
In Silico Activity Prediction and Docking Studies of the Binding Mechanisms of Levofloxacin Structure Derivatives to Active Receptor Sites of Bacterial Type IIA Topoisomerases

 ,
,  ,
, 
 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fluoroquinolone Samples
2.2. QSAR
2.3. PASS
2.4. Software of Molecular Docking
2.5. Equipment for Tribochemical Processing
2.6. Fourier Transform IR Spectroscopy
2.7. Optical Microscopy (OM)
2.8. Statistical Data Processing
3. Results and Discussion
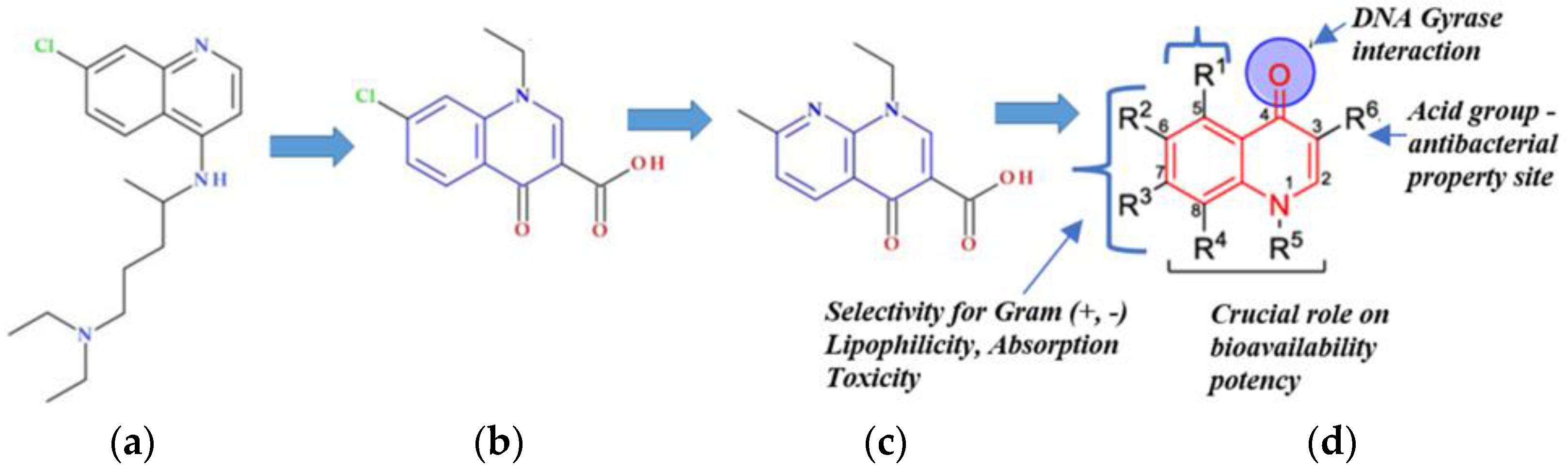
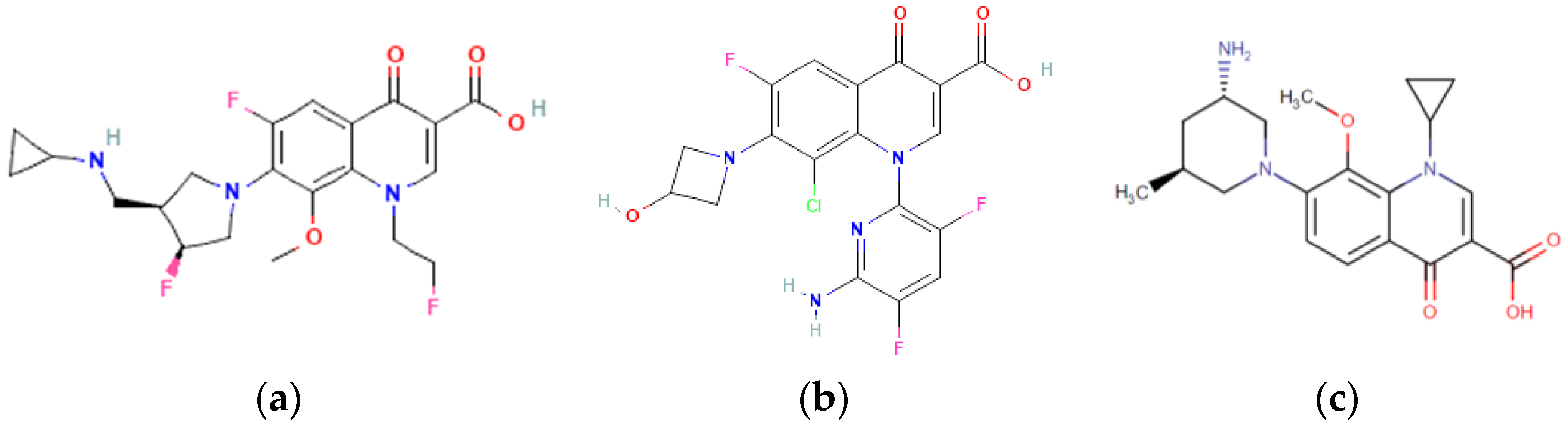
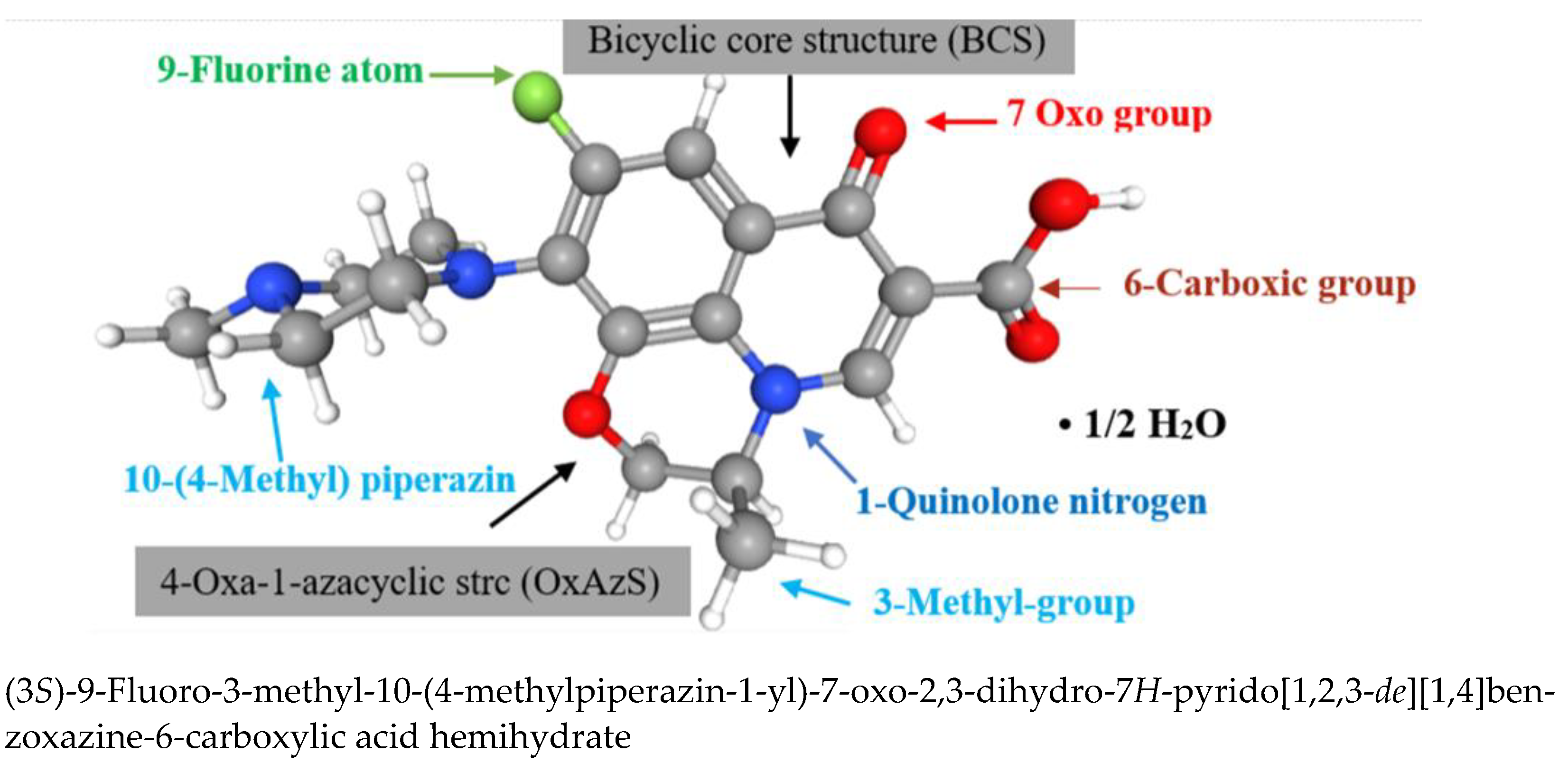
