Neuronal Lipoprotein Lipase Deficiency Alters Neuronal Function and Hepatic Metabolism

 ,
,
Abstract
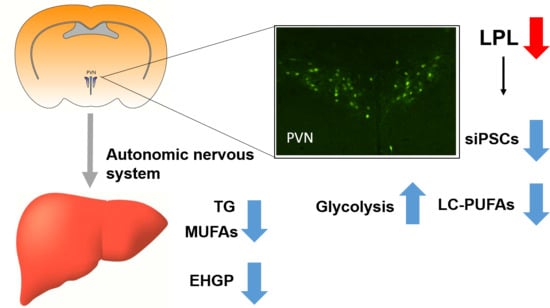
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
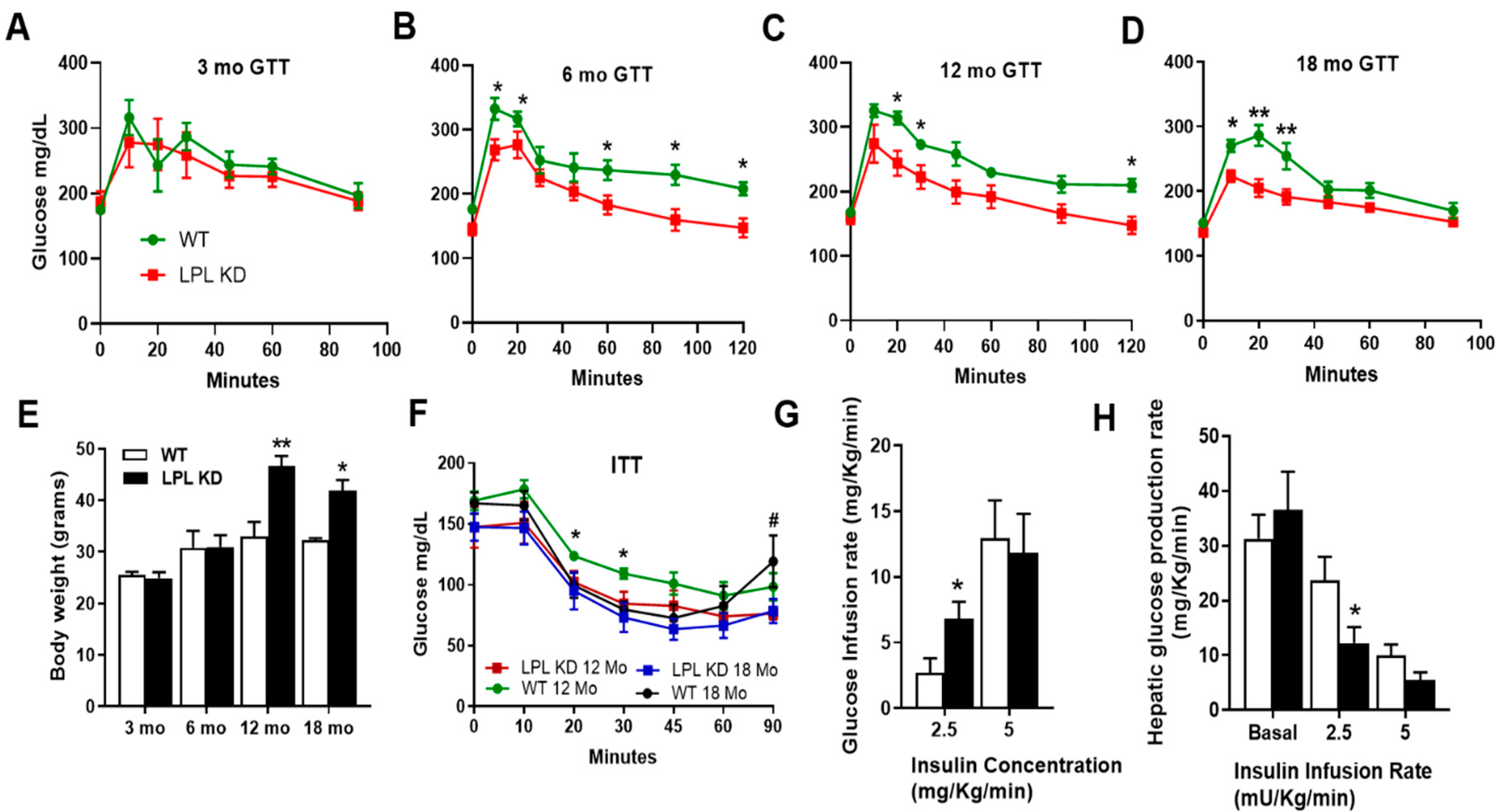
2.1. Mice with Neuron-Specific LPL Depletion Have Improved Glucose Tolerance
2.2. LPL KD Mice Have Reduced Hepatic Glucose Production
2.3. Neuronal LPL-KD Mice Have Reduced Hepatic Lipid Accumulation
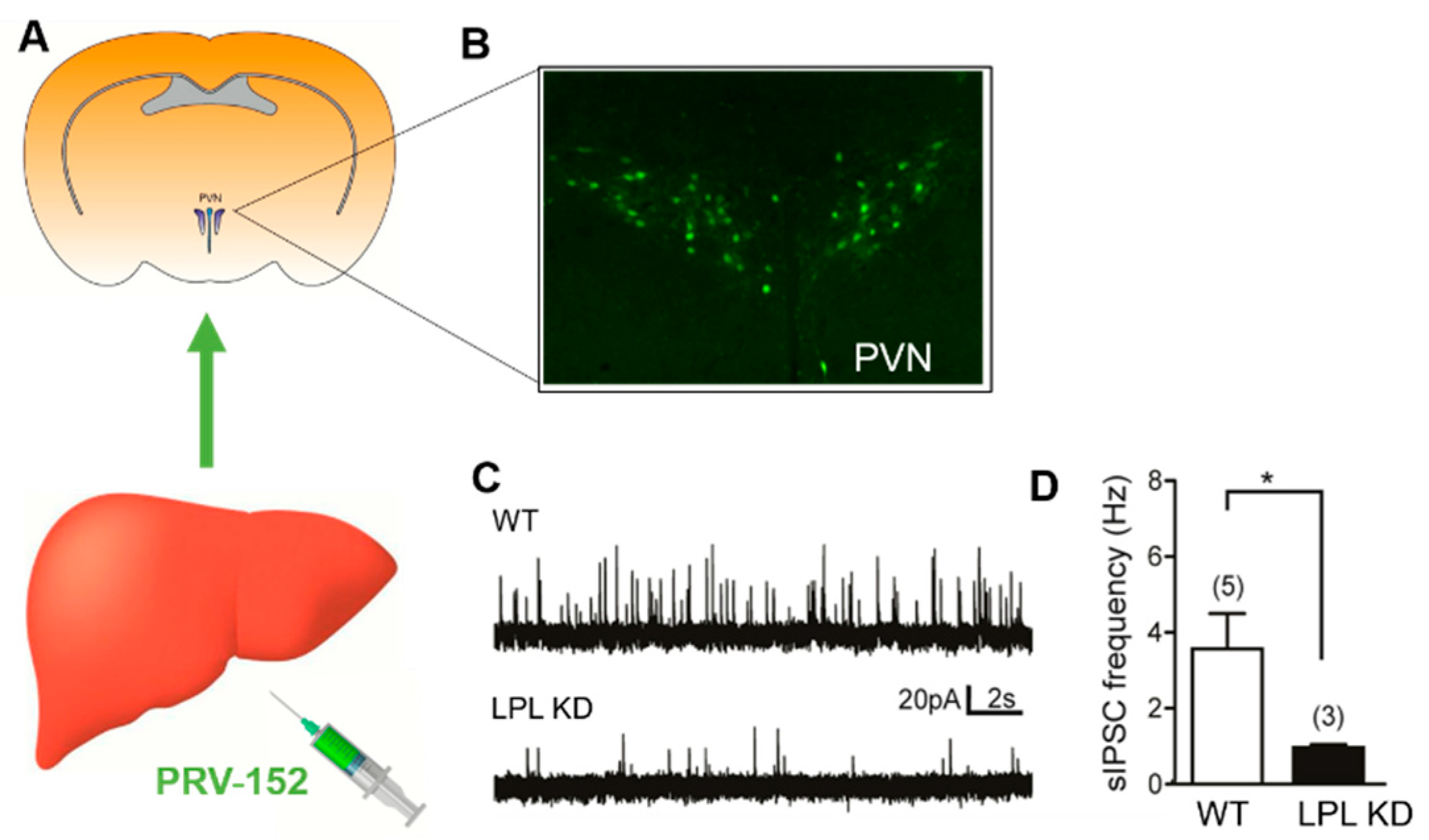
2.4. Liver-Related PVN Neurons of LPL KD Mice Have Reduced Inhibitory Synaptic Control
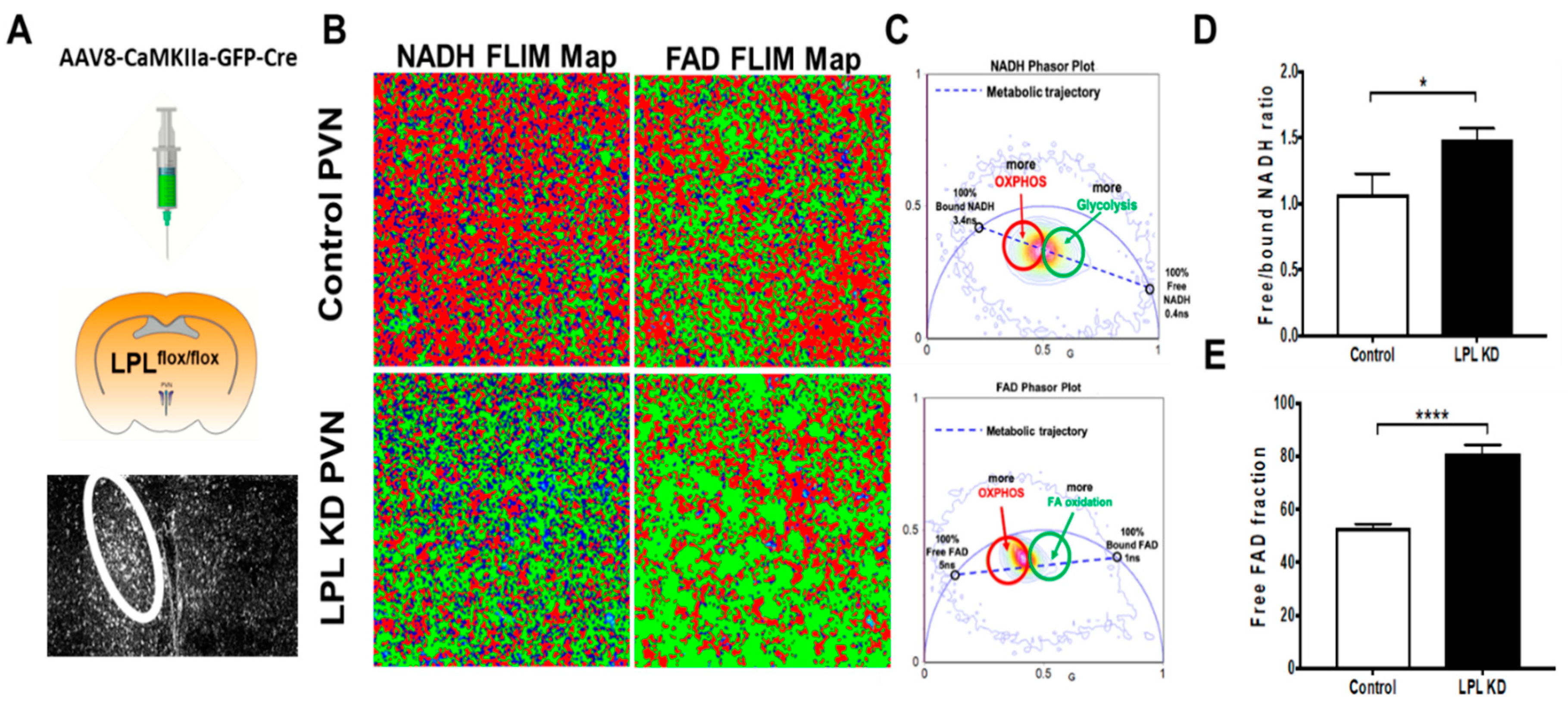
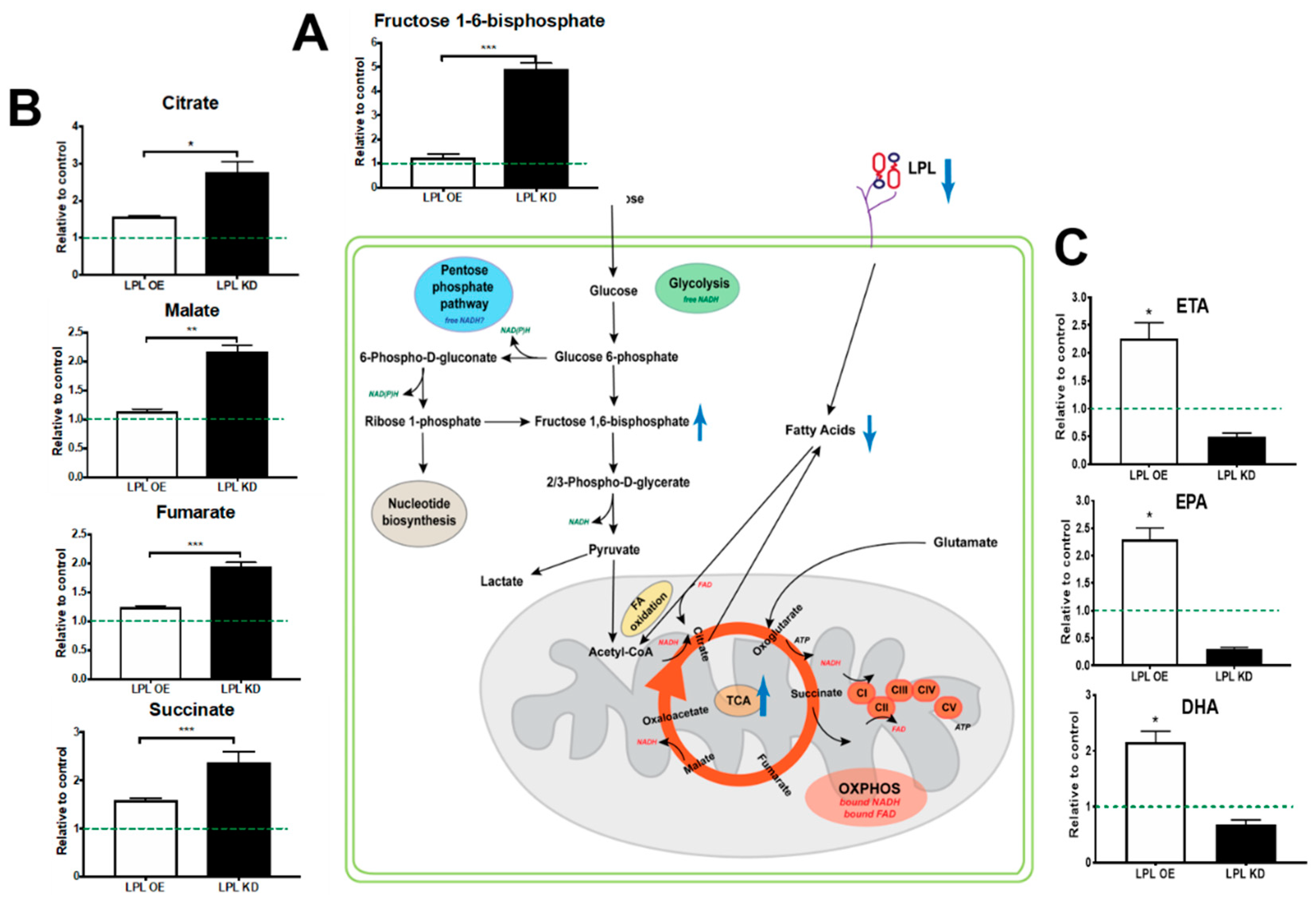
2.5. Loss of LPL in PVN Neurons Alters Metabolic Flux
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Glucose and Insulin Tolerance Tests
4.3. Hyperinsulinemic-Euglycemic Clamps
4.4. Analysis of Liver Lipids
4.5. Lipoprotein Profile
4.6. Quantitative Real-Time PCR
4.7. Identification of Liver-Related Neurons with PRV-152
4.8. Brain Slice Preparation
4.9. Whole-Cell Patch-Clamp Recordings
4.10. Stereotaxic AVV Injection
4.11. Fluorescence Lifetime Imaging Microscopy (FLIM)
4.12. Cell Culture Conditions and Reagents
4.13. Metabolomics
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bril, F.; Cusi, K. Management of Nonalcoholic Fatty Liver Disease in Patients With Type 2 Diabetes: A Call to Action. Diabetes Care 2017, 40, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2009, 51, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Yki-Järvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef]
- Ribeiro, I.M.R.; Antunes, V.R. The role of insulin at brain-liver axis in the control of glucose production. Am. J. Physiol. Liver Physiol. 2018, 315, G538–G543. [Google Scholar] [CrossRef]
- Lam, T.K.T.; Gutierrez-Juarez, R.; Pocai, A.; Bhanot, S.; Tso, P.; Schwartz, G.J.; Rossetti, L. Brain glucose metabolism controls the hepatic secretion of triglyceride-rich lipoproteins. Nat. Med. 2007, 13, 171–180. [Google Scholar] [CrossRef]
- Yue, J.T.Y.; Abraham, M.A.; Lapierre, M.P.; Mighiu, P.I.; Light, P.E.; Filippi, B.M.; Lam, T.K.T. A fatty acid-dependent hypothalamic–DVC neurocircuitry that regulates hepatic secretion of triglyceride-rich lipoproteins. Nat. Commun. 2015, 6, 5970. [Google Scholar] [CrossRef] [Green Version]
