
High Drug Loading of Amorphous Solid Dispersion by Hot Melt Extrusion: The Role of Magnesium Aluminometasilicate (Neusilin® US2)

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Physical Blends
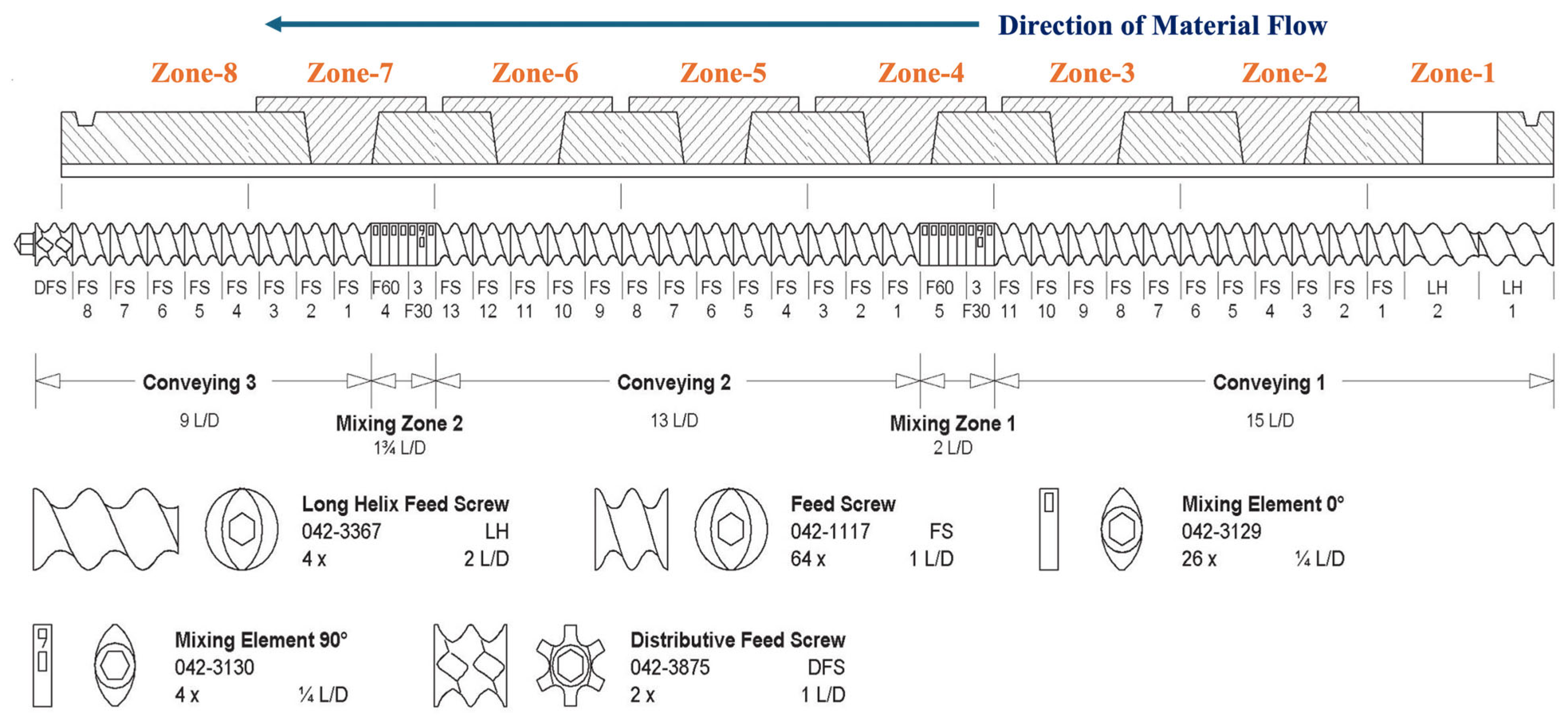
2.3. Hot Melt Extrusion (HME)
2.4. Modulated Differential Scanning Calorimetry (mDSC)
2.5. Powder X-Ray Diffraction (PXRD)
2.6. Downstream Processing
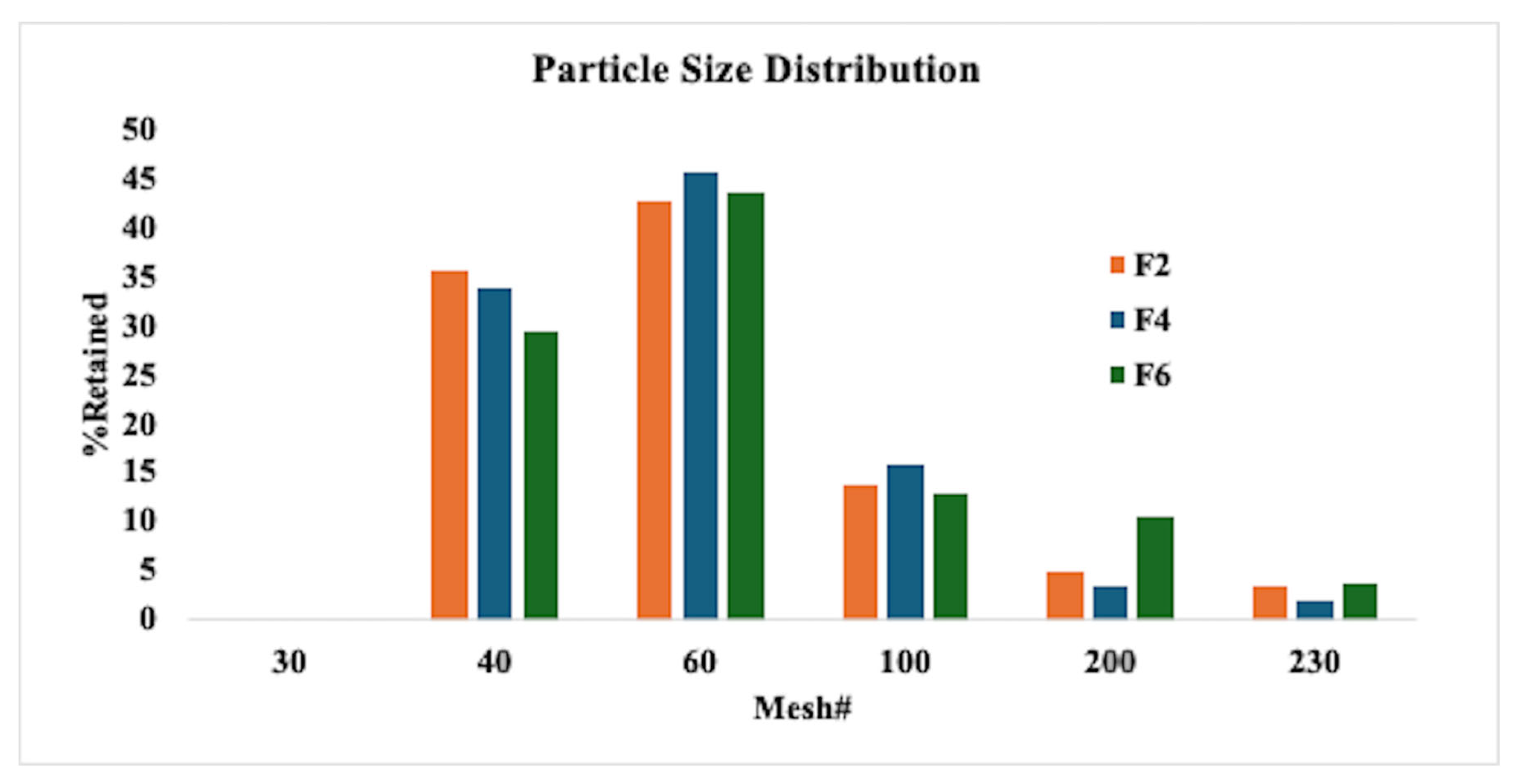
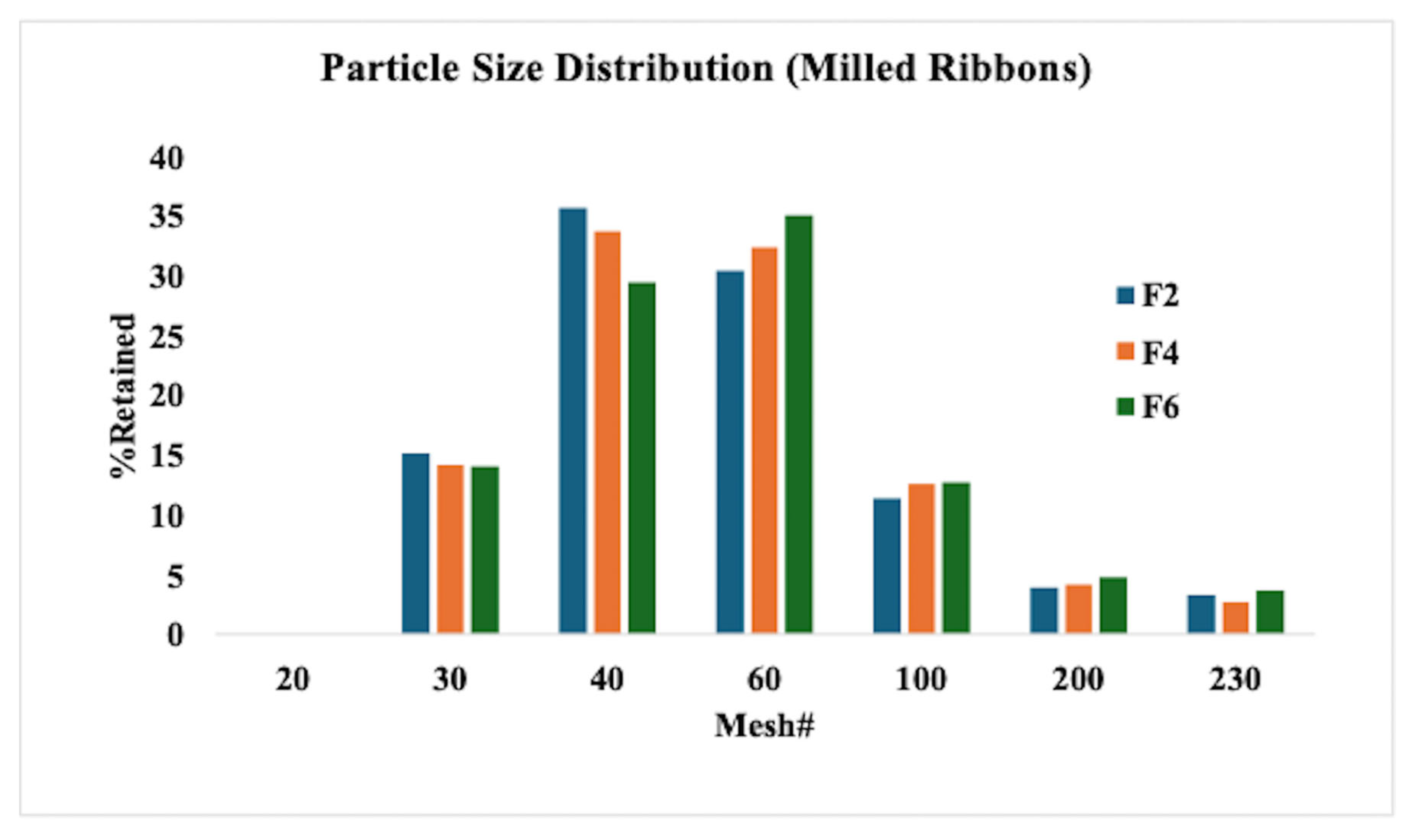
2.6.1. Milling and Particle Size Distribution
2.6.2. Blending for Roller Compaction Process (Intra-Granular Portion)
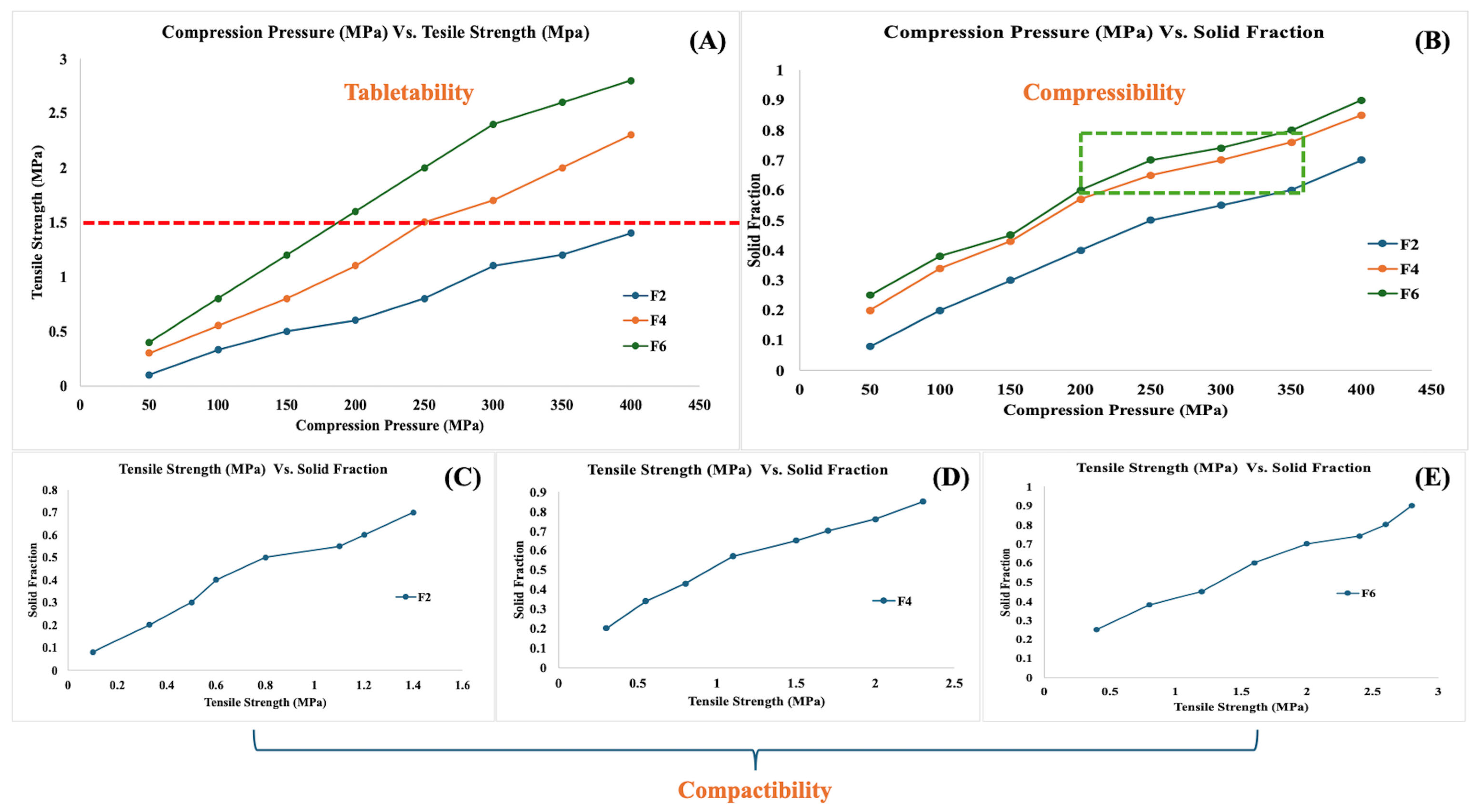
2.6.3. Compressibility, Tabletability, and Compactability (CTC) Profile
2.6.4. Dry Granulation by Roller Compaction
2.6.5. Characterization of Blend
2.6.6. Final Blending and Tablet Compression
2.7. Characterization of Tablets
2.7.1. Weight Variation, Thickness, Hardness, Friability, Disintegration
2.7.2. Assay and High-Performance Liquid Chromatography (HPLC)
2.7.3. In Vitro Drug Release
2.7.4. mDSC and Fourier Transform Infrared Spectroscopy (FTIR)
2.8. Stability Studies
3. Results and Discussion
3.1. Hot Melt Extrusion Process
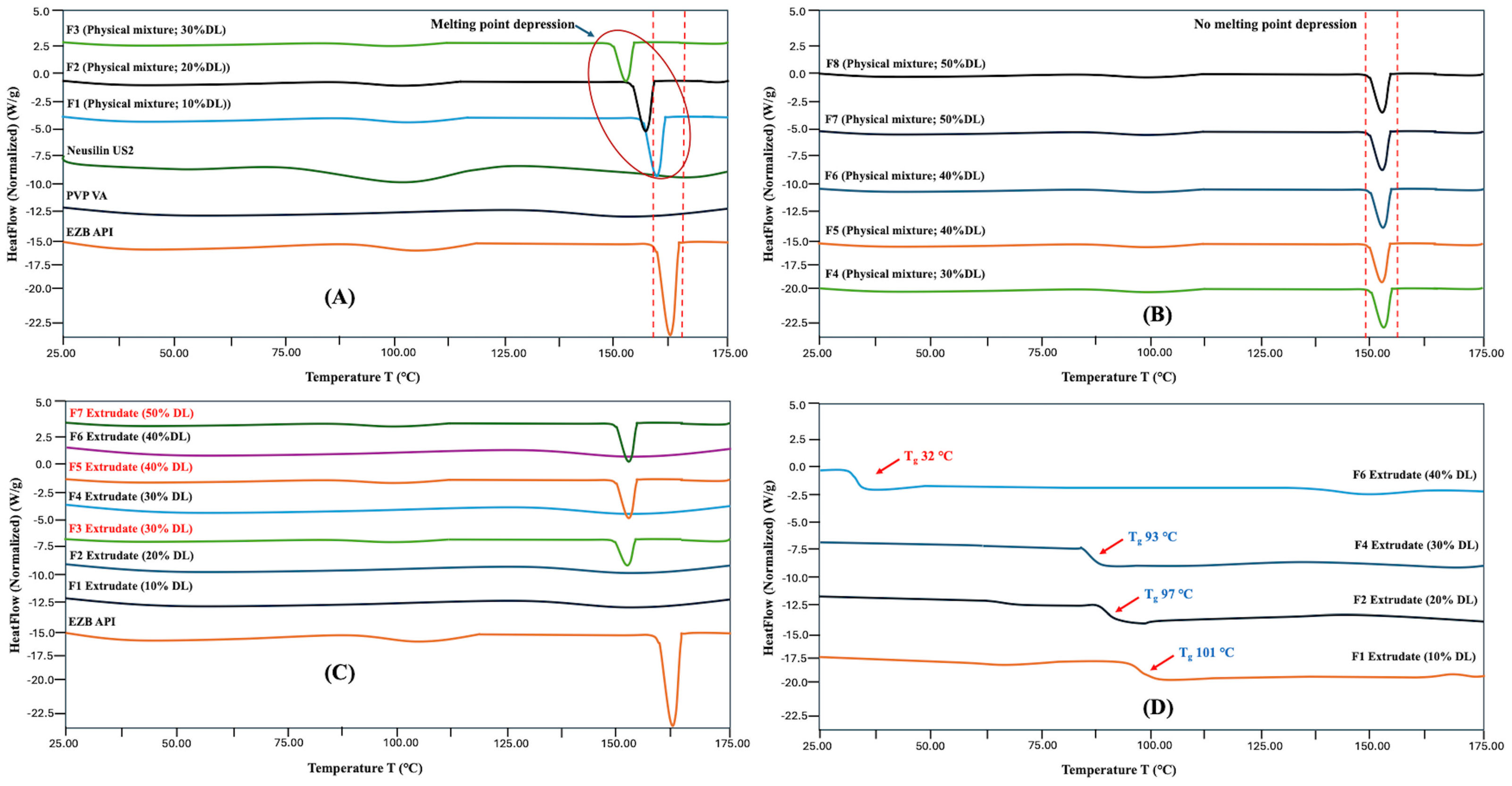
3.2. Characterization of HME Extrudates
3.3. CTC Profile
3.4. Roller Compaction and Characterization of Milled Ribbons
3.5. Compression of Tablets and Characterization
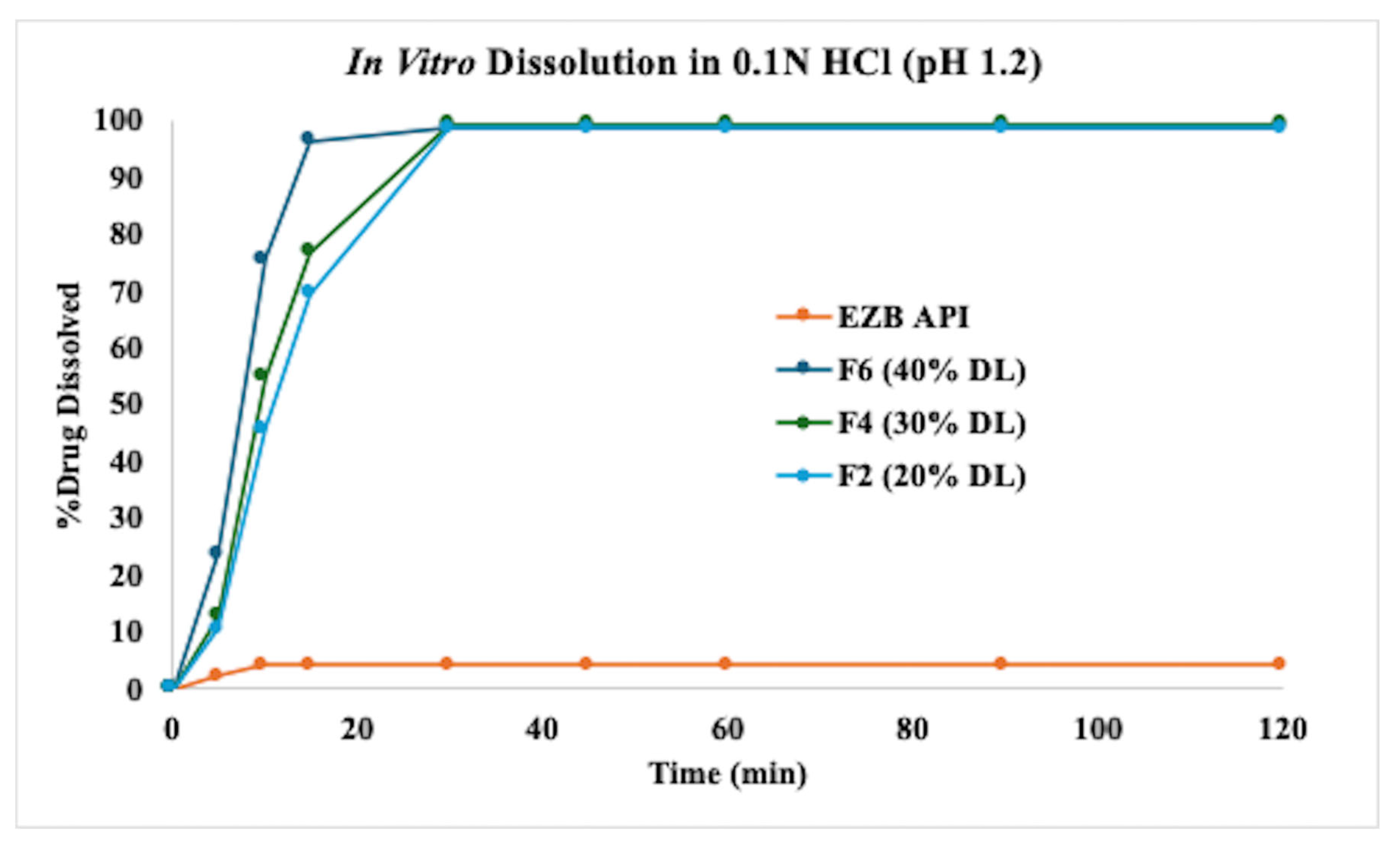
3.6. In Vitro Dissolution Profiles
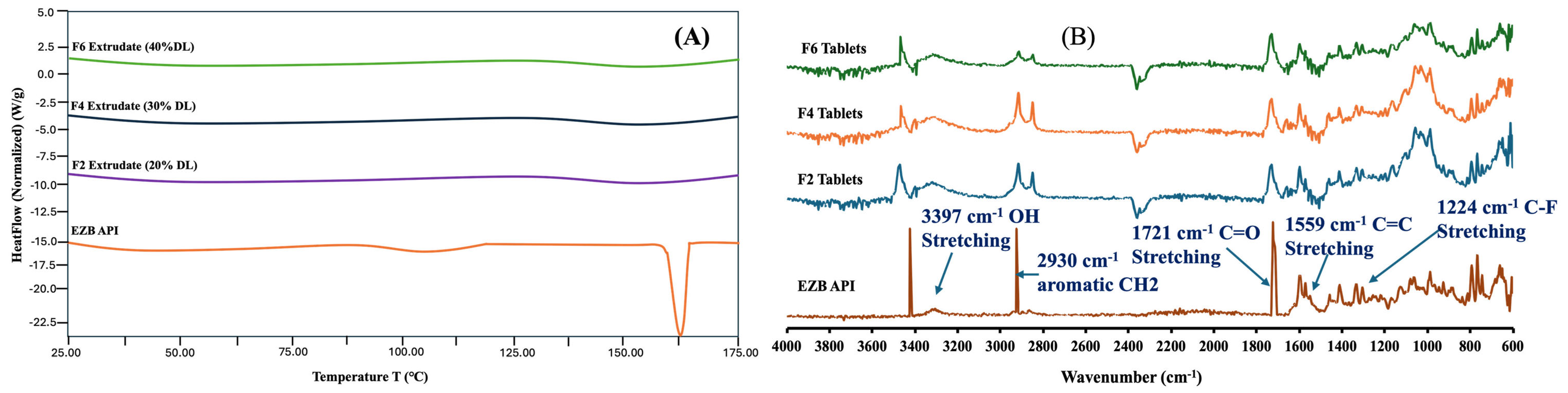
3.7. DSC and FTIR
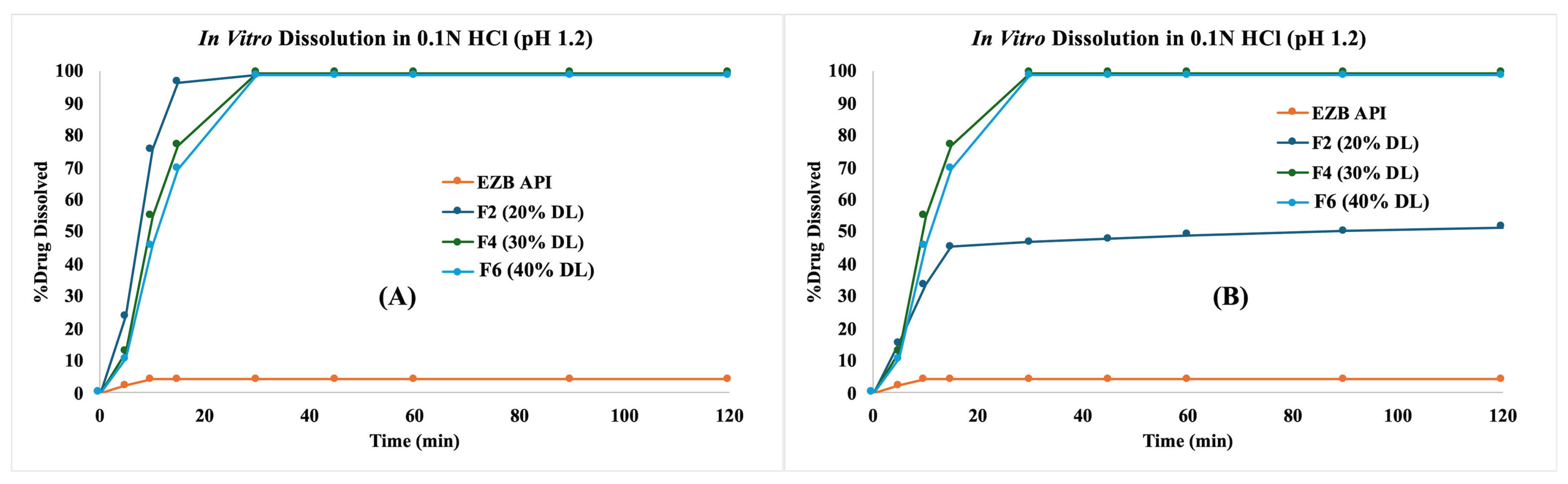
3.8. Stability Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yu, D.; Hoag, S.W. The Impact of Diluents on the Compaction, Dissolution, and Physical Stability of Amorphous Solid Dispersion Tablets. Int. J. Pharm. 2024, 654, 123924. [Google Scholar] [CrossRef] [PubMed]
- Deac, A.; Luebbert, C.; Qi, Q.; Courtney, R.M.; Indulkar, A.S.; Gao, Y.; Zhang, G.G.Z.; Sadowski, G.; Taylor, L.S. Dissolution Mechanisms of Amorphous Solid Dispersions: Application of Ternary Phase Diagrams to Explain Release Behavior. Mol. Pharm. 2024, 21, 1900–1918. [Google Scholar] [CrossRef] [PubMed]
- Bapat, P.; Paul, S.; Tseng, Y.C.; Taylor, L.S. Interplay of Drug-Polymer Interactions and Release Performance for HPMCAS-Based Amorphous Solid Dispersions. Mol. Pharm. 2024, 21, 1466–1478. [Google Scholar] [CrossRef] [PubMed]
- Moseson, D.E.; Tran, T.B.; Karunakaran, B.; Ambardekar, R.; Hiew, T.N. Trends in Amorphous Solid Dispersion Drug Products Approved by the U.S. Food and Drug Administration between 2012 and 2023. Int. J. Pharm. X 2024, 7, 100259. [Google Scholar] [CrossRef] [PubMed]
- Rusdin, A.; Mohd Gazzali, A.; Ain Thomas, N.; Megantara, S.; Aulifa, D.L.; Budiman, A.; Muchtaridi, M. Advancing Drug Delivery Paradigms: Polyvinyl Pyrolidone (PVP)-Based Amorphous Solid Dispersion for Enhanced Physicochemical Properties and Therapeutic Efficacy. Polymers 2024, 16, 286. [Google Scholar] [CrossRef] [PubMed]