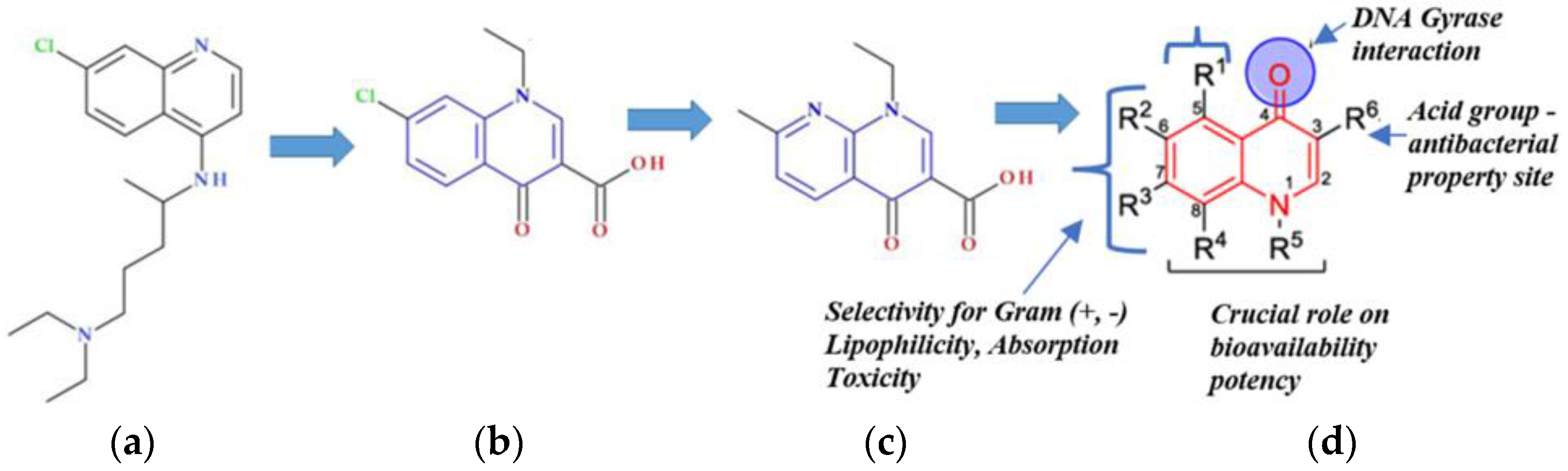
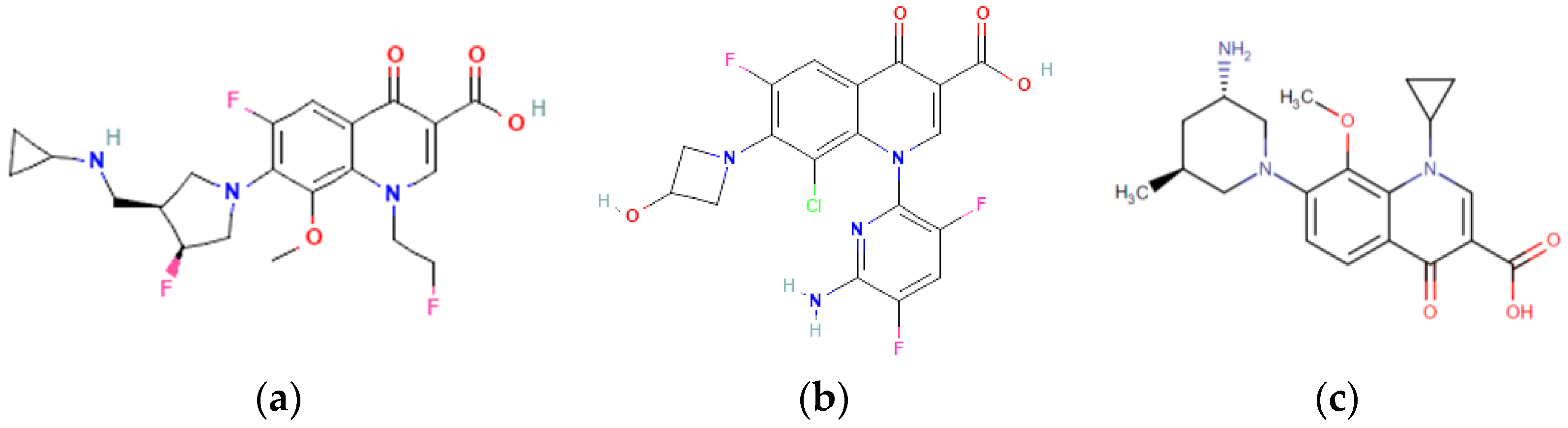
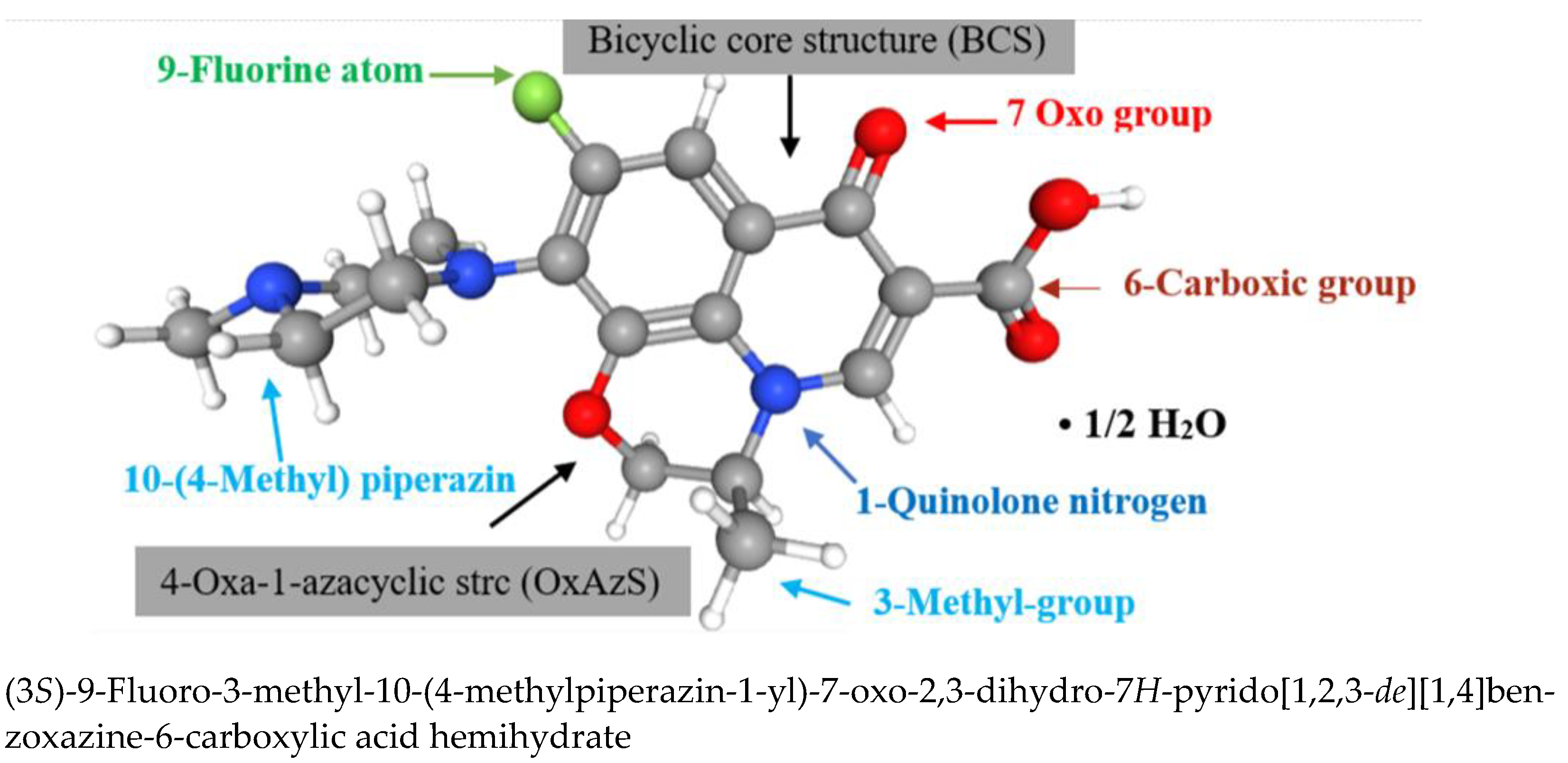
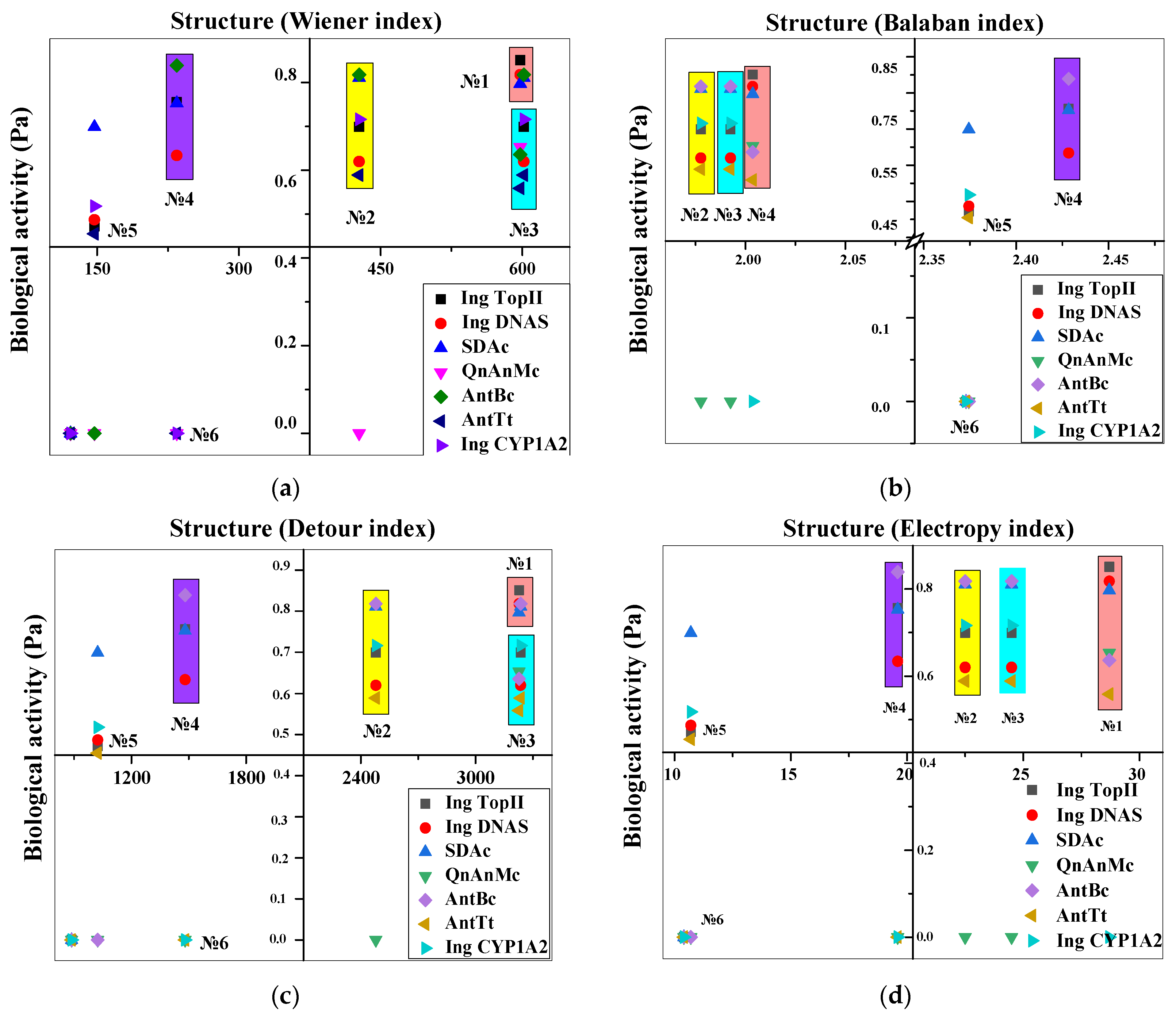
3.1. Structure-Activity Relationship Study
Experimenting In Silico (Chemicpen, PASS Online, ChemDescript)
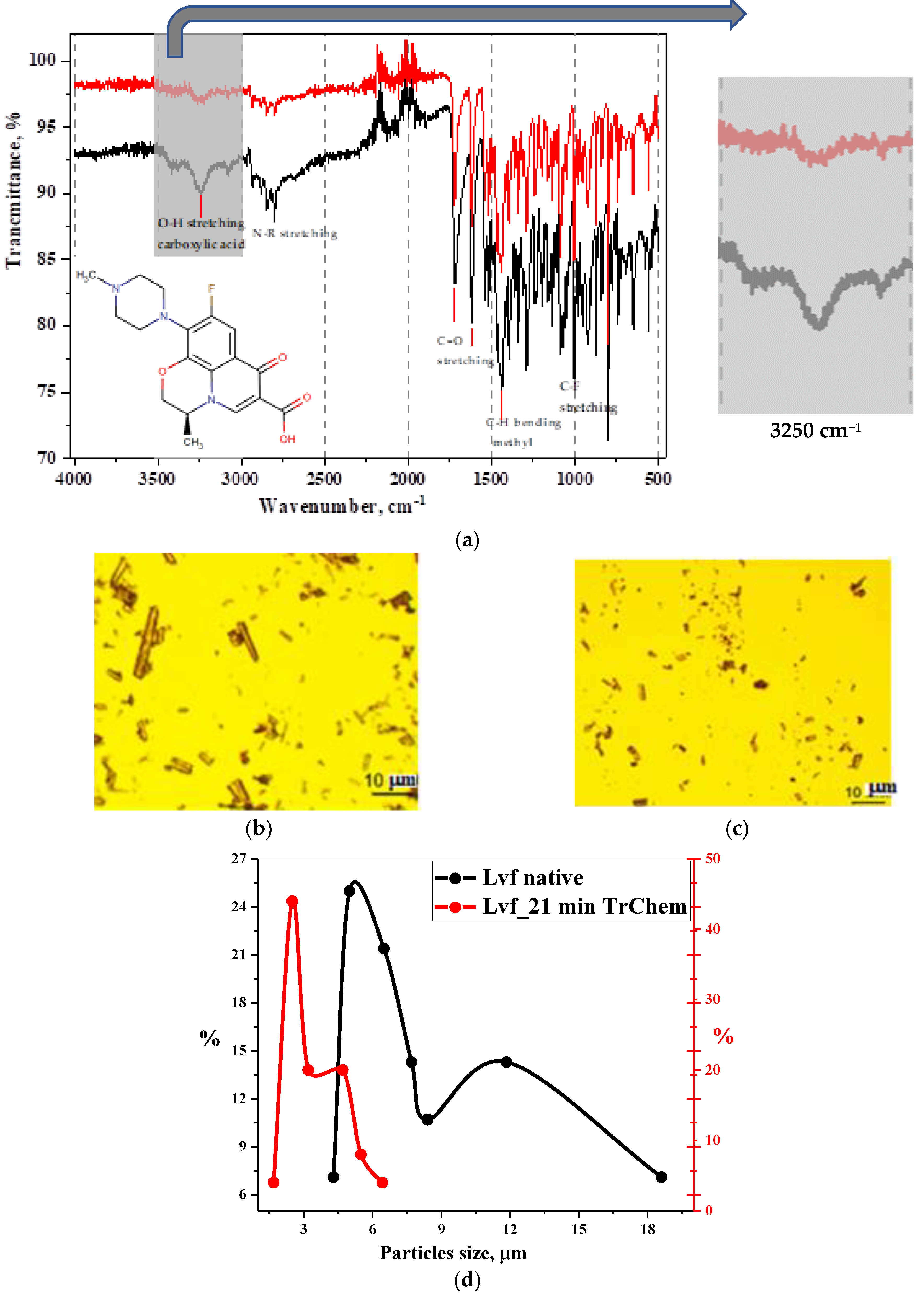
3.2. Experimentation Using Tribochemical Processes
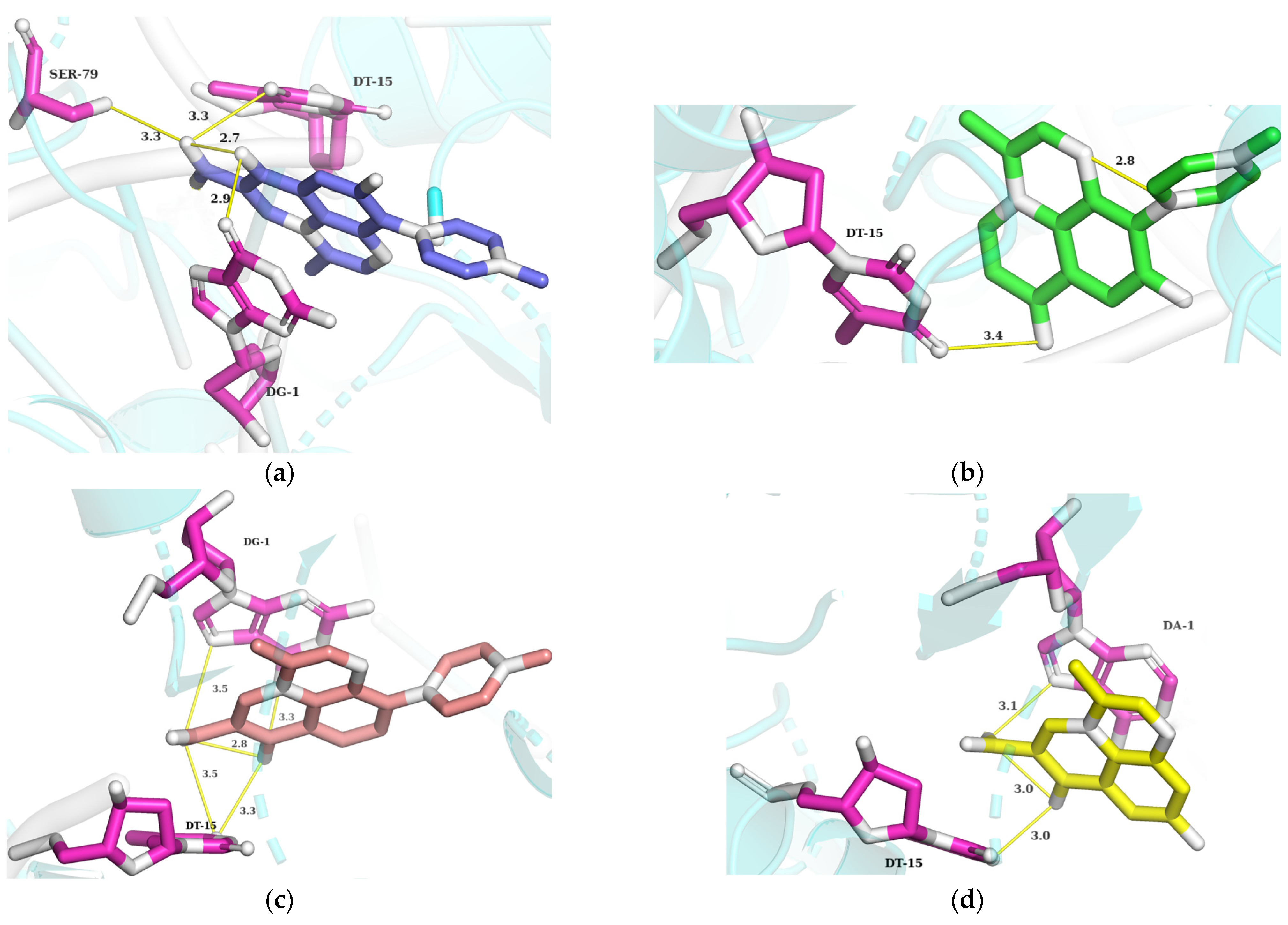
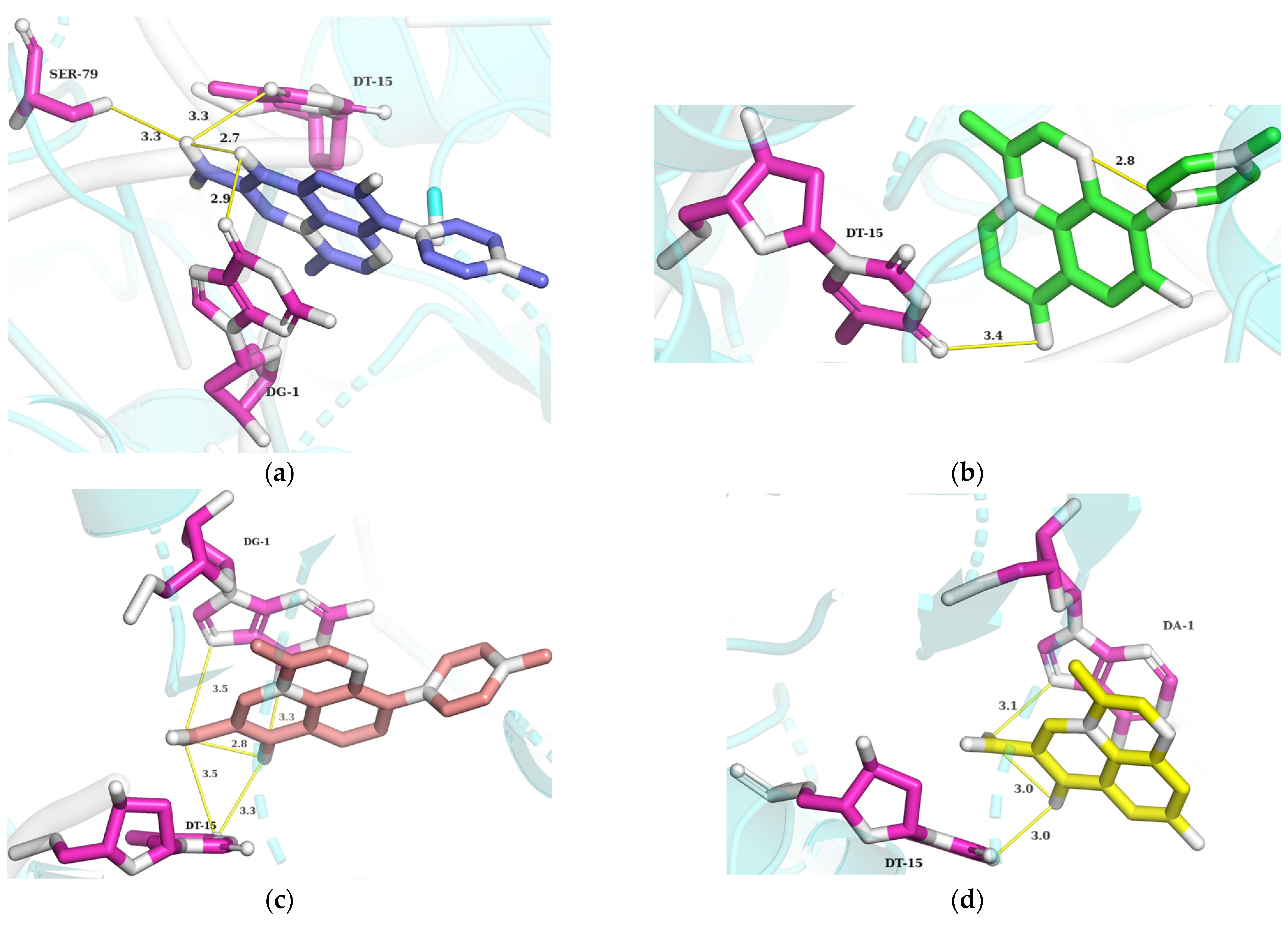
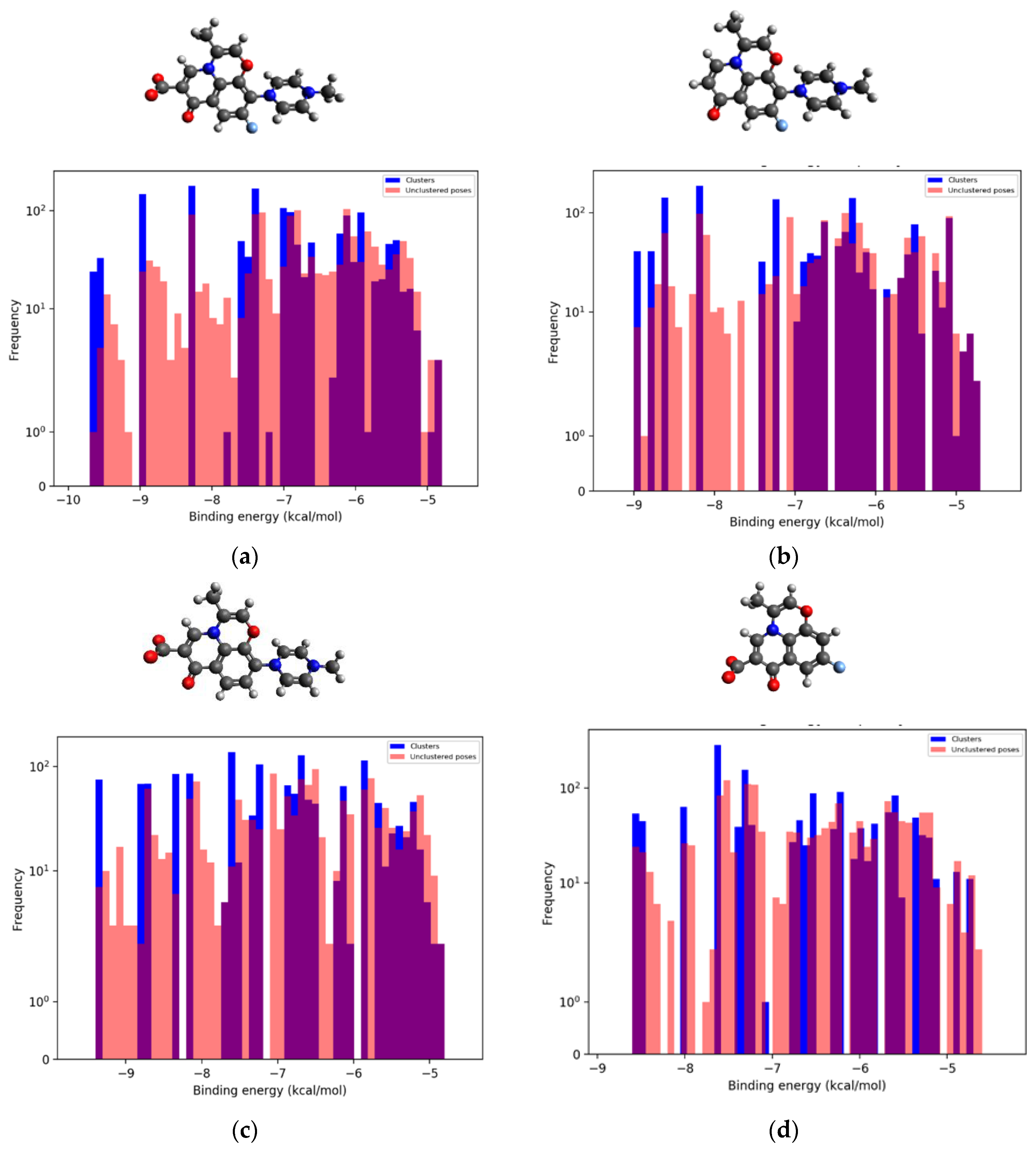
3.3. Binding Modes Prediction and Molecular Modeling
Visualization between RMS, Compounds of PMS, and DNA Gyrase II
