Targeted Metabolomic Profiling of Total Fatty Acids in Human Plasma by Liquid Chromatography-Tandem Mass Spectrometry

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Solvents
2.2. Standard Solutions
2.3. Study Samples
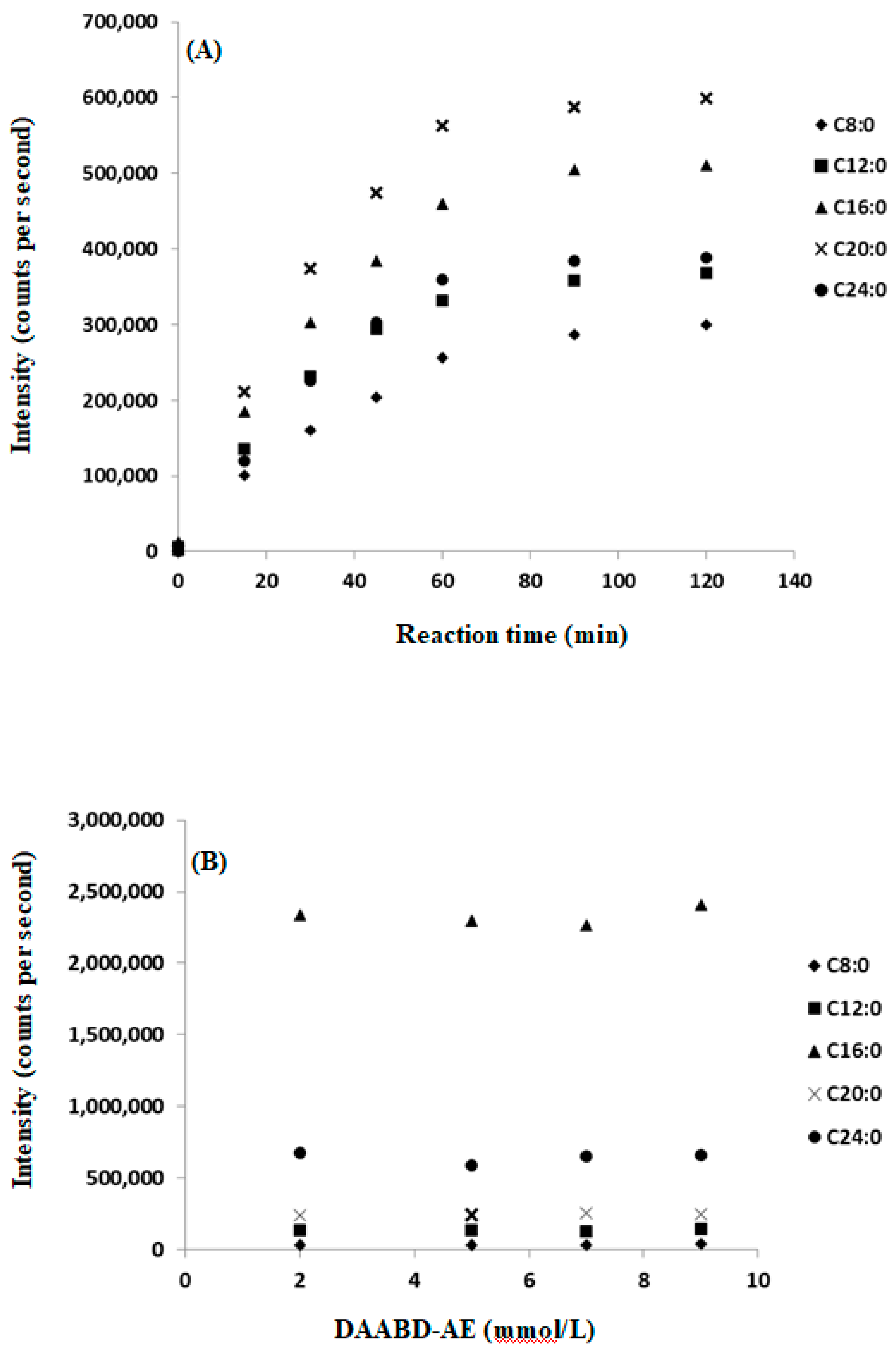
2.4. DAABD-AE Derivatization Reaction Optimization
2.5. Sample Preparation
2.6. LC-MS/MS System and Operating Conditions
2.7. Method Validation
3. Results and Discussion
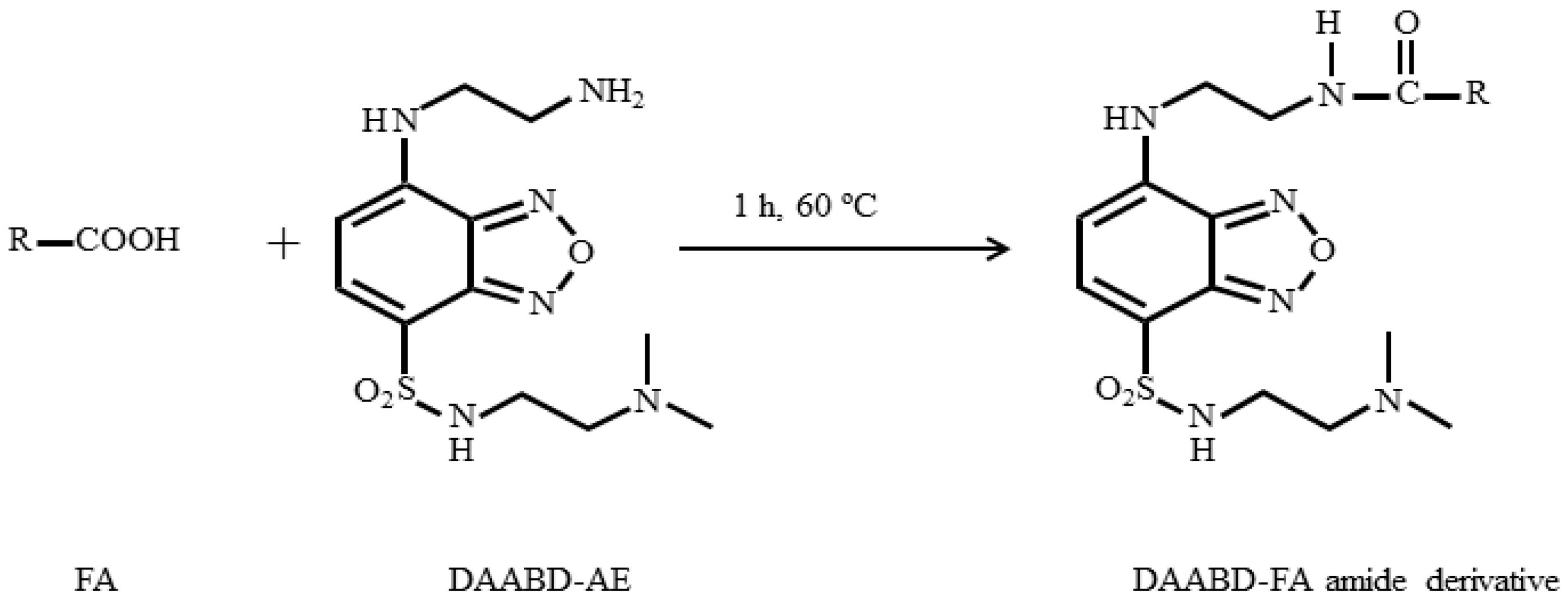
3.1. Derivatization of FAs with DAABD-AE
3.2. Method Development
3.2.1. Derivatization of FAs with DAABD-AE
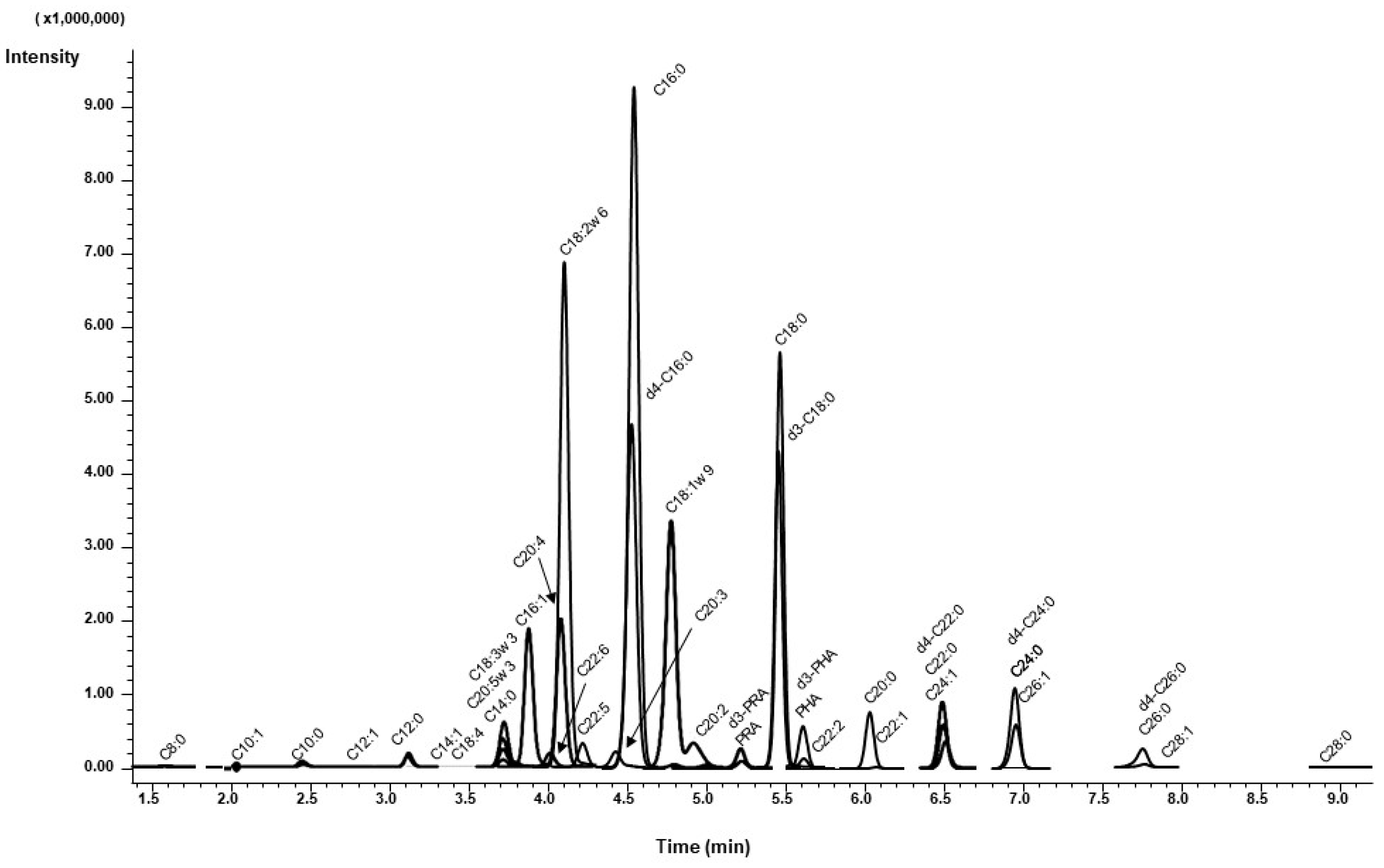
3.2.2. Chromatographic Separation
3.2.3. Linearity, LOD, and LOQ
3.2.4. Imprecision and Recovery
3.3. Determination of FAs Reference Intervals
3.4. Diagnostic Application on Samples from Patients with Inborn Errors of Metabolism
3.5. Method Comparison
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Compliance with Ethics Guidelines
References
- Calder, P.C. Functional Roles of Fatty Acids and Their Effects on Human Health. JPEN J. Parenter. Enter. Nutr. 2015, 39, 18s–32s. [Google Scholar] [CrossRef]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.; Haushalter, R.W.; Lee, D.J.; Markwick, P.R.; Bruegger, J.; Caldara-Festin, G.; Finzel, K.; Jackson, D.R.; Ishikawa, F.; O’Dowd, B.; et al. Trapping the dynamic acyl carrier protein in fatty acid biosynthesis. Nature 2014, 505, 427–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innis, S.M. Dietary (n-3) fatty acids and brain development. J. Nutr. 2007, 137, 855–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemaitre, R.N.; McKnight, B.; Sotoodehnia, N.; Fretts, A.M.; Qureshi, W.T.; Song, X.; King, I.B.; Sitlani, C.M.; Siscovick, D.S.; Psaty, B.M.; et al. Circulating Very Long-Chain Saturated Fatty Acids and Heart Failure: The Cardiovascular Health Study. J. Am. Heart Assoc. 2018, 7, e10019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.R.; Jeon, S.Y.; Lee, S.M. The association of cardiovascular risk factors with saturated fatty acids and fatty acid desaturase indices in erythrocyte in middle-aged Korean adults. Lipids Health Dis. 2015, 14, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, H.; Hayata, Y.; Kawamura, S.; Yamada, T.; Fujiwara, N.; Koike, K. Lipid Metabolic Reprogramming in Hepatocellular Carcinoma. Cancers Basel 2018, 10, 447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.Y.; Ann, D.K. When fats commit crimes: Fatty acid metabolism, cancer stemness and therapeutic resistance. Cancer Commun. Lond. 2018, 38, 47. [Google Scholar] [CrossRef] [Green Version]