- Song, B.J.; Elbert, A.; Rahman, T.; Orr, S.K.; Chen, C.T.; Febbraio, M.; Bazinet, R.P. Genetic Ablation of CD36 does not Alter Mouse Brain Polyunsaturated Fatty Acid Concentrations. Lipids 2010, 45, 291–299. [Google Scholar] [CrossRef]
- Laperrousaz, E.; Moullé, V.S.; Denis, R.G.P.; Kassis, N.; Berland, C.; Colsch, B.; Fioramonti, X.; Philippe, E.; Lacombe, A.; Vanacker, C.; et al. Lipoprotein lipase in hypothalamus is a key regulator of body weight gain and glucose homeostasis in mice. Diabetologia 2017, 60, 1314–1324. [Google Scholar] [CrossRef] [Green Version]
- Libby, A.E.; Wang, H.; Mittal, R.; Sungelo, M.; Potma, E.; Eckel, R.H. Lipoprotein lipase is an important modulator of lipid uptake and storage in hypothalamic neurons. Biochem. Biophys. Res. Commun. 2015, 465, 287–292. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Astarita, G.; Taussig, M.D.; Bharadwaj, K.G.; DiPatrizio, N.V.; Nave, K.-A.; Piomelli, D.; Goldberg, I.J.; Eckel, R. Deficiency of Lipoprotein Lipase in Neurons Modifies the Regulation of Energy Balance and Leads to Obesity. Cell Metab. 2011, 13, 105–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Taussig, M.D.; DiPatrizio, N.V.; Bruce, K.; Piomelli, D.; Eckel, R. Obesity development in neuron-specific lipoprotein lipase deficient mice is not responsive to increased dietary fat content or change in fat composition. Metabolism 2016, 65, 987–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Taussig, M.D.; DiPatrizio, N.V.; Astarita, G.; Piomelli, D.; Bergman, B.C.; Dell’Acqua, M.L.; Eckel, R.; Wang, H. Deficiency of Lipoprotein Lipase in Neurons Decreases AMPA Receptor Phosphorylation and Leads to Neurobehavioral Abnormalities in Mice. PLoS ONE 2015, 10, e0135113. [Google Scholar] [CrossRef] [Green Version]