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AntA | antimicrobial agents |
| ANOVA | one-way analysis of variance |
| MCh | mechanochemistry |
| MAct | mechanoactivation |
| TrbCh | tribochemical |
| QNs | quinolones |
| FlrQs | fluoroquinolones |
| SMComplex | supramolecular complex |
| FDA | Food and Drug Administration |
| EMA | The European Medicines Agency |
| ADR | adverse drug reactions |
| TrbCh | tribochemical |
| Lvf·Hh | levofloxacin hemihydrate |
| QSAR | quantitative structure-activity relationship |
| QRDR | Quinolone resistance determining region |
| MIC | minimal inhibitory concentrations |
| MRSA | Methicillin-Resistant Staphylococcus aureus |
| MDR | Penicillin-resistant and multi-drug-resistant Streptococcus pneumonia |
| RMS | real molecular structures |
| PMS | predicted molecular structures |
| PLD | predicted levofloxacin derivatives |
| dLvf | 11-decarboxylated levofloxacin |
| TI | topological index |
| FT-IR | Fourier transform IR spectroscopy |
| PASS | Prediction of Activity Spectra for Substances |
| MNA | Multilevel neighborhoods of atoms |
| LALLS | low-angle laser light scattering |
| OM | optical microscopy |
| DSA | dynamic strain aging |
| 2D-LS | two-dimensional dynamic backscattering |
| ChRS | chemometric reference sample |
| ETEC | enterotoxigenic Escherichia coli |
| RMSD | Root Mean Square Deviation |
References
- Vidyavathi, M.; Srividya, G. A review on ciprofloxacin: Dosage form perspective. Int. J. App. Pharm. 2018, 10, 6–10. [Google Scholar] [CrossRef]
- Center for Drug Evaluation and Research. FDA Updates Warnings for Oral and Injectable Fluoroquinolone. U.S. Food and Drug Administration. 2018. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-updates-warnings-oral-and-injectable-fluoroquinolone-antibiotics (accessed on 12 May 2016).