- Boden, G. Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes 1997, 46, 3–10. [Google Scholar] [CrossRef]
- Snowden, S.G.; Ebshina, A.A.; Hye, A.; An, Y.; Pletnikova, O.; Brien, R.O.; Troncosco, J.; Quigley, C.L.; Thambiesetty, M. Association Between Fatty Acid Metabolism in the Brain and Alzheimer Disease Neuropathology and Cognitive Performance: A Nontargeted Metabolomic Study. PLoS Med. 2017, 14. [Google Scholar] [CrossRef] [Green Version]
- Knottnerus, S.J.G.; Bleeker, J.C.; Wust, R.C.I.; Ferdinandusse, S.; Ijist, L.; Wijburg, F.A.; Wanders, R.J.A.; Visser, G.; Houtkooper, R.H. Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev. Endocr. Metab. Disord. 2018, 19, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Waterham, H.R.; Ferdinandusse, S.; Wanders, R.J. Human disorders of peroxisome metabolism and biogenesis. Biochim. Biophys. Acta 2016, 1863, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.J.; Choi, M.H.; Paik, M.J.; Yoon, H.R.; Chung, B.C. Gas chromatographic-mass spectrometric determination of plasma saturated fatty acids using pentafluorophenyldimethylsilyl derivatization. J. Chromatogr. B Biomed. Sci. Appl. 2000, 742, 37–46. [Google Scholar] [CrossRef]
- Lagerstedt, S.A.; Hinrichs, D.R.; Batt, S.M.; Magera, M.J.; Rinaldo, P.; McConnell, J.P. Quantitative determination of plasma c8-c26 total fatty acids for the biochemical diagnosis of nutritional and metabolic disorders. Mol. Genet. Metab. 2001, 73, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.R.; Paik, M.J.; Shin, H.S.; Yu, C.; Rinaldo, P. Analysis of plasma free fatty acid cyanomethyl derivatives by GC-NPD for the diagnosis of mitochondrial fatty acid oxidation disorders. Chromatographia 2020, 52, 211–216. [Google Scholar] [CrossRef]
- Durie, D.; Yeh, E.; McIntosh, N.; Fisher, L.; Bulman, D.E.; Birnboim, H.C.; Chakraborty, P.; Al-Dirbashi, O.Y. Quantification of DNA in Neonatal Dried Blood Spots by Adenine Tandem Mass Spectrometry. Anal. Chem. 2018, 90, 801–806. [Google Scholar] [CrossRef] [Green Version]
- Grebe, S.K.; Singh, R.J. LC-MS/MS in the Clinical Laboratory—Where to From Here? Clin. Biochem. Rev. 2011, 32, 5–31. [Google Scholar]
- Becher, F.; Ciccolini, J.; Imbs, D.C.; Marin, C.; Fournel, C.; Dupuis, C.; Fakhry, N.; Pourroy, B.; Ghettas, A.; Pruvost, A.; et al. A simple and rapid LC-MS/MS method for therapeutic drug monitoring of cetuximab: A GPCO-UNICANCER proof of concept study in head-and-neck cancer patients. Sci. Rep. 2017, 7, 2714. [Google Scholar] [CrossRef]