- Ayala, J.E.; Bracy, D.P.; McGuinness, O.P.; Wasserman, D.H. Considerations in the Design of Hyperinsulinemic-Euglycemic Clamps in the Conscious Mouse. Diabetes 2006, 55, 390–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelenik, T.; Kaul, K.; Séquaris, G.; Flögel, U.; Phielix, E.; Kotzka, J.; Knebel, B.; Fahlbusch, P.; Hörbelt, T.; Lehr, S.; et al. Mechanisms of Insulin Resistance in Primary and Secondary Nonalcoholic Fatty Liver. Diabetes 2017, 66, 2241–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Miyata, K.; Bhaskaran, M.D.; Derbenev, A.V.; Zsombok, A. Transient Receptor Potential Vanilloid Type 1–Dependent Regulation of Liver-Related Neurons in the Paraventricular Nucleus of the Hypothalamus Diminished in the Type 1 Diabetic Mouse. Diabetes 2012, 61, 1381–1390. [Google Scholar] [CrossRef] [Green Version]
- Gunion, M.W.; Taché, Y.; Rosenthal, M.J.; Miller, S.; Butler, B.; Zib, B. Bombesin microinfusion into the rat hypothalamic paraventricular nucleus increases blood glucose, free fatty acids and corticosterone. Brain Res. 1989, 478, 47–58. [Google Scholar] [CrossRef]
- Ionescu, E.; Coimbra, C.C.; Walker, C.D.; Jeanrenaud, B. Paraventricular nucleus modulation of glycemia and insulinemia in freely moving lean rats. Am. J. Physiol. Integr. Comp. Physiol. 1989, 257, R1370–R1376. [Google Scholar] [CrossRef]
- Kalsbeek, A.; La Fleur, S.; van Heijningen, C.; Buijs, R.M. Suprachiasmatic GABAergic Inputs to the Paraventricular Nucleus Control Plasma Glucose Concentrations in the Rat via Sympathetic Innervation of the Liver. J. Neurosci. 2004, 24, 7604–7613. [Google Scholar] [CrossRef] [Green Version]
- Magistretti, P.J.; Allaman, I. A Cellular Perspective on Brain Energy Metabolism and Functional Imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruinstroop, E.; Pei, L.; Ackermans, M.T.; Foppen, E.; Borgers, A.J.; Kwakkel, J.; Alkemade, A.; Fliers, E.; Kalsbeek, A. Hypothalamic Neuropeptide Y (NPY) Controls Hepatic VLDL-Triglyceride Secretion in Rats via the Sympathetic Nervous System. Diabetes 2012, 61, 1043–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Yu, Y.; Szabo, A.; Wu, Y.; Wang, H.; Camer, D.; Huang, X.F. Palmitic acid induces central leptin resistance and impairs hepatic glucose and lipid metabolism in male mice. J. Nutr. Biochem. 2015, 26, 541–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obici, S.; Feng, Z.; Karkanias, G.; Baskin, D.G.; Rossetti, L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat. Neurosci. 2002, 5, 566–572. [Google Scholar] [CrossRef]