- Dey, B.K.; Myrsing, E.; Amin, R.; Alam, F.; Rahman, M.; Gogol, P.; Afroz, T. A ripple effect of COVID-19 pandemic on shortage of medicinal products and its impact on patient care. Int. J. App. Pharm. 2021, 13, 364–370. [Google Scholar] [CrossRef]
- Rusu, A.; Munteanu, A.C.; Arbănași, E.M.; Uivarosi, V. Overview of side-effects of antibacterial fluoroquinolones: New drugs versus Old Drugs, a step forward in the safety profile? Pharmaceutics 2023, 15, 804. [Google Scholar] [CrossRef] [PubMed]
- Spencer, A.C.; Panda, S.S. DNA Gyrase as a Target for Quinolones. Biomedicines 2023, 11, 371. [Google Scholar] [CrossRef] [PubMed]
- Millanao, A.R.; Mora, A.Y.; Villagra, N.A.; Bucarey, S.A.; Hidalgo, A.A. Biological Effects of Quinolones: A Family of Broad-Spectrum Antimicrobial Agents. Molecules 2021, 26, 7153. [Google Scholar] [CrossRef] [PubMed]
- Ray, G.T.; Baxter, R.; DeLorenze, G.N. Hospital-level rates of fluoroquinolone use and the risk of hospital-acquired infection with ciprofloxacin-nonsusceptible pseudomonas aeruginosa. Clin. Infect. Dis. 2005, 41, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Kishii, R.; Yamaguchi, Y.; Takei, M. In vitro activities and spectrum of the novel fluoroquinolone lascufloxacin (KRP-AM1977). Antimicrob. Agents Chemother. 2017, 61, e00120-17. [Google Scholar] [CrossRef]
- Bhagwat, S.S.; Nandanwar, M.; Kansagara, A.; Patel, A.; Takalkar, S.; Chavan, R.; Periasamy, H.; Yeole, R.; Deshpande, P.; Bhavsar, S.; et al. Levonadifloxacin, a Novel Broad-Spectrum Anti-MRSA Benzoquinolizine Quinolone Agent: Review of Current. Drug Des. Dev. Ther. 2019, 13, 4351–4365. [Google Scholar] [CrossRef]
- Bridle, M.J.; Janesko, B.G. Computational Study of Fluoroquinolone Binding to Mg(H2O)N2+ and Its Applicability to Future Drug Design. Int. J. Quantum Chem. 2017, 117, e25428. [Google Scholar] [CrossRef]
- Bush, N.G.; Diez-Santos, I.; Abbott, L.R.; Maxwell, A. Quinolones: Mechanism, Lethality and Their Contributions to Antibiotic Resistance. Molecules 2020, 25, 5662. [Google Scholar] [CrossRef]
- Rusu, A.; Lungu, I.-A.; Moldovan, O.-L.; Tanase, C.; Hancu, G. Structural characterization of the millennial antibacterial (fluoro)quinolones—Shaping the fifth generation. Pharmaceutics 2021, 13, 1289. [Google Scholar] [CrossRef] [PubMed]
- US National Library of Medicine, National Center for Biotechnology Information, PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Lascufloxacin (accessed on 23 June 2023).
- Dayam, R.; Al-Mawsawi, L.Q.; Zawahir, Z.; Witvrouw, M.; Debyser, Z.; Neamati, N. Quinolone 3-carboxylic acid pharmacophore: Design of second generation HIV-1 integrase inhibitors. J. Med. Chem. 2008, 51, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Stern, E.; Muccioli, G.G.; Millet, R.; Goossens, J.-F.; Farce, A.; Chavatte, P.; Poupaert, J.H.; Lambert, D.M.; Depreux, P.; Hénichart, J.-P. Novel 4-oxo-1,4-dihydroquinoline-3-carboxamide derivatives as new CB2 cannabinoid receptors agonists: Synthesis, pharmacological properties and molecular modeling. J. Med. Chem. 2006, 49, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Manera, C.; Benetti, V.; Castelli, M.P.; Cavallini, T.; Lazzarotti, S.; Pibiri, F.; Saccomanni, G.; Tuccinardi, T.; Vannacci, A.; Martinelli, A.; et al. Design, synthesis, and biological evaluation of new 1,8-naphthyridin-4(1H)-on-3-carboxamide and quinolin-4(1H)-on-3-carboxamide derivatives as CB2 selective agonists. J. Med. Chem. 2006, 49, 5947–5957. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhang, Y.; Yang, J.; Wang, Y.; Zhang, J.; Zhao, Y.; Dong, W. Prediction of the pharmacokinetics and tissue distribution of levofloxacin in humans based on an extrapolated PBPK model. Eur. J. Drug Metab. Pharmacokinet. 2016, 41, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.R.; Bung, N.; Vangala, S.R.; Srinivasan, R.; Bulusu, G.; Roy, A. De novo structure-based drug design using Deep Learning. J. Chem. Inf. Model. 2022, 62, 5100–5109. [Google Scholar] [CrossRef] [PubMed]
- US National Library of Medicine, National Center for Biotechnology Information, PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Levofloxacin (accessed on 23 June 2023).
- Informer Technologies, Inc. Available online: https://chemicpen.software.informer.com/ (accessed on 21 October 2022).
