
Repurposing of Four Drugs as Anti-SARS-CoV-2 Agents and Their Interactions with Protein Targets

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Drug Database Construction and Ligand Preparation
2.2. Protein Preparation and Molecular Docking
2.3. Free Energy Binding Calculations (MM-GBSA)
2.4. Molecular Dynamics Simulation
2.5. Visualization
3. Results and Discussion
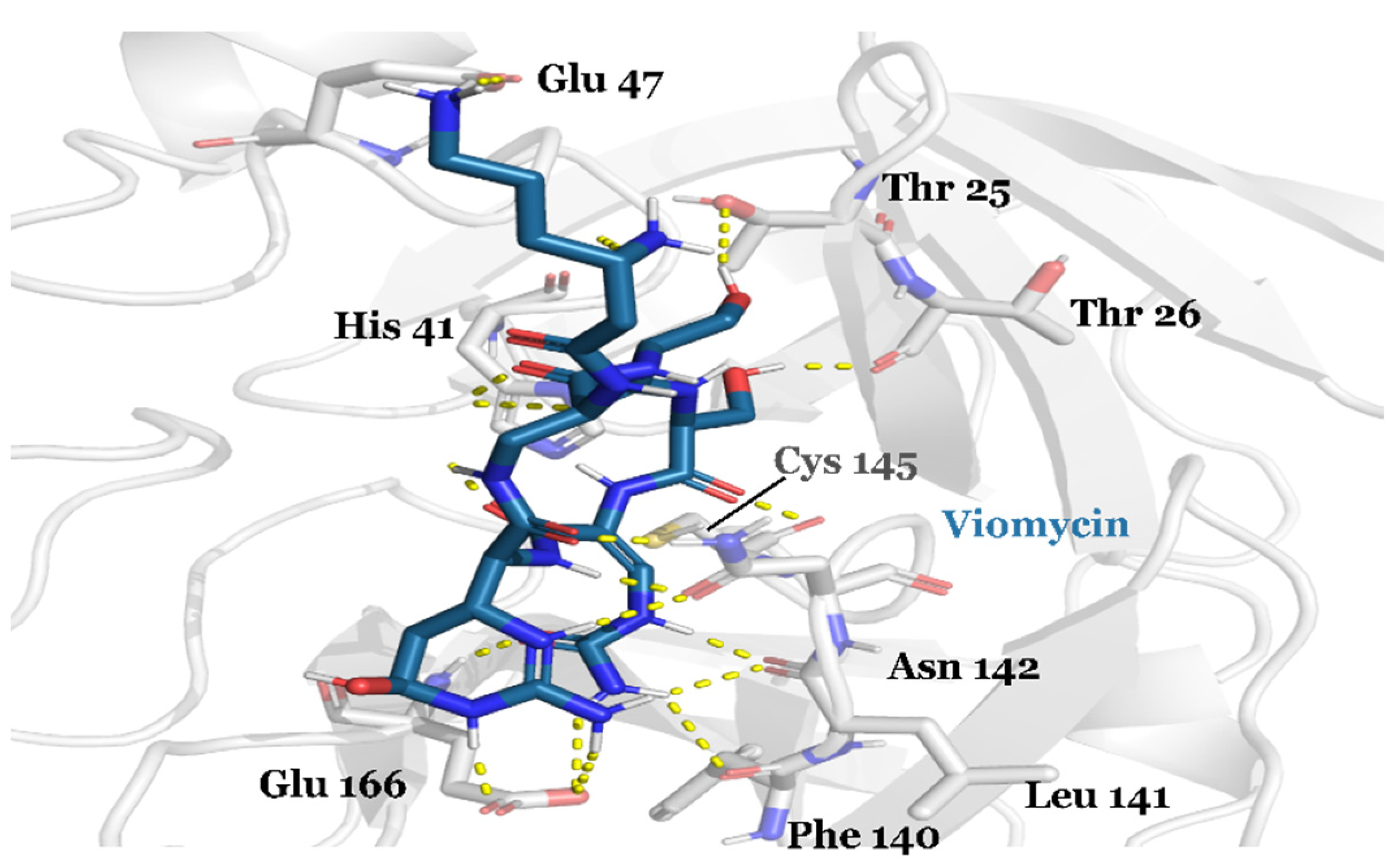
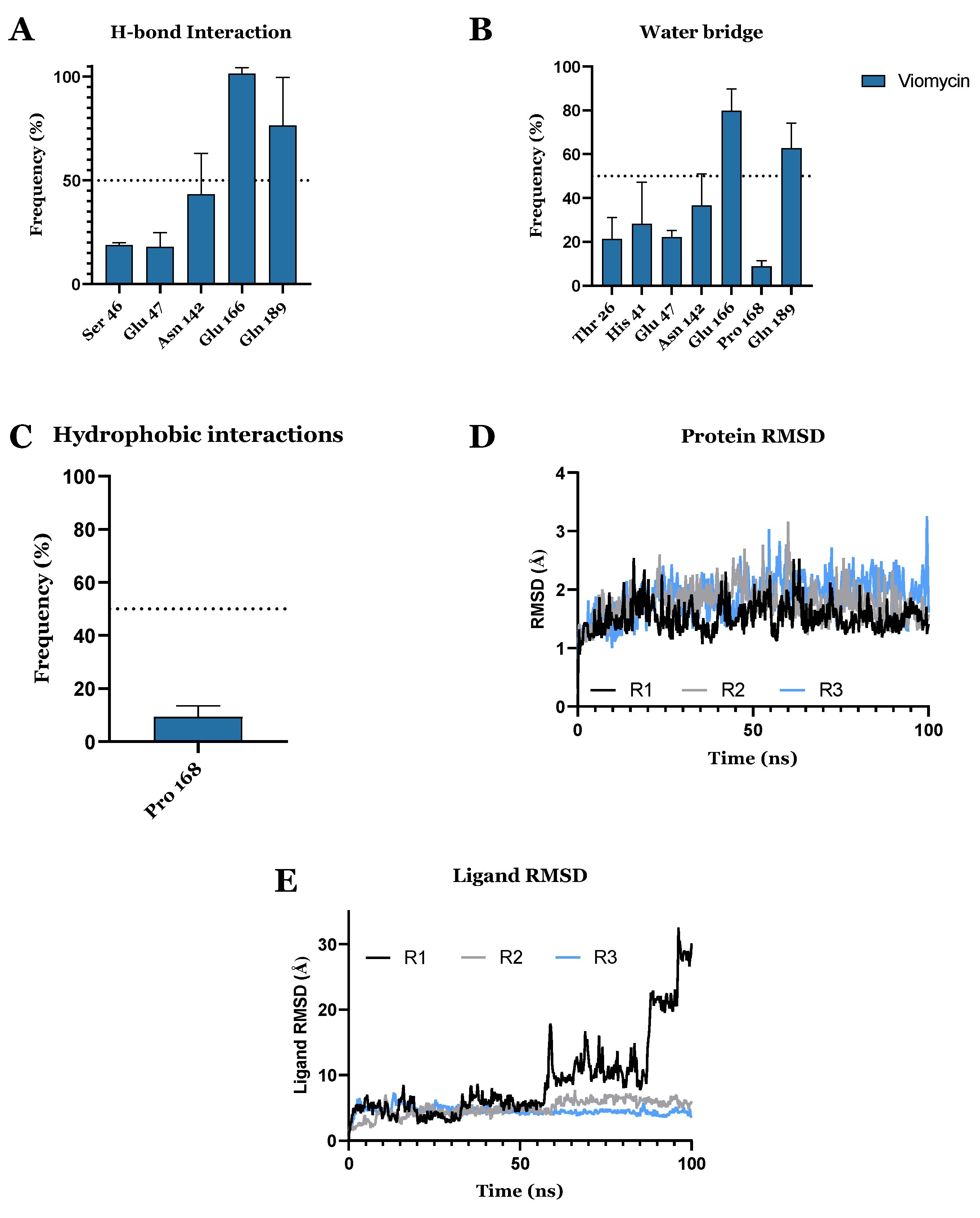
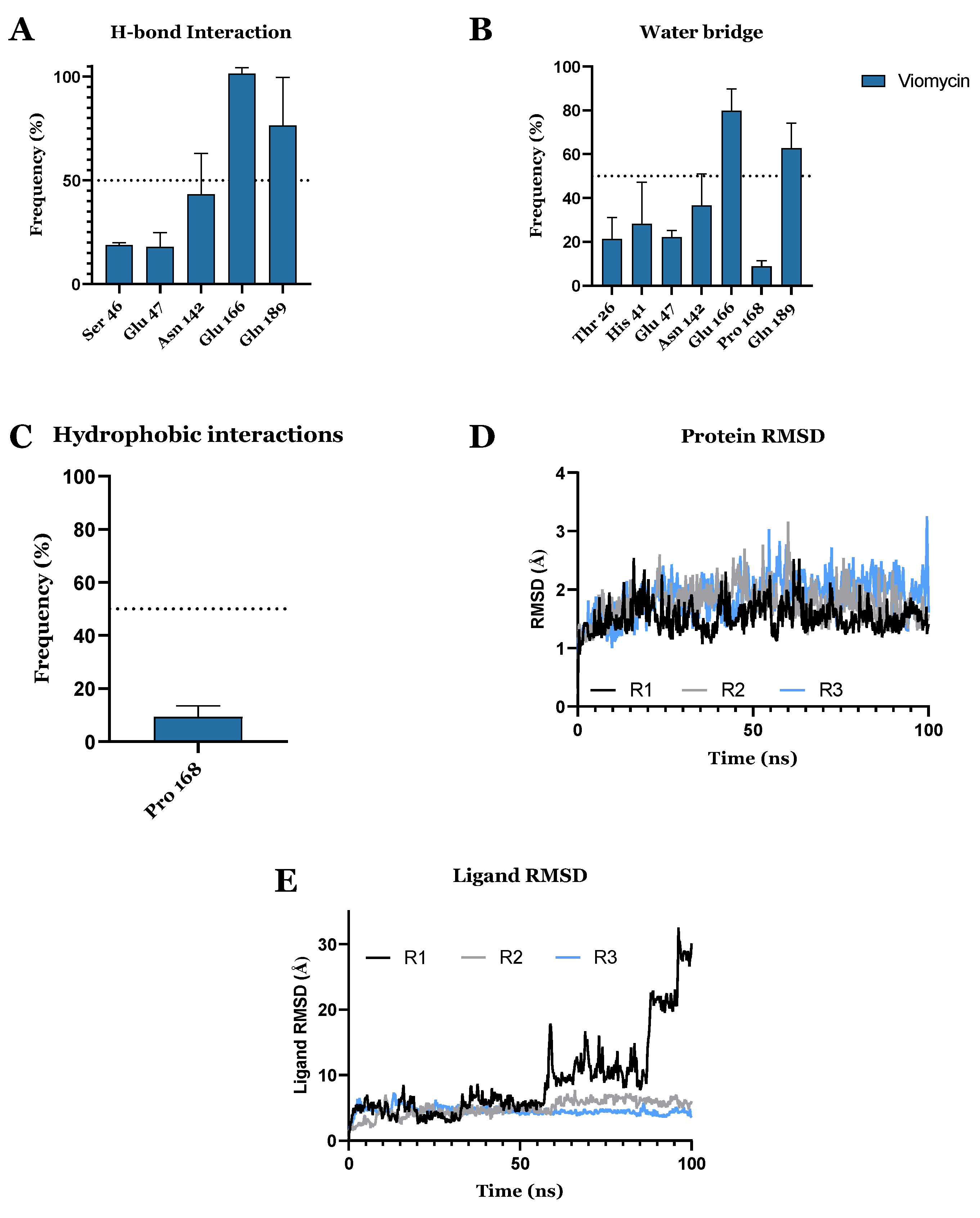