- Denis, G.V.; Hamilton, J.A. Healthy obese persons. Curr. Opin. Endocrinol. Diabetes Obes. 2013, 20, 369–376. [Google Scholar] [CrossRef] [Green Version]
- Blüher, M. The distinction of metabolically ‘healthy’ from ‘unhealthy’ obese individuals. Curr. Opin. Lipidol. 2010, 21, 38–43. [Google Scholar] [CrossRef]
- Samocha-Bonet, D.; Chisholm, D.; Tonks, K.; Campbell, L.; Greenfield, J. Insulin-sensitive obesity in humans—A ‘favorable fat’ phenotype? Trends Endocrinol. Metab. 2012, 23, 116–124. [Google Scholar] [CrossRef]
- Shimazu, T. Glycogen Synthetase Activity in Liver: Regulation by the Autonomic Nerves. Science 1967, 156, 1256–1257. [Google Scholar] [CrossRef]
- Shimazu, T. Regulation of glycogen metabolism in liver by the autonomic nervous systemV. Activation of glycogen synthetase by vagal stimulation. Biochim. Biophys. Acta Gen. Subj. 1971, 252, 28–38. [Google Scholar] [CrossRef]
- Sigala, B.; McKee, C.; Soeda, J.; Pazienza, V.; Morgan, M.; Lin, C.I.; Selden, C.; Borght, S.V.; Mazzoccoli, G.; Roskams, T.; et al. Sympathetic Nervous System Catecholamines and Neuropeptide Y Neurotransmitters are Upregulated in Human NAFLD and Modulate the Fibrogenic Function of Hepatic Stellate Cells. PLoS ONE 2013, 8, e72928. [Google Scholar] [CrossRef]
- Hurr, C.; Simonyan, H.; Morgan, D.A.; Rahmouni, K.; Young, C.N. Liver sympathetic denervation reverses obesity-induced hepatic steatosis. J. Physiol. 2019, 597, 4565–4580. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, J.D.; Zsombok, A. Brain-liver connections: Role of the preautonomic PVN neurons. Am. J. Physiol. Metab. 2016, 310, E183–E189. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.E.; Dobrinskikh, E.; Alfonso-Garcia, A.; Fast, A.; Janssen, R.C.; Soderborg, T.K.; Anderson, A.L.; Reisz, J.A.; D’Alessandro, A.; Frank, D.N.; et al. Pyrroloquinoline quinone prevents developmental programming of microbial dysbiosis and macrophage polarization to attenuate liver fibrosis in offspring of obese mice. Hepatol. Commun. 2018, 2, 313–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjit, S.; Dvornikov, A.; Dobrinskikh, E.; Wang, X.; Luo, Y.; Levi, M.; Gratton, E. Measuring the effect of a Western diet on liver tissue architecture by FLIM autofluorescence and harmonic generation microscopy. Biomed. Opt. Express 2017, 8, 3143–3154. [Google Scholar] [CrossRef] [Green Version]