- Reinstadler, V.; Lierheimer, S.; Boettcher, M.; Oberacher, H. A validated workflow for drug detection in oral fluid by non-targeted liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2019, 411, 867–876. [Google Scholar] [CrossRef] [Green Version]
- Al Dhahouri, N.; Langhans, C.D.; Al Hammadi, Z.; Okun, J.G.; Hoffmann, G.F.; Al-Jasmi, F.; Al-Dirbashi, O.Y. Quantification of methylcitrate in dried urine spots by liquid chromatography tandem mass spectrometry for the diagnosis of propionic and methylmalonic acidemias. Clin. Chim. Acta 2018, 487, 41–45. [Google Scholar] [CrossRef]
- Al-Dirbashi, O.Y.; Alfadhel, M.; Al-Thihli, K.; Al Dhahouri, N.; Langhans, C.D.; Al Hammadi, Z.; Al-Shamsi, A.; Hertecant, J.; Okun, J.G.; Hoffmann, G.F.; et al. Assessment of methylcitrate and methylcitrate to citrate ratio in dried blood spots as biomarkers for inborn errors of propionate metabolism. Sci. Rep. 2019, 9, 12366. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Ichikawa, T.; Inagaki, S.; Min, J.Z.; Fukushima, T.; Toyo’oka, T. Simple and practical derivatization procedure for enhanced detection of carboxylic acids in liquid chromatography-electrospray ionization-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2010, 52, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Bollinger, J.G.; Naika, G.S.; Sadilek, M.; Gelb, M.H. LC/ESI-MS/MS detection of FAs by charge reversal derivatization with more than four orders of magnitude improvement in sensitivity. J. Lipid. Res. 2013, 54, 3523–3530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jemal, M.; Zheng, O.; Teitz, D.S. High performance liquid chromatography mobile phase composition optimization for the quantitative determination of a carboxylic acid compound in human plasma by negative ion electrospray high performance liquid chromatography tandem mass spectrometry. Rapid Commun. Mass Spectrom. RCM 1998, 12. [Google Scholar] [CrossRef]
- Al-Dirbashi, O.Y.; Santa, T.; Rashed, M.S.; Al-Hassnan, Z.; Shimozawa, N.; Chedrawi, A.; Jacob, M.; Al-Mokhadab, M. Rapid UPLC-MS/MS method for routine analysis of plasma pristanic, phytanic, and very long chain fatty acid markers of peroxisomal disorders. J. Lipid Res. 2008, 49, 1855–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Franke, A.A. Improved LC-MS method for the determination of fatty acids in red blood cells by LC-orbitrap MS. Anal. Chem. 2011, 83, 3192–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leng, J.; Wang, H.; Zhang, L.; Zhang, J.; Guo, Y. A highly sensitive isotope-coded derivatization method and its application for the mass spectrometric analysis of analytes containing the carboxyl group. Anal. Chim. Acta 2013, 758, 114–121. [Google Scholar] [CrossRef]