- Randić, M.; Sabljić, A.; Nikolić, S.; Trinajstić, N. A rational selection of graph-theoretical indices in the QSAR. Int. J. Quan. Chem. 1998, 34, 267–285. [Google Scholar] [CrossRef]
- Balaban, A.T. Topological indices and their uses: A new approach for the coding of alkanes. J. Mol. Struct. THEOCHEM 1988, 165, 243–253. [Google Scholar] [CrossRef]
- Gopika, S.; Lakshmi, A.; Aiswarya, P.D.; Rajendran, S. On the Detour Based Indices. Math. Stat. Eng. Appl. 2022, 71, 951–966. [Google Scholar]
- Fang, W.; Liu, W.-H.; Liu, J.-B.; Chen, F.-Y.; Hong, Z.-M.; Xia, Z.-J. Maximum Detour–Harary Index for Some Graph Classes. Symmetry 2018, 10, 608. [Google Scholar] [CrossRef]
- Filimonov, D.A.; Lagunin, A.A.; Gloriozova, T.A. Prediction of the Biological Activity Spectra of Organic Compounds Using the Pass Online Web Resource. Chem. Heterocycl. Compd. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Fomenko, A.E.; Sobolev, B.N.; Filimonov, D.A.; Poroĭkov, V.V. Use of structural MNA descriptors for designing profiles of protein families. Biophysics 2003, 48, 595–605. [Google Scholar]
- Dmitriev, A.V.; Rudik, A.V.; Karasev, D.A.; Pogodin, P.V.; Lagunin, A.A.; Filimonov, D.A.; Poroikov, V.V. In silico prediction of drug–drug interactions mediated by cytochrome P450 isoforms. Pharmaceutics 2021, 13, 538. [Google Scholar] [CrossRef] [PubMed]
- Filimonov, D.A.; Druzhilovskiy, D.S.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Dmitriev, A.V.; Pogodin, P.V.; Poroikov, V.V. Computer-aided prediction of biological activity spectra for chemical compounds: Opportunities and limitation. Biomed. Chem. Res. Meth. 2018, 1, e00004. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Maschietto, F.; Allen, B.; Kyro, G.W.; Batista, V.S. MDiGest: A Python package for describing allostery from molecular dynamics simulations. J. Chem. Phys. 2023, 158, 215103. [Google Scholar] [CrossRef] [PubMed]
- Rosignoli, S.; di Paola, L.; Paiardini, A. PyPCN: Protein contact networks in PyMOL. Bioinformatics 2023, 39, btad675. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar]
- Sulimov, V.B.; Kutov, D.C.; Taschilova, A.S.; Ilin, I.S.; Tyrtyshnikov, E.E.; Sulimov, A.V. Docking Paradigm in Drug Design. Curr. Top. Med. Chem. 2021, 21, 507–546. [Google Scholar] [CrossRef]
- Vieira, T.F.; Sousa, S.F. Comparing AutoDock and Vina in Ligand/Decoy Discrimination for Virtual Screening. Appl. Sci. 2019, 9, 4538. [Google Scholar] [CrossRef]
- Zhou, X.; Ling, M.; Lin, Q.; Tang, S.; Wu, J.; Hu, H. Effectiveness Analysis of Multiple Initial States Simulated Annealing Algorithm, A Case Study on the Molecular Docking Tool AutoDock Vina. IEEE/ACM Trans. Comput. Biol. Bioinform. 2023, 1–12. Available online: https://ssrn.com/abstract=4120348;http://dx.doi.org/10.2139/ssrn.4120348 (accessed on 26 May 2022). [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Smith, J.C. Speed vs Accuracy: Effect on Ligand Pose Accuracy of Varying Box Size and Exhaustiveness in AutoDock Vina. Mol. Inform. 2023, 42, e2200188. [Google Scholar] [CrossRef] [PubMed]
- Uspenskaya, E.; Simutina, A.; Kuzmina, E.; Sukhanova, V.; Garaev, T.; Pleteneva, T.; Koldina, A.; Kolyabina, E.; Petrov, G.; Syroeshkin, A. Exploring the effects of cramped-impact-type mechanical action on active pharmaceutical ingredient (levofloxacin)—Prospects for pharmaceutical applications. Powders 2023, 2, 464–483. [Google Scholar] [CrossRef]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of activity spectra for Biologically Active Substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef] [PubMed]
- He, Q.Q.; Zhang, X.; Yang, L.M.; Zheng, Y.T.; Chen, F. Synthesis and biological evaluation of 5-fluoroquinolone-3-carboxylic acids as potential HIV-1 integrase inhibitors. J. Enzym. Inhib. Med. Chem. 2012, 28, 671–676. [Google Scholar] [CrossRef]
- Malík, M.; Tlustoš, P. Nootropics as cognitive enhancers: Types, dosage and side effects of smart drugs. Nutrients 2022, 14, 3367. [Google Scholar] [CrossRef]
- Stankevich, M.I.; Stankevich, I.V.; Zefirov, N.S. Topological indices in Organic Chemistry. Russ. Chem. Rev. 1998, 57, 191–208. [Google Scholar] [CrossRef]
- Kajdas, C. General approach to mechanochemistry and its relation to tribochemistry. Tribol. Eng. 2013, 11, 209–230. [Google Scholar]
- Maeda, Y.; Kiba, A.; Ohnishi, K.; Hikichi, Y. Implications of amino acid substitutions in GyrA at position 83 in terms of oxolinic acid resistance in field isolates of Burkholderia glumae, a causal agent of bacterial seedling rot and grain rot of Rice. Appl. Environ. Microbiol. 2004, 70, 5613–5620. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Zhao, H. Quinolone antibiotics: Resistance and therapy. Infect. Drug Resist. 2023, 16, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, A.; Tariq, A.; Iqbal, M.; Mirza, O.; Haque, A.; Walz, T.; Rahman, M. Mutational diversity in the quinolone resistance-determining regions of type-II topoisomerases of salmonella serovars. Antibiotics 2021, 10, 1455. [Google Scholar] [CrossRef] [PubMed]
- Mehla, K.; Ramana, J. Structural signature of Ser83Leu and Asp87Asn mutations in DNA gyrase from enterotoxigenic Escherichia coli and impact on quinolone resistance. Gene 2016, 576, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, H.; Xiao, Z.; Wang, F.; Wang, X.; Wang, Y. Combined 3D-QSAR, Molecular Docking and Molecular Dynamics Study on Derivatives of Peptide Epoxyketone and Tyropeptin-Boronic Acid as Inhibitors Against the β5 Subunit of Human 20S Proteasome. Int. J. Mol. Sci. 2011, 12, 1807–1835. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ji, J.; Song, F.; Hu, J. Intercellular receptor-ligand binding: Effect of protein-membrane interaction. J. Mol. Biol. 2023, 435, 167787. [Google Scholar] [CrossRef]
- Min, X.; Cheng, S.; Jincai, Y.; Qing, W.; Niu, H. Systematic Investigation of Docking Failures in Large-Scale Structure-Based Virtual Screening. ACS Omega 2022, 7, 39417–39428. [Google Scholar]
- Quiroga, R.; Villarreal, M. Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening. PLoS ONE 2016, 11, e0155183. [Google Scholar] [CrossRef]
- Sulimov, V.B.; Sulimov, A.V. Docking: Molecular Modeling for Drug Discovery; AINTELL: Moscow, Russia, 2017; p. 348. [Google Scholar]
- Suvannang, N.; Nantasenamat, C.; Isarankura-Na-Ayudhya, C.; Prachayasittikul, V. Molecular Docking of Aromatase Inhibitors. Molecules 2011, 16, 3597–3617. [Google Scholar] [CrossRef]