- Stringari, C.; Nourse, J.L.; Flanagan, L.A.; Gratton, E. Phasor Fluorescence Lifetime Microscopy of Free and Protein-Bound NADH Reveals Neural Stem Cell Differentiation Potential. PLoS ONE 2012, 7, e48014. [Google Scholar] [CrossRef] [Green Version]
- Drozdowicz-Tomsia, K.; Anwer, A.G.; Cahill, M.A.; Madlum, K.; Maki, A.M.; Baker, M.S.; Goldys, E.M. Multiphoton fluorescence lifetime imaging microscopy reveals free-to-bound NADH ratio changes associated with metabolic inhibition. J. Biomed. Opt. 2014, 19, 86016. [Google Scholar] [CrossRef] [Green Version]
- Ranjit, S.; Malacrida, L.; Stakic, M.; Gratton, E. Determination of the metabolic index using the fluorescence lifetime of free and bound NADH in the phasor approach. J. Biophotonics 2019, 12, e201900156. [Google Scholar] [CrossRef]
- Bolaños, J.P.; Almeida, A.; Moncada, S. Glycolysis: A bioenergetic or a survival pathway? Trends Biochem. Sci. 2010, 35, 145–149. [Google Scholar] [CrossRef]
- Herrero-Mendez, A.; Almeida, A.; Fernández, E.; Maestre, C.; Moncada, S.; Bolaños, J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C–Cdh1. Nature 2009, 11, 747–752. [Google Scholar] [CrossRef]
- Dienel, G.A. Brain lactate metabolism: The discoveries and the controversies. J. Cereb. Blood Flow Metab. 2012, 32, 1107–1138. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallrabe, H.; Svindrych, Z.; Alam, S.R.; Siller, K.H.; Wang, T.; Kashatus, D.; Hu, S.; Periasamy, A. Segmented cell analyses to measure redox states of autofluorescent NAD(P)H, FAD & Trp in cancer cells by FLIM. Sci. Rep. 2018, 8, 79. [Google Scholar] [PubMed] [Green Version]