3.1. Drug Candidates May Inhibit the Viral Protein Translation
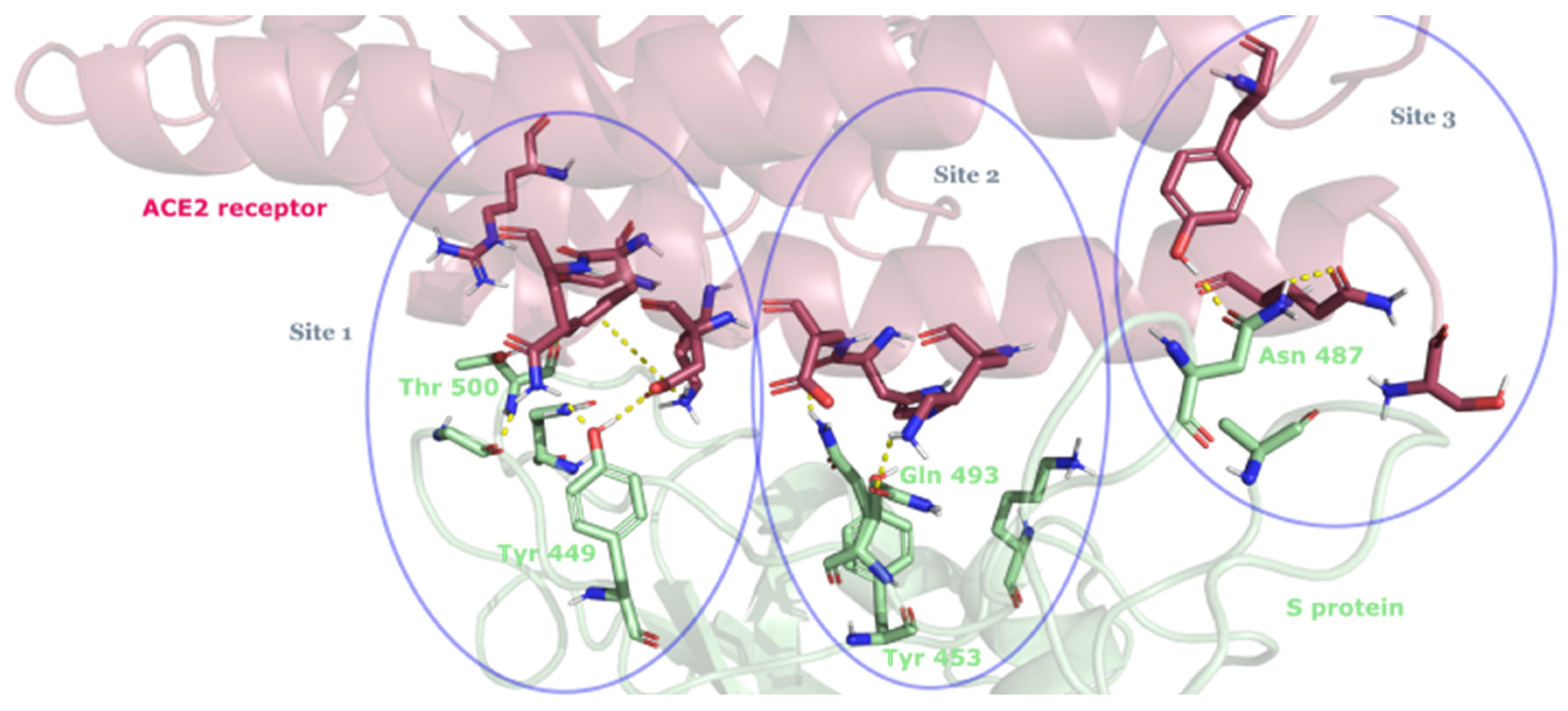