- Nyavanandi, D.; Mandati, P.; Narala, S.; Alzahrani, A.; Kolimi, P.; Vemula, S.K.; Repka, M.A. Twin Screw Melt Granulation: A Single Step Approach for Developing Self-Emulsifying Drug Delivery System for Lipophilic Drugs. Pharmaceutics 2023, 15, 2267. [Google Scholar] [CrossRef] [PubMed]
- Tambe, S.; Jain, D.; Meruva, S.K.; Rongala, G.; Juluri, A.; Nihalani, G.; Mamidi, H.K.; Nukala, P.K.; Bolla, P.K. Recent Advances in Amorphous Solid Dispersions: Preformulation, Formulation Strategies, Technological Advancements and Characterization. Pharmaceutics 2022, 14, 2203. [Google Scholar] [CrossRef] [PubMed]
- Mamidi, H.K.; Palekar, S.; Nukala, P.K.; Mishra, S.M.; Patki, M.; Fu, Y.; Supner, P.; Chauhan, G.; Patel, K. Process Optimization of Twin-Screw Melt Granulation of Fenofibrate Using Design of Experiment (DoE). Int. J. Pharm. 2021, 593, 120101. [Google Scholar] [CrossRef] [PubMed]
- Mamidi, H.K.; Rohera, B.D. Application of Thermodynamic Phase Diagrams and Gibbs Free Energy of Mixing for Screening of Polymers for Their Use in Amorphous Solid Dispersion Formulation of a Non-Glass-Forming Drug. J. Pharm. Sci. 2021, 110, 2703–2717. [Google Scholar] [CrossRef] [PubMed]
- Mamidi, H.K.; Mishra, S.M.; Rohera, B.D. Determination of Maximum Flowable Liquid-Loading Potential of Neusilin® US2 and Investigation of Compressibility and Compactibility of Its Liquisolid Blends with PEG (400). J. Drug Deliv. Sci. Technol. 2019, 54, 101285. [Google Scholar] [CrossRef]
- Palekar, S.; Mamidi, H.K.; Guo, Y.; Vartak, R.; Patel, K. Corroborating Various Material-Sparing Techniques with Hot Melt Extrusion for the Preparation of Triclabendazole Amorphous Solid Dispersions. Int. J. Pharm. 2023, 640, 122989. [Google Scholar] [CrossRef] [PubMed]
- Mamidi, H.; Palekar, S.; Patel, H.; Nukala, P.K.; Patel, K. Formulation Strategies for the Development of High Drug-Loaded Amorphous Solid Dispersions. Drug Discov. Today 2023, 28, 103806. [Google Scholar] [CrossRef] [PubMed]
- Kelsall, K.N.; Schubiner, R.O.; Schenck, L.; Frank, D.S.; Matzger, A.J. Enhancing the Acidity of Polymers for Improved Stabilization of Amorphous Solid Dispersions: Protonation of Weakly Basic Compounds. ACS Appl. Polym. Mater. 2024, 6, 1592–1598. [Google Scholar] [CrossRef]
- Nyavanandi, D.; Mandati, P.; Vidiyala, N.; Parupathi, P.; Kolimi, P.; Mamidi, H.K. Enhancing Patient-Centric Drug Development: Coupling Hot Melt Extrusion with Fused Deposition Modeling and Pressure-Assisted Microsyringe Additive Manufacturing Platforms with Quality by Design. Pharmaceutics 2024, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, X.; Foderà, V.; Larsson, P.; Schaal, Z.; Bergström, C.A.S.; Löbmann, K.; Kabedev, A. Analysis of Stabilization Mechanisms in β-Lactoglobulin-Based Amorphous Solid Dispersions by Experimental and Computational Approaches. Eur. J. Pharm. Sci. 2024, 192, 106639. [Google Scholar] [CrossRef] [PubMed]