- Broberger, C.; Visser, T.J.; Kuhar, M.J.; Hökfelt, T. Neuropeptide Y innervation and neuropeptide-Y-Y1-receptor-expressing neurons in the paraventricular hypothalamic nucleus of the mouse. Neuroendocrinology 1999, 70, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Pocai, A.; Lam, T.K.T.; Gutierrez-Juarez, R.; Obici, S.; Schwartz, G.J.; Bryan, J.; Aguilar-Bryan, L.; Rossetti, L. Hypothalamic KATP channels control hepatic glucose production. Nature 2005, 434, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Van Den Hoek, A.M.; van Heijningen, C.; der Elst, J.P.S.V.; Ouwens, D.M.; Havekes, L.M.; Romijn, J.A.; Kalsbeek, A.; Pijl, H. Intracerebroventricular Administration of Neuropeptide Y Induces Hepatic Insulin Resistance via Sympathetic Innervation. Diabetes 2008, 57, 2304–2310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obici, S.; Feng, Z.; Arduini, A.; Conti, R.; Rossetti, L. Inhibition of hypothalamic carnitine palmitoyltransferase-1 decreases food intake and glucose production. Nat. Med. 2003, 9, 756–761. [Google Scholar] [CrossRef]
- Loftus, T.M.; Jaworsky, D.E.; Frehywot, G.L.; Townsend, C.A.; Ronnett, G.V.; Lane, M.D.; Kuhajda, F.P. Reduced Food Intake and Body Weight in Mice Treated with Fatty Acid Synthase Inhibitors. Science 2000, 288, 2379–2381. [Google Scholar] [CrossRef]
- Bazan, N.G. Synaptic lipid signaling: Significance of polyunsaturated fatty acids and platelet-activating factor. J. Lipid Res. 2003, 44, 2221–2233. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Higashimori, T.; Park, S.Y.; Choi, H.; Dong, J.; Kim, Y.J.; Noh, H.L.; Cho, Y.R.; Cline, G.; Kim, Y.B.; et al. Differential effects of interleukin-6 and -10 on skeletal muscle and liver insulin action in vivo. Diabetes 2004, 53, 1060–1067. [Google Scholar] [CrossRef] [Green Version]
- Libby, A.E.; Bales, E.; Orlicky, D.J.; McManaman, J.L. Perilipin-2 Deletion Impairs Hepatic Lipid Accumulation by Interfering with Sterol Regulatory Element-binding Protein (SREBP) Activation and Altering the Hepatic Lipidome. J. Boil. Chem. 2016, 291, 24231–24246. [Google Scholar] [CrossRef] [Green Version]