- Wang, M.; Han, R.H.; Han, X. Fatty acidomics: Global analysis of lipid species containing a carboxyl group with a charge-remote fragmentation-assisted approach. Anal. Chem. 2013, 85, 9312–9320. [Google Scholar] [CrossRef] [Green Version]
- Cai, T.; Ting, H.; Xin-Xiang, Z.; Jiang, Z.; Jin-Lan, Z. HPLC-MRM relative quantification analysis of fatty acids based on a novel derivatization strategy. Analyst 2014, 139, 6154–6159. [Google Scholar] [CrossRef]
- Sun, F.; Choi, A.A.; Wu, R. Systematic Analysis of Fatty Acids in Human Cells with a Multiplexed Isobaric Tag (TMT)-Based Method. J. Proteome. Res. 2018, 17, 1606–1614. [Google Scholar] [CrossRef]
- Volpato, M.; Spencer, J.A.; Race, A.D.; Munarini, A.; Belluzzi, A.; Cockbain, A.J.; Hull, M.A.; Loadman, P.M. A liquid chromatography-tandem mass spectrometry method to measure fatty acids in biological samples. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1055–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Gao, Z.; Wu, Y.; Shrestha, R.; Imai, H.; Uemura, N.; Hirano, K.I.; Chiba, H.; Hui, S.P. Development of a simultaneous quantitation for short-, medium-, long-, and very long-chain fatty acids in human plasma by 2-nitrophenylhydrazine-derivatization and liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2019, 1126–1127. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.C.; Adamec, J.; Regnier, F.E. Enhancement of the LC/MS analysis of fatty acids through derivatization and stable isotope coding. Anal. Chem. 2007, 79, 5150–5157. [Google Scholar] [CrossRef] [PubMed]
- Vreken, P.; van Lint, A.E.M.; Bootsma, A.H.; Overmars, H.; Wanders, R.J.; van Gennip, A.H. Rapid stable isotope dilution analysis of very-long-chain fatty acids, pristanic acid and phytanic acid using gas chromatography-electron impact mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 1998, 713. [Google Scholar] [CrossRef]
- Al-Dirbashi, O.Y.; Kolker, S.; Ng, D.; Fisher, L.; Rupar, T.; Lepage, N.; Rashed, M.S.; Santa, T.; Goodman, S.I.; Geraghty, M.T.; et al. Diagnosis of glutaric aciduria type 1 by measuring 3-hydroxyglutaric acid in dried urine spots by liquid chromatography tandem mass spectrometry. J. Inherit. Metab. Dis. 2011, 34, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Al-Dirbashi, O.Y.; McIntosh, N.; McRoberts, C.; Fisher, L.; Rashed, M.S.; Makhseed, N.; Geraghty, M.T.; Santa, T.; Chakraborty, P. Analysis of methylcitrate in dried blood spots by liquid chromatography-tandem mass spectrometry. JIMD Rep. 2014, 16, 65–73. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Retention Time (min) | IS * | Precursor Ion (m/z) | Linear Range (μmol/L) | LOD # | LOQ § |