- Petrova, S.P.; Mohamed, M.A.; Wu, H.; Taylor, L.S.; Edgar, K.J. Threading the Needle: Achieving Simplicity and Performance in Cellulose Alkanoate ω-Carboxyalkanoates for Amorphous Solid Dispersion. Carbohydr. Polym. 2024, 333, 121988. [Google Scholar] [CrossRef] [PubMed]
- Petersen, E.F.; Larsen, B.S.; Nielsen, R.B.; Pijpers, I.; Versweyveld, D.; Holm, R.; Tho, I.; Snoeys, J.; Nielsen, C.U. Co-Release of Paclitaxel and Encequidar from Amorphous Solid Dispersions Increase Oral Paclitaxel Bioavailability in Rats. Int. J. Pharm. 2024, 654, 123965. [Google Scholar] [CrossRef] [PubMed]
- Pisay, M.; Padya, B.S.; Mutalik, S.; Koteshwara, K.B. Stability Challenges of Amorphous Solid Dispersions of Drugs: A Critical Review on Mechanistic Aspects. Crit. Rev. Ther. Drug Carrier Syst. 2024, 41, 45–94. [Google Scholar] [CrossRef] [PubMed]
- Ugur, B.E.; Caggiano, N.J.; Monson, S.; Bechtold, A.G.; Seo, Y.; Prud’homme, R.K.; Priestley, R.D.; Webb, M.A. Molecular Interactions Underlying Dissolution Trends in Cannabidiol-Polymer Amorphous Solid Dispersions. Macromolecules 2024, 57, 8287–8297. [Google Scholar] [CrossRef]
- Tripathi, D.; Manjunatha Prabhu, B.H.; Sahoo, J.; Kumari, J. Navigating the Solution to Drug Formulation Problems at Research and Development Stages by Amorphous Solid Dispersion Technology. Recent. Adv. Drug Deliv. Formul. 2024, 18, 79–99. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, N.; Charbe, N.; Plano, D.; Al Shoyaib, A.; Pal, A.; Boyce, H.; Zhao, L.; Wu, F.; Polli, J.; Dressman, J.; et al. A Physiologically Based Biopharmaceutics Modeling (PBBM) Framework for Characterizing Formulation-Dependent Food Effects: Paving the Road towards Fed Virtual BE Studies for Itraconazole Amorphous Solid Dispersions. Eur. J. Pharm. Sci. 2025, 209, 107047. [Google Scholar] [CrossRef] [PubMed]
- Wdowiak, K.; Tajber, L.; Miklaszewski, A.; Cielecka-Piontek, J. Sweeteners Show a Plasticizing Effect on PVP K30—A Solution for the Hot-Melt Extrusion of Fixed-Dose Amorphous Curcumin-Hesperetin Solid Dispersions. Pharmaceutics 2024, 16, 659. [Google Scholar] [CrossRef] [PubMed]
- Nyavanandi, D.; Narala, S.; Repka, M.A.; Nyavanandi, D.; Narala, S.; Repka, M.A. Hot-Melt Extrusion Paired Fused Deposition Modeling 3D Printing: Development of Pharmaceutical Medications; Springer: New York, NY, USA, 2024; pp. 169–194. [Google Scholar] [CrossRef]
- Bolla, G.; Sarma, B.; Nangia, A.K. Crystal Engineering of Pharmaceutical Cocrystals in the Discovery and Development of Improved Drugs. Chem. Rev. 2022, 122, 11514–11603. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Zhang, S.; Wang, L.; Tao, X. Recent Advances in Polymorph Discovery Methods of Organic Crystals. Cryst. Growth Des. 2023, 23, 637–654. [Google Scholar] [CrossRef]