- Orlicky, D.J.; Libby, A.E.; Bales, E.S.; Mcmahan, R.H.; Monks, J.; Rosa, F.G.; McManaman, J.L. Perilipin-2 promotes obesity and progressive fatty liver disease in mice through mechanistically distinct hepatocyte and extra-hepatocyte actions. J. Physiol. 2019, 597, 1565–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, A.E.; Bales, E.S.; Monks, J.; Orlicky, D.J.; McManaman, J.L. Perilipin-2 deletion promotes carbohydrate-mediated browning of white adipose tissue at ambient temperature. J. Lipid Res. 2018, 59, 1482–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, K.D.; Cagampang, F.R.; Argenton, M.; Zhang, J.; Ethirajan, P.L.; Burdge, G.C.; Bateman, A.C.; Clough, G.F.; Poston, L.; Hanson, M.A.; et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology 2009, 50, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, A.J.; Lyons, T.J.; Zheng, D.; Otvos, J.D.; Lackland, D.T.; McGee, D.; Garvey, W.T.; Klein, R.L. The DCCT/EDIC Research Group Serum Lipoproteins in the Diabetes Control and Complications Trial/Epidemiology of Diabetes Intervention and Complications Cohort: Associations with gender and glycemia. Diabetes Care 2003, 26, 810–818. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Molinas, A.J.; Miyata, K.; Qiao, X.; Zsombok, A. Overactivity of Liver-Related Neurons in the Paraventricular Nucleus of the Hypothalamus: Electrophysiological Findings in db/db Mice. J. Neurosci. 2017, 37, 11140–11150. [Google Scholar] [CrossRef] [Green Version]
- Zsombok, A.; Gao, H.; Miyata, K.; Issa, A.; Derbenev, A.V. Immunohistochemical localization of transient receptor potential vanilloid type 1 and insulin receptor substrate 2 and their co-localization with liver-related neurons in the hypothalamus and brainstem. Brain Res. 2011, 1398, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Krashes, M.J.; Shah, B.P.; Madara, J.C.; Olson, D.P.; Strochlic, D.E.; Garfield, A.S.; Vong, L.; Pei, H.; Watabe-Uchida, M.; Uchida, N.; et al. An excitatory paraventricular nucleus to AgRP neuron circuit that drives hunger. Nature 2014, 507, 238–242. [Google Scholar] [CrossRef]
- Berezin, M.; Achilefu, S. Fluorescence Lifetime Measurements and Biological Imaging. Chem. Rev. 2010, 110, 2641–2684. [Google Scholar] [CrossRef] [Green Version]
- Digman, M.A.; Caiolfa, V.R.; Zamai, M.; Gratton, E. The Phasor Approach to Fluorescence Lifetime Imaging Analysis. Biophys. J. 2007, 94, L14–L16. [Google Scholar] [CrossRef] [Green Version]