|---|---|---|---|---|---|---|
| Octanoic acid, C8:0 | 1.6 | (A) | 455.3 | 0.75–75 | 9.9 | 33.0 |
| Decenoic acid, C10:1 | 2.0 | (B) | 481.3 | - | ||
| Decanoic acid, C10:0 | 2.5 | (B) | 483.3 | 3.75–375 | 14.0 | 46.7 |
| Lauroleic acid, C12:1 | 2.8 | (C) | 509.2 | - | ||
| Lauric acid, C12:0 | 3.2 | (C) | 511.2 | 7.5–750 | 12.7 | 42.3 |
| Tetradecadienoic acid, C14:2 | 3.0 | (D) | 535.4 | - | ||
| Myristoleic acid, C14:1 | 3.4 | (D) | 537.4 | - | ||
| Myristic acid, C14:0 | 3.8 | (D) | 539.4 | 7.5–750 | 9.0 | 30.0 |
| Hexadecenoic acid, C16:1w9 | 3.9 | (E) | 565.4 | 7.5–750 | 8.6 | 28.7 |
| Palmitic acid, C16:0 | 4.7 | (E) | 567.3 | 60–6000 | 4.2 | 14.0 |
| Stearidonic acid, C18:4 | 3.5 | (F) | 587.4 | - | ||
| α-Linolenic acid, C18:3w3 | 3.8 | (F) | 589.4 | 3.75–375 | 5.1 | 17.0 |
| Linoleic acid, C18:2w6 | 4.2 | (F) | 591.4 | - | ||
| Oleic acid, C18:1w9 | 4.9 | (F) | 593.4 | - | 5.9 | 19.7 |
| Stearic acid, C18:0 | 5.5 | (F) | 595.4 | 30–3000 | 6.8 | 22.7 |
| EPA, C20:5w3 | 3.8 | (H) | 613.4 | 1.9–188 | 14.0 | 46.7 |
| Arachidonic acid, C20:4w6 | 4.2 | (H) | 615.4 | 22.5–2250 | 8.6 | 28.7 |
| h-γ-Linolenic acid, C20:3w6 | 4.5 | (H) | 617.4 | - | ||
| Eicosadienoic acid, C20:2 | 5.1 | (H) | 619.4 | - | ||
| Gondoic acid, C20:1 | 5.6 | (H) | 621.4 | - | ||
| Arachidic acid, C20:0 | 6.1 | (H) | 623.4 | 0.75–75 | 9.0 | 30.0 |
| Pristanic acid, C19:0 branched | 5.3 | (G) | 609.2 | 0.75–75 | 11.4 | 38.0 |
| Phytanic acid, C20:0 branched | 5.7 | (H) | 623.2 | 0.75–75 | 9.6 | 32.0 |
| DHA, C22:6w3 | 4.1 | (I) | 639.2 | - | ||
| DPA, C22:5w3 | 4.6 | (I) | 641.2 | 0.75–75 | 14.3 | 47.7 |
| DTA, C22:4w6 | 4.9 | (I) | 643.2 | - | ||
| Docosatrienoic acid, C22:3 | 5.5 | (I) | 645.2 | - | ||
| Docosadienoic acid, C22:2 | 5.8 | (I) | 647.2 | - | ||
| Docosenoic acid, C22:1 | 6.2 | (I) | 649.2 | - | ||
| Docosanoic acid, C22:0 | 6.6 | (I) | 651.2 | 3.75–375 | 10.3 | 34.3 |
| Nervonic acid, C24:1 | 6.6 | (J) | 677.3 | - | ||
| Tetracosanoic acid, C24:0 | 7.1 | (J) | 679.3 | 3.75–375 | 9.8 | 32.7 |
| Hexacosenoic acid, C26:1 | 7.1 | (K) | 705.3 | - | ||
| Hexacosanoic acid, C26:0 | 7.9 | (K) | 707.3 | 0.15–15 | 15.1 | 50.3 |
| Octacosenoic acid, C28:1 | 7.9 | (K) | 733.3 | - | ||
| Montanic acid, C28:0 | 9.1 | (K) | 735.3 | - |