- Suresh, K.; Mehta, T.; Thakrar, V.; Sharma, R.G. Innovative Strategies in Generic Drug Development: The Role of Polymorph, Amorphous, Pseudopolymorph, and Cocrystal Solid Forms. Cryst. Growth Des. 2025, 25, 1282–1292. [Google Scholar] [CrossRef]
- Wu, D.; Zhang, B.; Yao, Q.; Hou, B.; Zhou, L.; Xie, C.; Gong, J.; Hao, H.; Chen, W. Evaluation on Cocrystal Screening Methods and Synthesis of Multicomponent Crystals: A Case Study. Cryst. Growth Des. 2021, 21, 4531–4546. [Google Scholar] [CrossRef]
- Baumgartner, A.; Dobaj, N.; Planinšek, O. Investigating the Influence of Processing Conditions on Dissolution and Physical Stability of Solid Dispersions with Fenofibrate and Mesoporous Silica. Pharmaceutics 2024, 16, 575. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Ono, T.; Hayashi, Y.; Kumada, S.; Onuki, Y. Use of Time-Domain NMR for 1H T1 Relaxation Measurement and Fitting Analysis in Homogeneity Evaluation of Amorphous Solid Dispersion. J. Pharm. Sci. 2024, 113, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, X.; Tozzetti, M.; Arnous, A.; Leng, D.; Foderà, V.; Löbmann, K. Investigating the Influence of Protein Secondary Structure on the Dissolution Behavior of β-Lactoglobulin-Based Amorphous Solid Dispersions. Int. J. Pharm. 2024, 653, 123887. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Hu, E.; Zhang, C.; Gao, Y.; Qian, S.; Heng, W.; Zhang, J.; Wei, Y. Hot “Dissolving” Extrusion of Lurasidone with Natural Liquid Small Molecule for Amorphous Solid Dispersion Based Self-Assembled Submicron Emulsion. Adv. Healthc. Mater. 2024, 13, 2302488. [Google Scholar] [CrossRef] [PubMed]
- Leon, A.S.C.; Waterman, K.C.; Wang, G.; Wang, L.; Cai, T.; Zhang, X. Accelerated Stability Modeling of Recrystallization from Amorphous Solid Dispersions: A Griseofulvin/HPMC-AS Case Study. Int. J. Pharm. 2024, 657, 124189. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Wang, H.; Jia, J.; Song, D.; Yang, L.; Tang, X.; He, Z.; Fu, Q. Moisture-Induced Limited Recrystallization May Not Reduce the Dissolution of Amorphous Solid Dispersions: A Case of Nitrendipine. J. Ind. Eng. Chem. 2024, 137, 235–242. [Google Scholar] [CrossRef]
- Song, S.; Yao, X.; Wang, C.; Sun, C.C.; Siegel, R.A. Delaying the First Nucleation Event of Amorphous Solid Dispersions above the Polymer Overlap Concentration (C*): PVP and PVPVA in Posaconazole. J. Pharm. Sci. 2025, 114, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Rosiak, N.; Tykarska, E.; Cielecka-Piontek, J. Enhanced Antioxidant and Neuroprotective Properties of Pterostilbene (Resveratrol Derivative) in Amorphous Solid Dispersions. Int. J. Mol. Sci. 2024, 25, 2774. [Google Scholar] [CrossRef] [PubMed]