- Novichikhina, N.P.; Shestakov, A.S.; Medvedeva, S.M.; Lagutina, A.M.; Krysin, M.Y.; Podoplelova, N.A.; Panteleev, M.A.; Ilin, I.S.; Sulimov, A.V.; Tashchilova, A.S.; et al. New Hybrid Tetrahydropyrrolo[3,2,1-ij]quinolin-1-ylidene-2-thioxothiazolidin-4-ones as New Inhibitors of Factor Xa and Factor XIa: Design, Synthesis, and In Silico and Experimental Evaluation. Molecules 2023, 28, 3851. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Weight, g·mol−1 | Log Po/w | pKa (Strongest Acidic) | Water Solubility, mg·mL−1 | Toxicity in Mice LD50, mg·kg−1 | Microbiologic Activity/ Indications |
|---|---|---|---|---|---|
| NALIDIXIC ACID (FlrQ-1G) | |||||
| 232.2 | 1.6 | 8.60 | 0.1 | 4000 | Enterobacteriaceae/Uncomplicated urinary tract infections, not for use in systemic infections * |
| CIPROFLOXACIN (FlrQ-2G) | |||||
| 331.3 | 0.3 | 6.10 | <1.00 | 2000 | Enterobacteriaceae, atypical pathogens; Pseudomonas aeruginosa, Pneumoccoci/ * and also complicated urinary tract and catheter-related infections, gastroenteritis |
| LEVOFLOXACIN (FlrQ-3G) | |||||
| 361.4 | 0.7 | 5.50 | 1.44 | 1800 | Enterobacteriaceae, atypical pathogens, streptococci. Pneumoccoci MIC90: 0.25–0.5 mg·L−1/* and also community-acquired pneumonia in hospitalized patients or if atypical pathogens are strongly suspected |
| MOXIFLOXACIN (FlrQ-4G) | |||||
| 401.4 | 2.9 | 5.49 | 1.15 | 100 | Enterobacteriaceae, P. aeruginosa, atypical pathogens, MSSA, streptococci, anaerobes, Pneumoccoci. Consider for treatment of intra-abdominal infections |
| LEVONADIFLOXACIN (FlrQ-5G) | |||||
| 360.4 | 0.87 ** | 5.94 ** | 0.63 ** | 535 ** | Anti-MDR, MRSA, MDR S. pneumoniae, pathogenies ESKAPE, P. aeruginosa и S. aureus strains/hospital-acquired and nosocomial pneumonia, diabetic foot ulcer infections and skin and soft tissue infections, acute otitis eterna (swimmer’s ear) |
| Topological Index | Definition | Equation |
|---|---|---|
| Wiener (W) | the shortest distances sum between all pairs of vertices in G graph | where dij is the shortest distance between vertices i and j |
| Balaban (J) | the average distance-sum connectivity index | where n and m are the cardinalities of the vertex and the edge set of G, respectively, and w(u) (resp. w(v)) denotes the sum of distances from u (resp. v) to all the other vertices of G |
| Detour (Ip) | the sum of the upper triangle of the detour | where the i,j-th entry ∆ij denotes the longest path between vertices i and j of the underlying graph (i, j = 1, 2, ... N) where N denotes the number of vertices |
| Electropy (Ie) | the sum of the squares of the atomic nuclear charges divided by the square of the number of atoms in the molecule minus one | pa and pi represent the probabilities for the occurrence of an a priori event and i—posteriori event. The larger the Ie index value, the more electropositive the molecule is. |
| № | Lvf Structure Derivatives | Prediction Spectra of Biological Activity (Pa) | ||||||
|---|---|---|---|---|---|---|---|---|
| Ing TopII 1* | Ing DNAS 2* | SDAc 3* | QnAnMc 4* | AntBc 5* | AntTt 6* | Ing CYP1 A2 7* | ||
| 1 | basic | 0.851 | 0.818 | 0.797 | 0.653 | 0.636 | 0.559 | 0 |
| 2 | 6-decarboxylated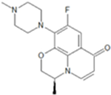 | 0.699 | 0.620 | 0.811 | 0.351 | 0.818 | 0.589 | 0.716 |
| 3 | 9-defluorinated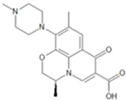 | 0.699 | 0.620 | 0.811 | 0.351 | 0.818 | 0.589 | 0.716 |
| 4 | 10-depyperazine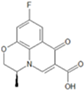 | 0.756 | 0.634 | 0.753 | 0.495 | 0.575 | 0.347 | 0.467 |
| 5 | 4-benzoxazine-BCS | 0.472 | 0.487 | 0.699 | 0.095 | 0.426 | 0.455 | 0.518 |
| 6 | 5-dehydro-4-benzoxazine-BCS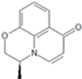 | 0.302 | 0.405 | 0.638 | 0.035 | 0.271 | 0.453 | 0.424 |
| Ligand | Steric Interaction | Non-Steric Interaction | Affinity Values, kcal/mol | |||
|---|---|---|---|---|---|---|
| Gauss 1 | Gauss 2 | Repulsion | Hydrophobic Attraction | Non-Directional Hydrogen Bond | ||
| basic Lvf | 7.50 | 91.97 | 0.16 | 0.29 | 0.08 | −9.7 |
| 9-defluorinated | 7.67 | 91.74 | 0.17 | 0.31 | 0.11 | −9.4 |
| 6-decarboxylated | 7.35 | 92.17 | 0.12 | 0.33 | 0.03 | −8.9 |
| 10-depyperazine | 8.66 | 90.84 | 0.19 | 0.19 | 0.12 | −8.6 |
| Poses in Cluster | Best Pose | Binding Site Coordinates | Kb·105, M−1 |
|---|---|---|---|
| basic levofloxacin | 1.31 | ||
| 24 | 592 | (−22.18, 50.92, −37.92) | |
| 33 | 1201 | (−39.00, 54.45, −38.88) | |
| 69 | 232 | (−18.02, 26.50, −36.81) | |
| 9-defluorinated | 1.30 | ||
| 36 | 982 | (−22.25; 51.62, −38.84) | |
| 39 | 591 | (−39.10, 54.89, −38.22) | |
| 68 | 173 | (−18.43, 27.23, −36.66) | |
| 6-decarboxylated | 1.29 | ||
| 41 | 1228 | (−39,16, 54.97, −38.91) | |
| 41 | 1001 | (−22.00, 51.04, −38.77) | |
| 66 | 258 | (−17.54, 26.57, −36.31) | |
| 10-depyperazine | 1.28 | ||
| 54 | 1223 | (−38.84, 54.74, −37.11) | |
| 45 | 592 | (−22.03, 51.80, −37.31) | |
| 37 | 985 | (−18.27, 55.28, −25.49) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uspenskaya, E.V.; Sukhanova, V.A.; Kuzmina, E.S.; Pleteneva, T.V.; Levitskaya, O.V.; Garaev, T.M.; Syroeshkin, A.V. In Silico Activity Prediction and Docking Studies of the Binding Mechanisms of Levofloxacin Structure Derivatives to Active Receptor Sites of Bacterial Type IIA Topoisomerases. Sci. Pharm. 2024, 92, 1. https://doi.org/10.3390/scipharm92010001
Uspenskaya EV, Sukhanova VA, Kuzmina ES, Pleteneva TV, Levitskaya OV, Garaev TM, Syroeshkin AV. In Silico Activity Prediction and Docking Studies of the Binding Mechanisms of Levofloxacin Structure Derivatives to Active Receptor Sites of Bacterial Type IIA Topoisomerases. Scientia Pharmaceutica. 2024; 92(1):1. https://doi.org/10.3390/scipharm92010001
Chicago/Turabian StyleUspenskaya, Elena V., Vasilisa A. Sukhanova, Ekaterina S. Kuzmina, Tatyana V. Pleteneva, Olga V. Levitskaya, Timur M. Garaev, and Anton V. Syroeshkin. 2024. "In Silico Activity Prediction and Docking Studies of the Binding Mechanisms of Levofloxacin Structure Derivatives to Active Receptor Sites of Bacterial Type IIA Topoisomerases" Scientia Pharmaceutica 92, no. 1: 1. https://doi.org/10.3390/scipharm92010001
APA StyleUspenskaya, E. V., Sukhanova, V. A., Kuzmina, E. S., Pleteneva, T. V., Levitskaya, O. V., Garaev, T. M., & Syroeshkin, A. V. (2024). In Silico Activity Prediction and Docking Studies of the Binding Mechanisms of Levofloxacin Structure Derivatives to Active Receptor Sites of Bacterial Type IIA Topoisomerases. Scientia Pharmaceutica, 92(1), 1. https://doi.org/10.3390/scipharm92010001