| Compound | Sample | Concentration (μ mol/L) | Intra-Day (n = 15) | Inter-Day (n = 20) | Recovery (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | CV (%) | Mean | SD | CV (%) | ||||
| C8:0 | QC 1 | 38.0 | 36.6 | 2.4 | 6.6 | 36.3 | 0.6 | 1.8 | 98.7 |
| QC 2 | 72.4 | 73.4 | 2.5 | 3.4 | 73.4 | 2.5 | 3.4 | ||
| C10:0 | QC 1 | 86.2 | 88.2 | 3.9 | 4.4 | 87.3 | 2.0 | 2.3 | 101.0 |
| QC 2 | 282.4 | 281.6 | 9.1 | 3.2 | 284.3 | 3.5 | 1.2 | ||
| C12:0 | QC 1 | 209.7 | 201.7 | 7.9 | 3.9 | 200.9 | 3.7 | 1.8 | 98.5 |
| QC 2 | 580.5 | 588.7 | 11.7 | 2.0 | 584.9 | 5.3 | 0.9 | ||
| C14:0 | QC 1 | 315.2 | 308.2 | 21.6 | 7.0 | 314.6 | 3.7 | 1.2 | 99.2 |
| QC 2 | 693.1 | 685.6 | 8.0 | 1.2 | 695.7 | 7.1 | 1.0 | ||
| C16:1 | QC 1 | 406.6 | 397.8 | 25.0 | 6.3 | 398.5 | 3.9 | 1.0 | 98.8 |
| QC 2 | 675.7 | 671.9 | 28.8 | 4.3 | 675.7 | 8.8 | 1.3 | ||
| C16:0 | QC 1 | 3663.1 | 3610.3 | 367.6 | 10.2 | 3697.7 | 46.0 | 1.2 | 99.5 |
| QC 2 | 5538.9 | 5620.3 | 213.7 | 3.8 | 5367.8 | 534.3 | 10.0 | ||
| C18:3w3 | QC 1 | 124.8 | 129.9 | 7.5 | 5.8 | 127.6 | 8.5 | 6.7 | 100.8 |
| QC 2 | 330.8 | 325.6 | 21.9 | 6.7 | 324.8 | 3.8 | 1.2 | ||
| C18:0 | QC 1 | 1655.4 | 1639.7 | 113.3 | 6.9 | 1640.6 | 35.1 | 2.1 | 99.4 |
| QC 2 | 2902.7 | 2889.7 | 39.8 | 1.4 | 2895.6 | 63.8 | 2.2 | ||
| C20:5w3 | QC 1 | 94.9 | 94.6 | 2.5 | 2.6 | 95.8 | 2.5 | 2.6 | 101.1 |
| QC 2 | 177.2 | 181.7 | 6.5 | 3.6 | 179.7 | 2.4 | 1.3 | ||
| C20:4 | QC 1 | 1340.2 | 1546.2 | 67.3 | 4.4 | 1621.3 | 145.4 | 9.0 | 106.4 |
| QC 2 | 2214.1 | 1965.4 | 41.8 | 2.1 | 2224.7 | 36.7 | 1.7 | ||
| C20:0 | QC 1 | 32.5 | 33.5 | 0.4 | 1.3 | 31.2 | 1.3 | 4.3 | 94.5 |
| QC 2 | 75.4 | 67.8 | 2.0 | 2.9 | 67.0 | 2.5 | 3.7 | ||
| Pristanic | QC 1 | 16.6 | 16.9 | 0.6 | 3.4 | 16.4 | 1.0 | 6.2 | 98.5 |
| QC 2 | 55.9 | 55.1 | 2.3 | 4.2 | 53.4 | 2.0 | 3.8 | ||
| Phytanic | QC 1 | 16.8 | 17.0 | 1.0 | 5.7 | 16.3 | 0.4 | 2.3 | 98.0 |
| QC 2 | 56.2 | 55.0 | 2.0 | 3.6 | 54.1 | 1.5 | 2.8 | ||
| C22:5w6 | QC 1 | 42.6 | 42.9 | 3.9 | 9.1 | 43.3 | 1.2 | 2.7 | 101.5 |
| QC 2 | 70.2 | 72.0 | 3.2 | 4.4 | 71.2 | 1.9 | 2.7 | ||
| C22:0 | QC 1 | 124.9 | 124.0 | 2.0 | 1.6 | 123.3 | 1.4 | 1.2 | 99.2 |
| QC 2 | 317.7 | 317.5 | 12.6 | 4.0 | 314.4 | 5.1 | 1.6 | ||
| C24.0 | QC 1 | 126.0 | 127.6 | 2.6 | 2.1 | 124.2 | 1.6 | 1.3 | 100.8 |
| QC 2 | 309.1 | 315.8 | 7.2 | 2.3 | 313.4 | 2.3 | 0.7 | ||
| C26.0 | QC 1 | 3.8 | 3.7 | 0.1 | 3.0 | 3.7 | 0.1 | 1.5 | 97.8 |
| QC 2 | 11.8 | 11.4 | 0.2 | 1.9 | 11.7 | 0.2 | 1.5 | ||
| Compound | <1 Month | 1–12 Month | 1–18 Year | >18 Year | ||||
|---|---|---|---|---|---|---|---|---|