- Aulifa, D.L.; Al Shofwan, A.A.; Megantara, S.; Fakih, T.M.; Budiman, A. Elucidation of Molecular Interactions Between Drug–Polymer in Amorphous Solid Dispersion by a Computational Approach Using Molecular Dynamics Simulations. Adv. Appl. Bioinform. Chem. 2024, 17, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Schoeman, C.; van Niekerk, S.; Liebenberg, W.; Hamman, J. Cyclodextrin Inclusion Complex and Amorphous Solid Dispersions as Formulation Approaches for Enhancement of Curcumin’s Solubility and Nasal Epithelial Membrane Permeation. Future J. Pharm. Sci. 2024, 10, 85. [Google Scholar] [CrossRef]
- Trenkenschuh, E.; Blattner, S.M.; Hirsh, D.; Hoffmann, R.; Luebbert, C.; Schaefer, K. Development of Ternary Amorphous Solid Dispersions Manufactured by Hot-Melt Extrusion and Spray-Drying—Comparison of In Vitro and In Vivo Performance. Mol. Pharm. 2024, 21, 1309–1320. [Google Scholar] [CrossRef] [PubMed]
- Mora-Castaño, G.; Millán-Jiménez, M.; Niederquell, A.; Schönenberger, M.; Shojaie, F.; Kuentz, M.; Caraballo, I. Amorphous Solid Dispersion of a Binary Formulation with Felodipine and HPMC for 3D Printed Floating Tablets. Int. J. Pharm. 2024, 658, 124215. [Google Scholar] [CrossRef] [PubMed]
- Almeida, H.; Teixeira, N.; Sarmento, B.; Vasconcelos, T. Freeze-Drying Cycle Optimization of an Amorphous Solid Dispersion of Resveratrol. Eur. J. Pharm. Sci. 2024, 200, 106855. [Google Scholar] [CrossRef] [PubMed]
- Samsoen, S.; Dudognon, É.; Le Fer, G.; Fournier, D.; Woisel, P.; Affouard, F. Impact of the Polymer Dispersity on the Properties of Curcumin/Polyvinylpyrrolidone Amorphous Solid Dispersions. Int. J. Pharm. 2024, 653, 123895. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Fina, F.; Rossignolo, F.; Kim, S.H.; Lee, H.; Jeong, K.; Xu, X.; Pignaffo, C.; Yang, C.; Koo, J.; et al. Evaluation of a Spray-Dried Amorphous Solid Dispersion Formulation of ID11916, a New Molecular Entity with Dual Inhibition Mechanisms Targeting the Androgen Receptor and Phosphodiesterase Type-5. J. Pharm. Investig. 2024, 54, 317–327. [Google Scholar] [CrossRef]
- He, R.; Lamm, M.S.; Brunskill, A.; Axnanda, S.; Li, Y. Impact of Processing Methods on the Physico-Chemical Properties of Posaconazole Amorphous Solid Dispersions. Pharm. Res. 2024, 41, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Rosiak, N.; Tykarska, E.; Cielecka-Piontek, J. Myricetin Amorphous Solid Dispersions—Antineurodegenerative Potential. Molecules 2024, 29, 1287. [Google Scholar] [CrossRef] [PubMed]
- Huzjak, T.; Jakasanovski, O.; Berginc, K.; Puž, V.; Zajc-Kreft, K.; Jeraj; Janković, B. Overcoming Drug Impurity Challenges in Amorphous Solid Dispersion with Rational Development of Biorelevant Dissolution-Permeation Method. Eur. J. Pharm. Sci. 2024, 192, 106655. [Google Scholar] [CrossRef] [PubMed]