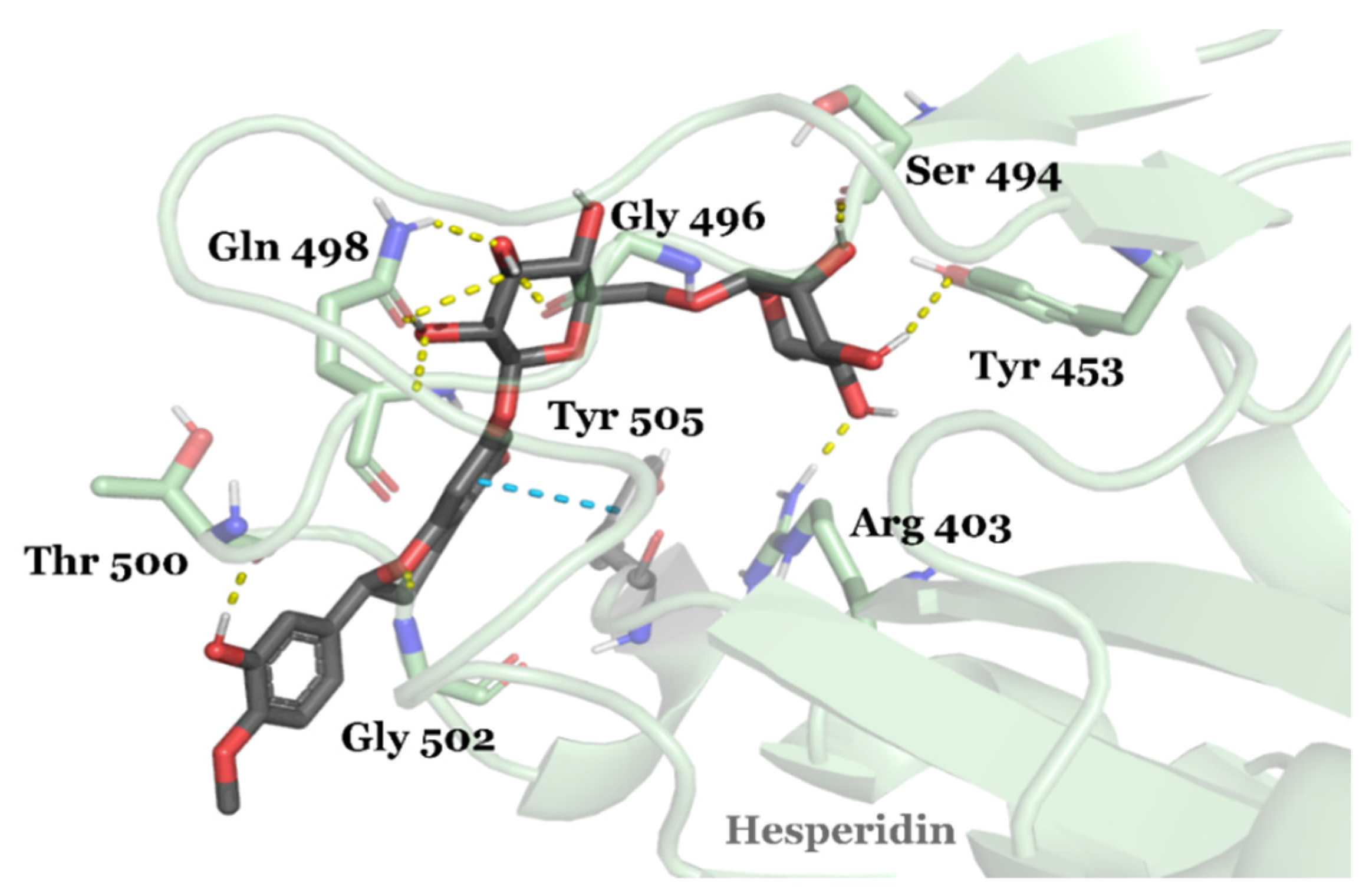
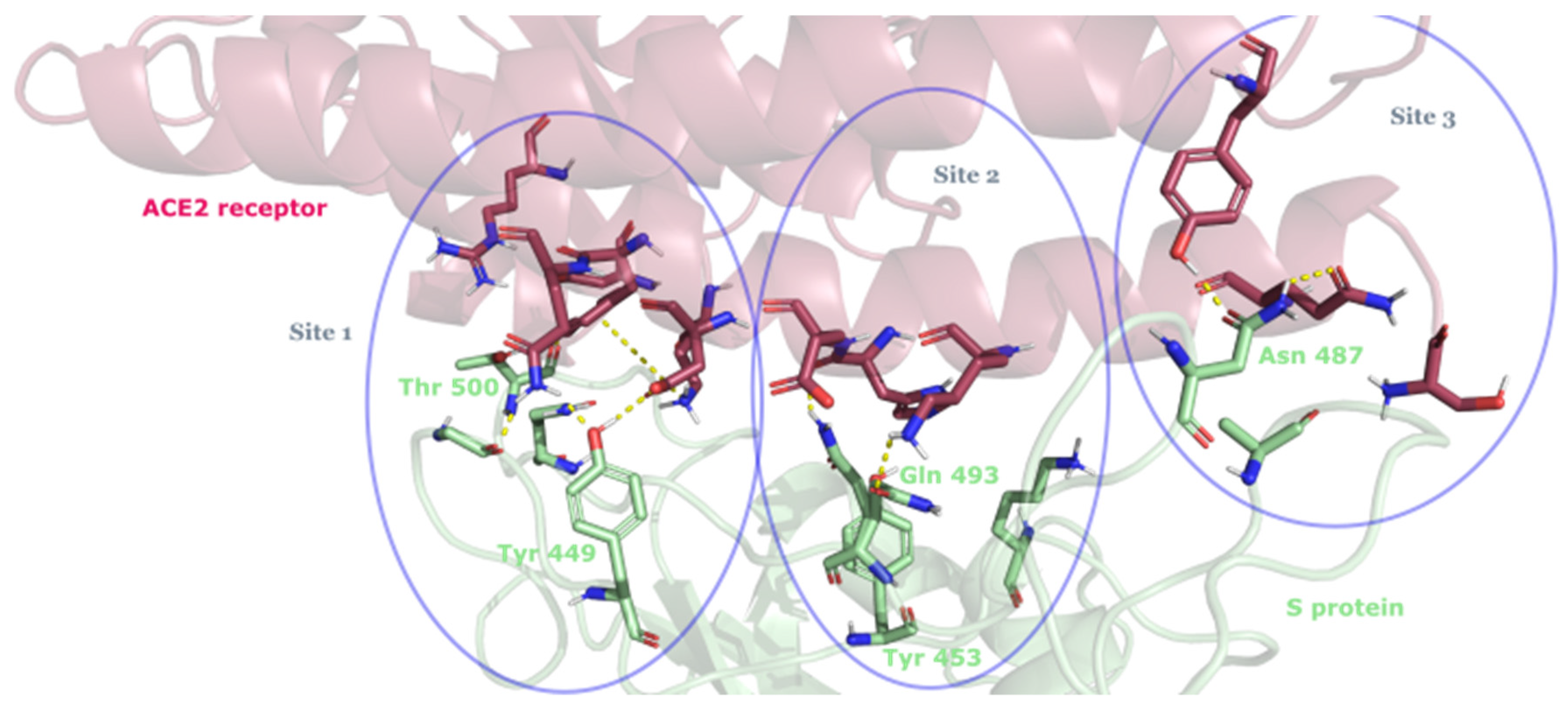
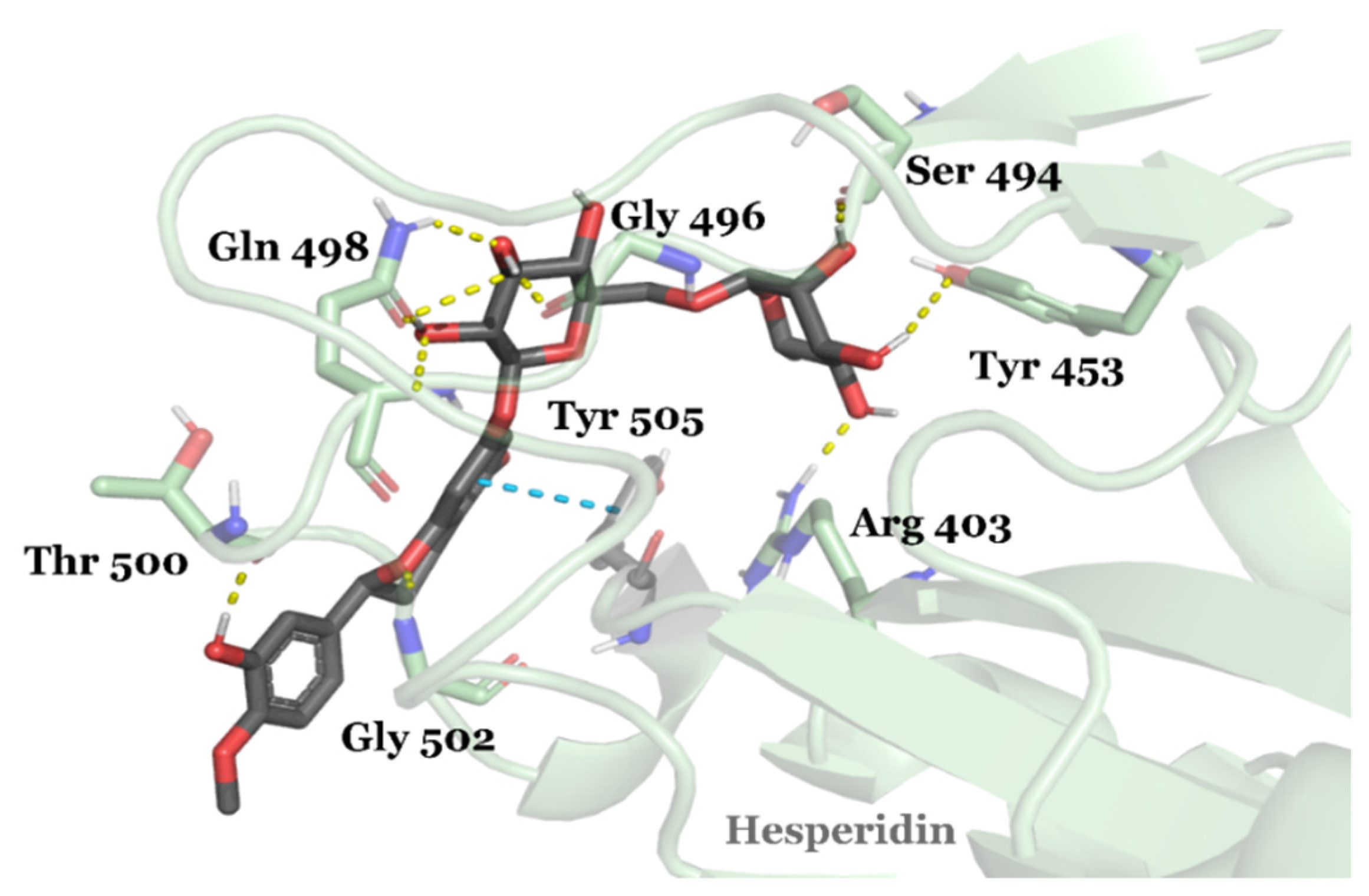