| Low | High | Low | High | Low | High | Low | High | |
| Octanoic acid, C8:0 | 22 | 53 | 21 | 60 | 22 | 62 | 18 | 41 |
| Decenoic acid, C10:1 | 0.2 | 1.7 | 0.2 | 2.3 | 0.1 | 1.3 | 0.1 | 1.1 |
| Decanoic acid, C10:0 | 12 | 46 | 9 | 60 | 10 | 57 | 9 | 41 |
| Lauroleic acid, C12:1 | 0.1 | 4.0 | 0.2 | 2.8 | 0.2 | 1.6 | 0.2 | 2.4 |
| Lauric acid, C12:0 | 21 | 165 | 19 | 211 | 25 | 202 | 35 | 152 |
| Tetradecadienoic acid, C14:2 | 0.1 | 4.1 | 0.2 | 9.4 | 0.2 | 4.3 | 0.1 | 3.6 |
| Myristoleic acid, C14:1 | 0.7 | 6.8 | 0.8 | 10.1 | 1.0 | 20.5 | 1.2 | 14.2 |
| Myristic acid, C14:0 | 35 | 367 | 43 | 327 | 37 | 293 | 40 | 337 |
| Hexadecenoic acid, C16:1w9 | 163 | 654 | 74 | 517 | 49 | 590 | 72 | 514 |
| Palmitic acid, C16:0 | 1304 | 3654 | 1289 | 3595 | 554 | 3411 | 1238 | 3999 |
| Stearidonic acid, C18:4 | 0.0 | 21.1 | 0.1 | 17.5 | 0.2 | 27.2 | 0 | 29 |
| α-Linolenic acid, C18:3w3 | 0.5 | 46.9 | 1.6 | 66.8 | 5.7 | 62.7 | 3 | 44 |
| Linoleic acid, C18:2w6 | 216 | 1750 | 620 | 2544 | 655 | 2193 | 672 | 2961 |
| Oleic acid, C18:1w9 | 925 | 3250 | 1237 | 4943 | 857 | 4041 | 816 | 4433 |
| Stearic acid, C18:0 | 562 | 1410 | 580 | 1553 | 253 | 1414 | 511 | 1507 |
| EPA, C20:5w3 | 10 | 118 | 7 | 73 | 9 | 92 | 6 | 88 |
| Arachidonic acid, C20:4w6 | 622 | 1652 | 303 | 1316 | 122 | 1155 | 275 | 1576 |
| h-γ-Linolenic acid, C20:3w6 | 30 | 111 | 17 | 113 | 29 | 149 | 23 | 131 |
| Eicosadienoic acid, C20:2 | 4 | 33 | 5 | 38 | 5 | 23 | 5 | 22 |
| Gondoic acid, C20:1 | 6 | 36 | 8 | 49 | 5 | 38 | 6 | 35 |
| Arachidic acid, C20:0 | 11 | 37 | 5 | 40 | 5 | 28 | 5 | 33 |
| Pristanic acid, C19:0 branched | 1.1 | 3.0 | 1.1 | 2.8 | 1.2 | 3.0 | 1.3 | 3.0 |
| Phytanic acid, C20:0 branched | 1.6 | 3.6 | 1.8 | 4.9 | 1.7 | 10.3 | 1.8 | 8.0 |
| DHA, C22:6w3 | 9 | 60 | 15 | 64 | 5 | 45 | 4 | 39 |
| DPA, C22:5w3 | 13 | 88 | 7 | 73 | 8 | 52 | 4 | 43 |
| DTA, C22:4w6 | 19 | 68 | 14 | 64 | 14 | 53 | 9 | 61 |
| Docosatrienoic acid, C22:3 | 1 | 7 | 0 | 5 | 1 | 6 | 0 | 4 |
| Docosadienoic acid, C22:2 | 3 | 15 | 3 | 14 | 2 | 11 | 2 | 10 |
| Docosenoic acid, C22:1 | 15 | 45 | 14 | 35 | 12 | 38 | 11 | 39 |
| Docosanoic acid, C22:0 | 27 | 60 | 21 | 100 | 28 | 70 | 28 | 77 |
| Nervonic acid, C24:1 | 77 | 257 | 82 | 220 | 54 | 210 | 68 | 267 |
| Tetracosanoic acid, C24:0 | 18 | 59 | 17 | 70 | 21 | 65 | 17 | 76 |
| Hexacosenoic acid, C26:1 | 0.9 | 3.2 | 0.3 | 2.0 | 0.2 | 1.9 | 0.2 | 1.9 |
| Hexacosanoic acid, C26:0 | 0.3 | 1.0 | 0.2 | 1.0 | 0.2 | 1.2 | 0.3 | 1.1 |
| Octacosenoic acid, C28:1 | 0.01 | 0.10 | 0.00 | 0.16 | 0.00 | 0.10 | 0.01 | 0.09 |