- Lennernäs, H.; Brisander, M.; Liljebris, C.; Jesson, G.; Andersson, P. Enhanced Bioavailability and Reduced Variability of Dasatinib and Sorafenib with a Novel Amorphous Solid Dispersion Technology Platform. Clin. Pharmacol. Drug Dev. 2024, 13, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Podgorbunskikh, E.; Kuskov, T.; Rychkov, D.; Lomovskii, O.; Bychkov, A. Mechanical Amorphization of Chitosan with Different Molecular Weights. Polymers 2022, 14, 4438. [Google Scholar] [CrossRef] [PubMed]
- Szafraniec-Szczęsny, J.; Antosik-Rogóż, A.; Kurek, M.; Gawlak, K.; Górska, A.; Peralta, S.; Knapik-Kowalczuk, J.; Kramarczyk, D.; Paluch, M.; Jachowicz, R. How Does the Addition of Kollidon®VA64 Inhibit the Recrystallization and Improve Ezetimibe Dissolution from Amorphous Solid Dispersions? Pharmaceutics 2021, 13, 147. [Google Scholar] [CrossRef] [PubMed]
- Knapik, J.; Wojnarowska, Z.; Grzybowska, K.; Hawelek, L.; Sawicki, W.; Wlodarski, K.; Markowski, J.; Paluch, M. Physical Stability of the Amorphous Anticholesterol Agent (Ezetimibe): The Role of Molecular Mobility. Mol. Pharm. 2014, 11, 4280–4290. [Google Scholar] [CrossRef] [PubMed]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric Amorphous Solid Dispersions: A Review of Amorphization, Crystallization, Stabilization, Solid-State Characterization, and Aqueous Solubilization of Biopharmaceutical Classification System Class II Drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.R.; Mutalik, S.; Reddy, S. Once-Daily Sustained-Release Matrix Tablets of Nicorandil: Formulation and in Vitro Evaluation. AAPS PharmSciTech 2003, 4, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Bhimani, D.; Patel, J.; Patel, D. Solid-State Characterization and Dissolution Properties of Ezetimibe-Cyclodextrins Inclusion Complexes. J. Incl. Phenom. Macrocycl. Chem. 2008, 60, 241–251. [Google Scholar] [CrossRef]
- Tres, F.; Treacher, K.; Booth, J.; Hughes, L.P.; Wren, S.A.C.; Aylott, J.W.; Burley, J.C. Indomethacin-Kollidon VA64 Extrudates: A Mechanistic Study of PH-Dependent Controlled Release. Mol. Pharm. 2016, 13, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Parmar, T.; Kadu, P.; Kale, P. Critical Strategies for Drug Precipitation Inhibition: A Review with the Focus on Poorly Soluble Drugs. Curr. Drug Deliv. 2022, 20, 497–507. [Google Scholar] [CrossRef]
- Sugandha, K.; Kaity, S.; Mukherjee, S.; Isaac, J.; Ghosh, A. Solubility Enhancement of Ezetimibe by a Cocrystal Engineering Technique. Cryst. Growth Des. 2014, 14, 4475–4486. [Google Scholar] [CrossRef]
- Martins, G.A.G.; Murakami, F.S.; Sangoi, M.S.; Todeschini, V.S.; Bernardi, L.R.; Oliveira, P. Characterization, Purity Determination and Decomposition Kinetics of Ezetimibe Under Non-Isothermal Conditions. Curr. Pharm. Anal. 2018, 15, 327–332. [Google Scholar] [CrossRef]