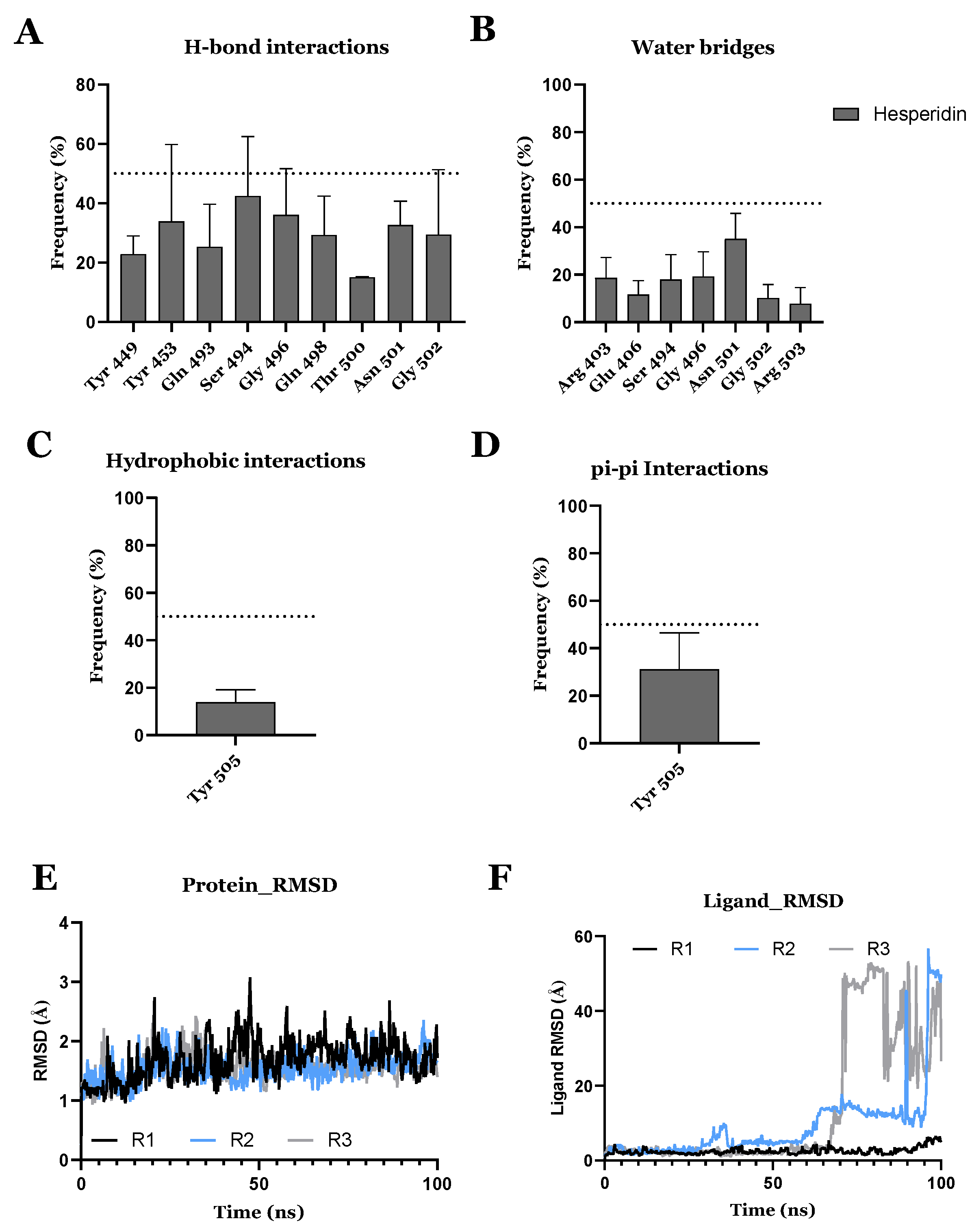
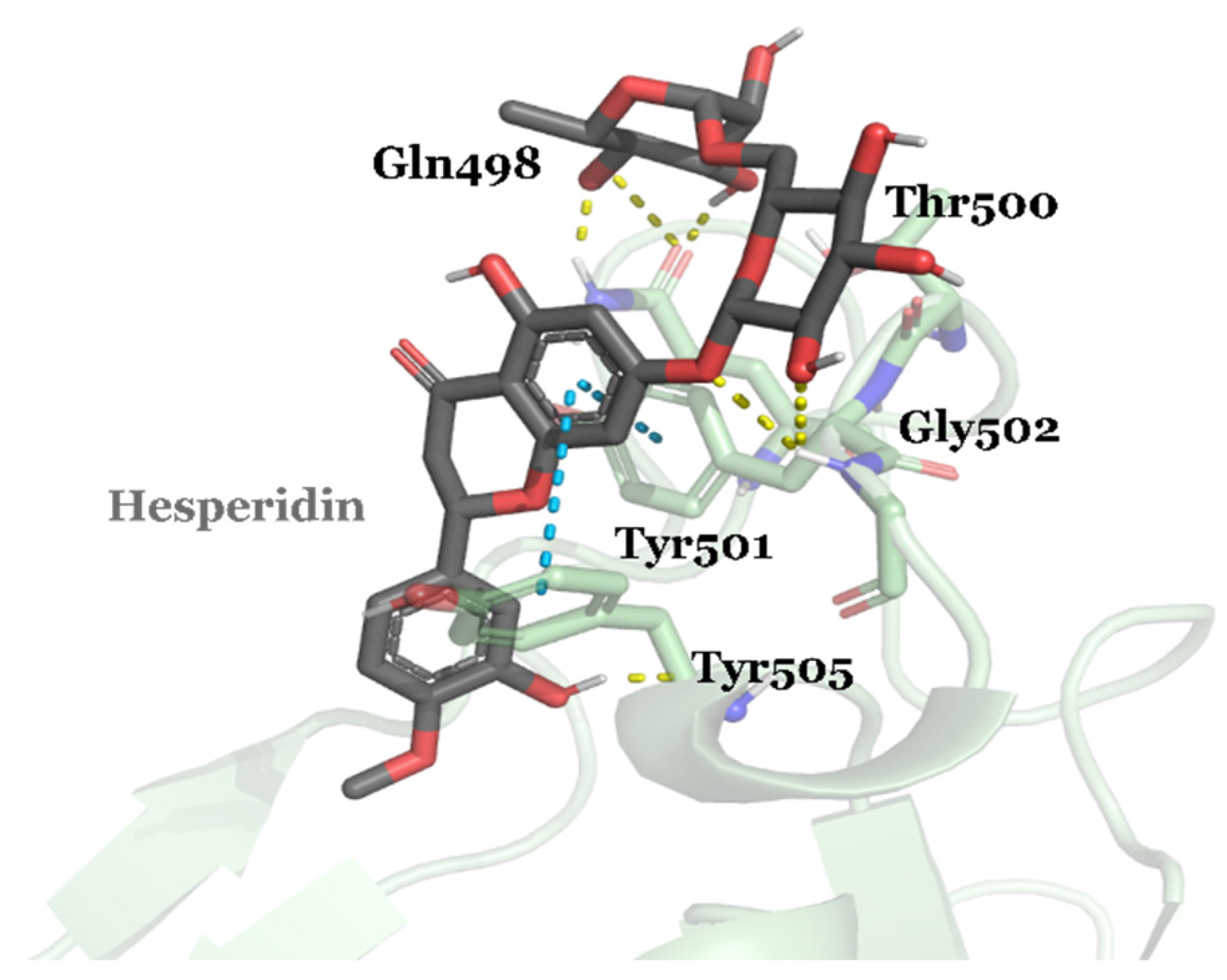
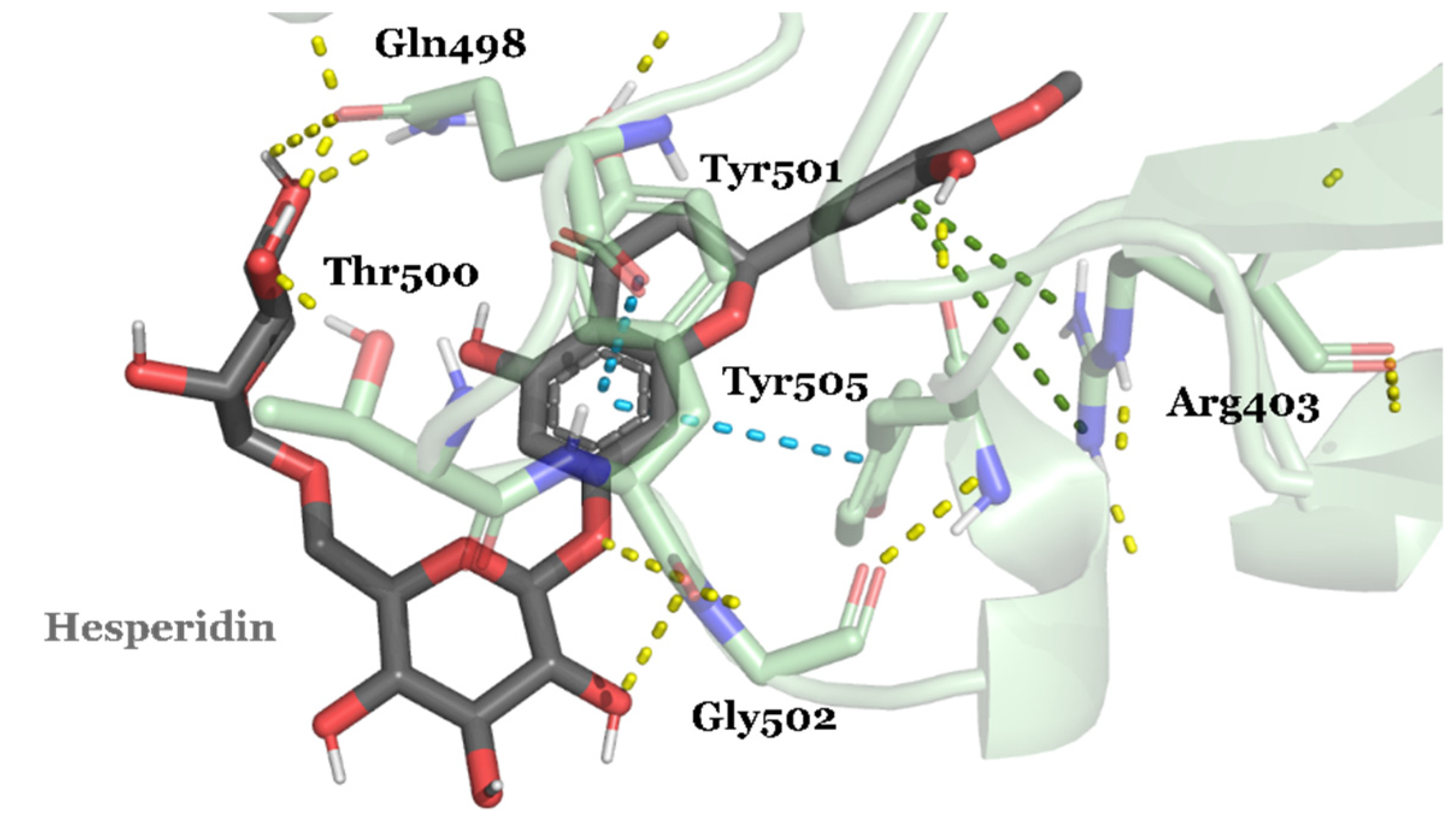