| Montanic acid, C28:0 | 0.07 | 0.19 | 0.04 | 0.18 | 0.03 | 0.20 | 0.03 | 0.15 |
| Total fatty acids (mmol/L) | 4.1 | 14.1 | 4.4 | 16.2 | 2.8 | 14.4 | 3.9 | 16.6 |
| Total saturated fatty acids (mmol/L) | 2.0 | 5.9 | 2.0 | 6.0 | 1.0 | 5.6 | 1.9 | 6.3 |
| Total MUFA (mmol/L) | 1.2 | 4.3 | 1.4 | 5.8 | 1.0 | 4.9 | 1.0 | 5.3 |
| Total PUFA (mmol/L) | 0.9 | 4.0 | 1.0 | 4.4 | 0.9 | 3.9 | 1.0 | 5.0 |
| Patient | 1 | 2 | 3 | 4 | 5 | Reference Interval |
|---|---|---|---|---|---|---|
| Age | 10 day | 10.8 year | 2.6 year | 22.2 year | 2 year | |
| Sex | F | M | F | M | F | |
| Diagnosis | Severe PBD | X-ALD | RD | AMN | Mild PBD | |
| PRA | 1.6 | 1.9 | 3.7 | 1.7 | 4.2 | 1.2–3.0 |
| PHA | 4.3 | 4.3 | 23.2 | 4.7 | 15.2 | 1.7–10.3 |
| C22:0 | 46.9 | 40.1 | 47.0 | 52.3 | 25.1 | 21–100 |
| C24:0 | 63.8 | 57.0 | 28.0 | 86.7 | 24.5 | 17–76 |
| C26:0 | 17.4 | 2.2 | 0.9 | 3.0 | 2.1 | 0.2–1.2 |
| C28:0 | 1.82 | 0.38 | 0.19 | 0.38 | 0.20 | 0.03–0.2 |
| C24:0/C22:0 | 1.36 | 1.42 | 0.76 | 1.66 | 0.98 | ≤1.20 |
| C26:0/C22:0 | 0.372 | 0.054 | 0.019 | 0.057 | 0.082 | ≤0.022 |
| C28:0/C22:0 | 0.0388 | 0.0094 | 0.0041 | 0.0073 | 0.0080 | ≤0.0045 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aidaros, A.A.; Sharma, C.; Langhans, C.-D.; G. Okun, J.; Hoffmann, G.F.; Dasouki, M.; Chakraborty, P.; Aljasmi, F.; Y. Al-Dirbashi, O. Targeted Metabolomic Profiling of Total Fatty Acids in Human Plasma by Liquid Chromatography-Tandem Mass Spectrometry. Metabolites 2020, 10, 400. https://doi.org/10.3390/metabo10100400
Aidaros AA, Sharma C, Langhans C-D, G. Okun J, Hoffmann GF, Dasouki M, Chakraborty P, Aljasmi F, Y. Al-Dirbashi O. Targeted Metabolomic Profiling of Total Fatty Acids in Human Plasma by Liquid Chromatography-Tandem Mass Spectrometry. Metabolites. 2020; 10(10):400. https://doi.org/10.3390/metabo10100400
Chicago/Turabian StyleAidaros, Anas Al, Charu Sharma, Claus-Dieter Langhans, Jürgen G. Okun, Georg F. Hoffmann, Majed Dasouki, Pranesh Chakraborty, Fatma Aljasmi, and Osama Y. Al-Dirbashi. 2020. "Targeted Metabolomic Profiling of Total Fatty Acids in Human Plasma by Liquid Chromatography-Tandem Mass Spectrometry" Metabolites 10, no. 10: 400. https://doi.org/10.3390/metabo10100400
APA StyleAidaros, A. A., Sharma, C., Langhans, C.-D., G. Okun, J., Hoffmann, G. F., Dasouki, M., Chakraborty, P., Aljasmi, F., & Y. Al-Dirbashi, O. (2020). Targeted Metabolomic Profiling of Total Fatty Acids in Human Plasma by Liquid Chromatography-Tandem Mass Spectrometry. Metabolites, 10(10), 400. https://doi.org/10.3390/metabo10100400