- Prajapati, P.; Pandey, J.; Shimpi, M.R.; Srivastava, A.; Tandon, P.; Velaga, S.P.; Sinha, K. Combined Spectroscopic and Quantum Chemical Studies of Ezetimibe. J. Mol. Struct. 2016, 1125, 193–203. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation# | EZB (%w/w) | Kollidon VA 64 (%) | Neusilin US2 (%) |
|---|---|---|---|
| F-001 | 10 | 90 | - |
| F-002 | 20 | 80 | - |
| F-003 | 30 | 70 | - |
| F-004 | 30 | 60 | 10 |
| F-005 | 40 | 50 | 10 |
| F-006 | 40 | 45 | 15 |
| F-007 | 50 | 35 | 15 |
| F-008 | 50 | 30 | 20 |
| Materials | %w/w | ||
|---|---|---|---|
| F2 | F4 | F6 | |
| Intragranular | |||
| HME milled extrudates | 50.00 | 33.33 | 25.00 |
| Avicel PH102 (microcrystalline cellulose) | 29.00 | 40.00 | 45.00 |
| SuperTab 21AN (lactose anhydrous) | 14.00 | 19.67 | 23.00 |
| Aerosil 200 (colloidal silicon dioxide) | 1.00 | 1.00 | 1.00 |
| AC-DI-SOL (Croscarmellose sodium) | 3.00 | 3.00 | 3.00 |
| Magnesium stearate | 0.50 | 0.50 | 0.50 |
| Total Intragranular Portion | 97.50 | 97.50 | 97.50 |
| Extragranular | |||
| AC-DI-SOL (Croscarmellose sodium) | 2.00 | 2.00 | 2.00 |
| Magnesium stearate | 0.50 | 0.50 | 0.50 |
| Total Extragranular Portion | 2.50 | 2.50 | 2.50 |
| Total | 100.00 | 100.00 | 100.00 |
| Process Parameters | F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 |
|---|---|---|---|---|---|---|---|---|
| Zone 1 (C) | 25 | 25 | 25 | 25 | 25 | 25 | 25 | 25 |
| Zone 2–8 (C) | 170 | 170 | 170 | 175 | 175 | 180 | 180 | upto 240 |
| Die (C) | 165 | 165 | 165 | 170 | 170 | 175 | 175 | upto 235 |
| Feed Rate (g/min) | 2.5–3.5 | 2.5–3.5 | 2.5–3.5 | 2.5–3.5 | 2.5–3.5 | 2.5–3.5 | 2.5–3.5 | 2.5–3.5 |
| Screw Speed (RPM) | 50 | 50 | 50 | 50 | 50 | 50 | 50 | 50 |
| Process torque (%) | 45–50 | 40–45 | 35–40 | 40–45 | 35–40 | 45–50 | 40–45 | 100 |
| Die Nozzle (mm) | 2.5 | 2.5 | 2.5 | 2.5 | 2.5 | 2.5 | 2.5 | 2.5 |
| Successful Extrusion (Yes/No) | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No |
| Formulation | Roll Force (kN) | Roll Speed (rpm) | Screw Speed (rpm) | Screw Current (Amps) | True Density (g/cm) | Envelop Density (g/cm) | Solid Fraction |
|---|---|---|---|---|---|---|---|
| F2 | 13 | 1 | 35–45 | 0.5–0.6 | 1.53 | 0.94 | 0.61 |
| F4 | 13 | 1 | 30–35 | 0.35–0.45 | 1.54 | 1.12 | 0.73 |
| F6 | 13 | 1 | 20–25 | 0.25–0.30 | 1.54 | 1.3 | 0.84 |
| Formulation | Manufacturing Stage | Carr’s Index (%) | Hausner’s Ratio | Flow Function (FF) |
|---|---|---|---|---|
| F2 | HME Milled Powder | 36.45 (Very poor flow) | 1.49 | 2.45 (Poor flow) |
| Pre-Roller Compaction Blend | 27.32 (Poor flow) | 1.37 | 5.66 (Intermediate flow) | |
| Milled Ribbons | 18.36 (Fair flow) | 1.21 | 7.45 (Intermediate flow) | |
| F4 | HME Milled Powder | 36.11 (Very poor flow) | 1.49 | 3.11 (Poor flow) |
| Pre-Roller Compaction Blend | 22.51 (Passable flow) | 1.31 | 8.67 (Intermediate flow) | |
| Milled Ribbons | 16.23 (Fair flow) | 1.22 | 12.08 (Good flow) | |