- Marwan, A.I.; Shabeka, U.; Reisz, J.A.; Zheng, C.; Serkova, N.J.; Dobrinskikh, E. Unique Heterogeneous Topological Pattern of the Metabolic Landscape in Rabbit Fetal Lungs following Tracheal Occlusion. Fetal Diagn. Ther. 2018, 45, 145–154. [Google Scholar] [CrossRef]
- Dobrinskikh, E.; Al-Juboori, S.I.; Shabeka, U.; Reisz, J.A.; Zheng, C.; Marwan, A.I. Heterogeneous Pulmonary Response After Tracheal Occlusion: Clues to Fetal Lung Growth. J. Surg. Res. 2019, 239, 242–252. [Google Scholar] [CrossRef] [PubMed]
- McCurdy, C.E.; Schenk, S.; Hetrick, B.; Houck, J.; Drew, B.G.; Kaye, S.; Lashbrook, M.; Bergman, B.C.; Takahashi, D.L.; Dean, T.A.; et al. Maternal obesity reduces oxidative capacity in fetal skeletal muscle of Japanese macaques. JCI Insight 2016, 1, e86612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melamud, E.; Vastag, L.; Rabinowitz, J.D. Metabolomic Analysis and Visualization Engine for LC−MS Data. Anal. Chem. 2010, 82, 9818–9826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Wishart, D.S. MSEA: A web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 2010, 38, W71–W77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Wishart, D.S. MetPA: A web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 2010, 26, 2342–2344. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruce, K.D.; Dobrinskikh, E.; Wang, H.; Rudenko, I.; Gao, H.; Libby, A.E.; Gorkhali, S.; Yu, T.; Zsombok, A.; Eckel, R.H. Neuronal Lipoprotein Lipase Deficiency Alters Neuronal Function and Hepatic Metabolism. Metabolites 2020, 10, 385. https://doi.org/10.3390/metabo10100385
Bruce KD, Dobrinskikh E, Wang H, Rudenko I, Gao H, Libby AE, Gorkhali S, Yu T, Zsombok A, Eckel RH. Neuronal Lipoprotein Lipase Deficiency Alters Neuronal Function and Hepatic Metabolism. Metabolites. 2020; 10(10):385. https://doi.org/10.3390/metabo10100385
Chicago/Turabian StyleBruce, Kimberley D., Evgenia Dobrinskikh, Hong Wang, Ivan Rudenko, Hong Gao, Andrew E. Libby, Sachi Gorkhali, Tian Yu, Andrea Zsombok, and Robert H. Eckel. 2020. "Neuronal Lipoprotein Lipase Deficiency Alters Neuronal Function and Hepatic Metabolism" Metabolites 10, no. 10: 385. https://doi.org/10.3390/metabo10100385