | 2.17 | ↑ | na | - | |
| RPLC ESI (+) | 2 | 705.5341 | 6.75 | ↓ | PC 30:0 | 2 |
| 729.5347 | 6.48 | ↓ | PC 32:2 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neef, S.K.; Janssen, N.; Winter, S.; Wallisch, S.K.; Hofmann, U.; Dahlke, M.H.; Schwab, M.; Mürdter, T.E.; Haag, M. Metabolic Drug Response Phenotyping in Colorectal Cancer Organoids by LC-QTOF-MS. Metabolites 2020, 10, 494. https://doi.org/10.3390/metabo10120494
Neef SK, Janssen N, Winter S, Wallisch SK, Hofmann U, Dahlke MH, Schwab M, Mürdter TE, Haag M. Metabolic Drug Response Phenotyping in Colorectal Cancer Organoids by LC-QTOF-MS. Metabolites. 2020; 10(12):494. https://doi.org/10.3390/metabo10120494
Chicago/Turabian StyleNeef, Sylvia K., Nicole Janssen, Stefan Winter, Svenja K. Wallisch, Ute Hofmann, Marc H. Dahlke, Matthias Schwab, Thomas E. Mürdter, and Mathias Haag. 2020. "Metabolic Drug Response Phenotyping in Colorectal Cancer Organoids by LC-QTOF-MS" Metabolites 10, no. 12: 494. https://doi.org/10.3390/metabo10120494
APA StyleNeef, S. K., Janssen, N., Winter, S., Wallisch, S. K., Hofmann, U., Dahlke, M. H., Schwab, M., Mürdter, T. E., & Haag, M. (2020). Metabolic Drug Response Phenotyping in Colorectal Cancer Organoids by LC-QTOF-MS. Metabolites, 10(12), 494. https://doi.org/10.3390/metabo10120494