| F6 | HME Milled Powder | 36.21 (Very poor flow) | 1.47 | 2.91 (Poor flow) |
| Pre-Roller Compaction Blend | 19.32 (Fair flow) | 1.20 | 9.77 (Intermediate flow) | |
| Milled Ribbons | 14.27 (Free flow) | 1.14 | 14.89 (Good flow) |
| Formulation | Weight Variation (mg) | Thickness (mm) | Hardness (kp) | Friability (%) | Disintegration (s) | Assay (%) |
|---|---|---|---|---|---|---|
| F2 | 100.12 ± 0.72 | 2.49 ± 0.21 | 7.2 ± 1.7 | 0.43 | 180 | 98.23 ± 0.54 |
| F4 | 99.23 ± 1.21 | 2.55 ± 0.12 | 7.3 ± 1.3 | 0.34 | 240 | 98.67 ± 0.34 |
| F6 | 101.45 ± 1.67 | 2.63 ± 0.08 | 7.5 ± 0.9 | 0.41 | 270 | 99.31 ± 0.69 |
| Stability Time Point | Formulation | Weight Variation (mg) | Hardness (kp) | Friability (%) | Disintegration (s) | Assay (%) |
|---|---|---|---|---|---|---|
| Initial (0 M) | F2 | 100.12 ± 0.72 | 7.2 ± 1.7 | 0.43 | 180 | 98.23 ± 0.54 |
| F4 | 99.23 ± 1.21 | 7.3 ± 1.3 | 0.34 | 240 | 98.67 ± 0.34 | |
| F6 | 101.45 ± 1.67 | 7.5 ± 0.9 | 0.41 | 270 | 99.31 ± 0.69 | |
| 3 M | F2 | 98.34 ± 1.21 | 6.8 ± 0.7 | 0.54 | 196 | 99.15 ± 1.23 |
| F4 | 99.45 ± 0.67 | 7.2 ± 1.6 | 0.41 | 235 | 100.45 ± 0.67 | |
| F6 | 100.33 ± 1.09 | 7.1 ± 1.1 | 0.45 | 260 | 97.63 ± 0.32 | |
| 6 M | F2 | 98.11 ± 0.11 | 6.4 ± 1.6 | 0.57 | 187 | 97.11 ± 1.31 |
| F4 | 100.01 ± 0.47 | 6.8 ± 0.5 | 0.44 | 238 | 101.21 ± 0.87 | |
| F6 | 99.65 ± 2.45 | 6.7 ± 1.2 | 0.41 | 265 | 100.09 ± 1.67 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Published by MDPI on behalf of the Österreichische Pharmazeutische Gesellschaft. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vidiyala, N.; Sunkishala, P.; Parupathi, P.; Mandati, P.; Mantena, S.K.; Kasu, R.R.R.; Nyavanandi, D. High Drug Loading of Amorphous Solid Dispersion by Hot Melt Extrusion: The Role of Magnesium Aluminometasilicate (Neusilin® US2). Sci. Pharm. 2025, 93, 30. https://doi.org/10.3390/scipharm93030030
Vidiyala N, Sunkishala P, Parupathi P, Mandati P, Mantena SK, Kasu RRR, Nyavanandi D. High Drug Loading of Amorphous Solid Dispersion by Hot Melt Extrusion: The Role of Magnesium Aluminometasilicate (Neusilin® US2). Scientia Pharmaceutica. 2025; 93(3):30. https://doi.org/10.3390/scipharm93030030
Chicago/Turabian StyleVidiyala, Nithin, Pavani Sunkishala, Prashanth Parupathi, Preethi Mandati, Srujan Kumar Mantena, Raghu Rami Reddy Kasu, and Dinesh Nyavanandi. 2025. "High Drug Loading of Amorphous Solid Dispersion by Hot Melt Extrusion: The Role of Magnesium Aluminometasilicate (Neusilin® US2)" Scientia Pharmaceutica 93, no. 3: 30. https://doi.org/10.3390/scipharm93030030
APA StyleVidiyala, N., Sunkishala, P., Parupathi, P., Mandati, P., Mantena, S. K., Kasu, R. R. R., & Nyavanandi, D. (2025). High Drug Loading of Amorphous Solid Dispersion by Hot Melt Extrusion: The Role of Magnesium Aluminometasilicate (Neusilin® US2). Scientia Pharmaceutica, 93(3), 30. https://doi.org/10.3390/scipharm93030030