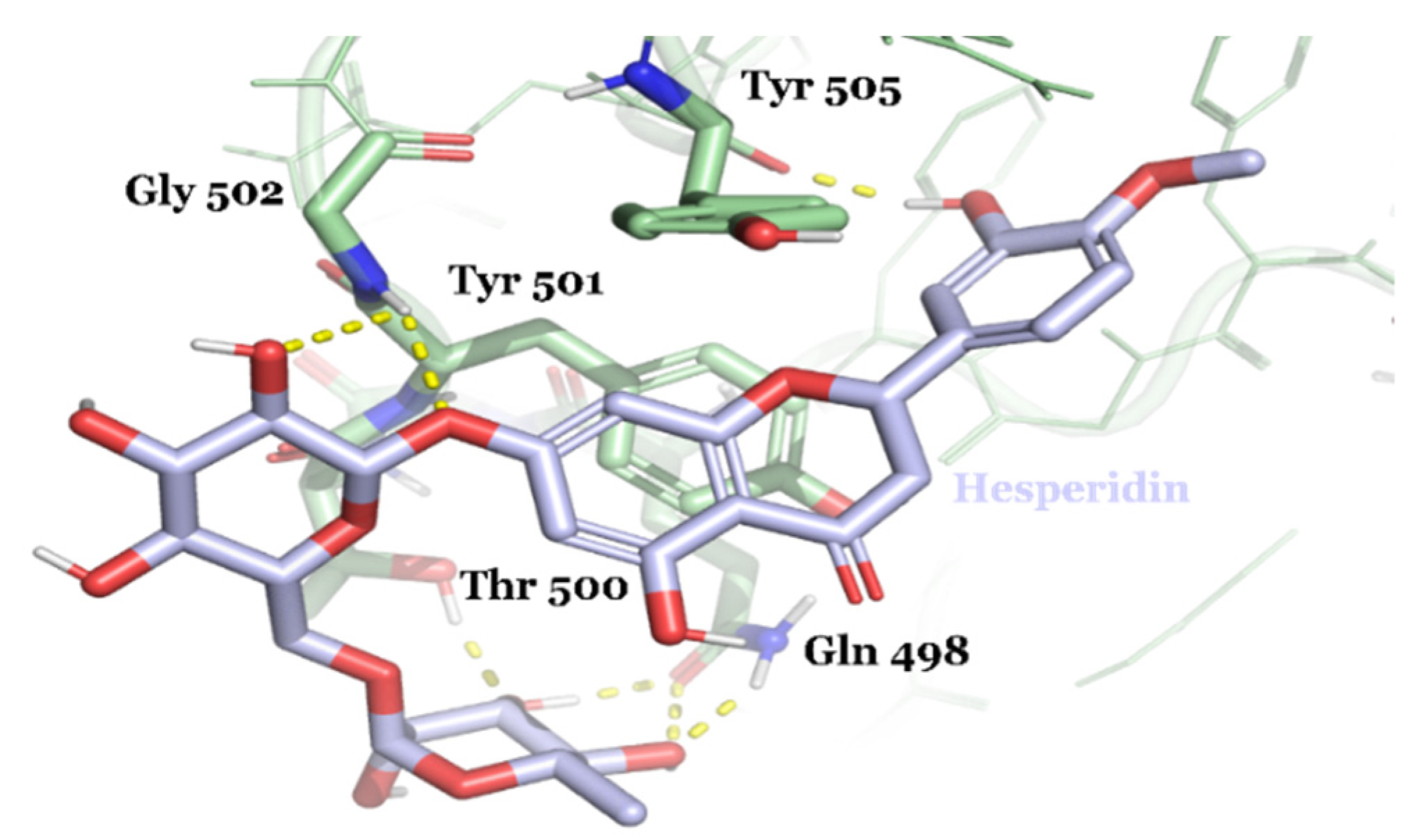
3.2. Drug Candidates May Inhibit SARS-CoV-2 Entry into the Host Cells
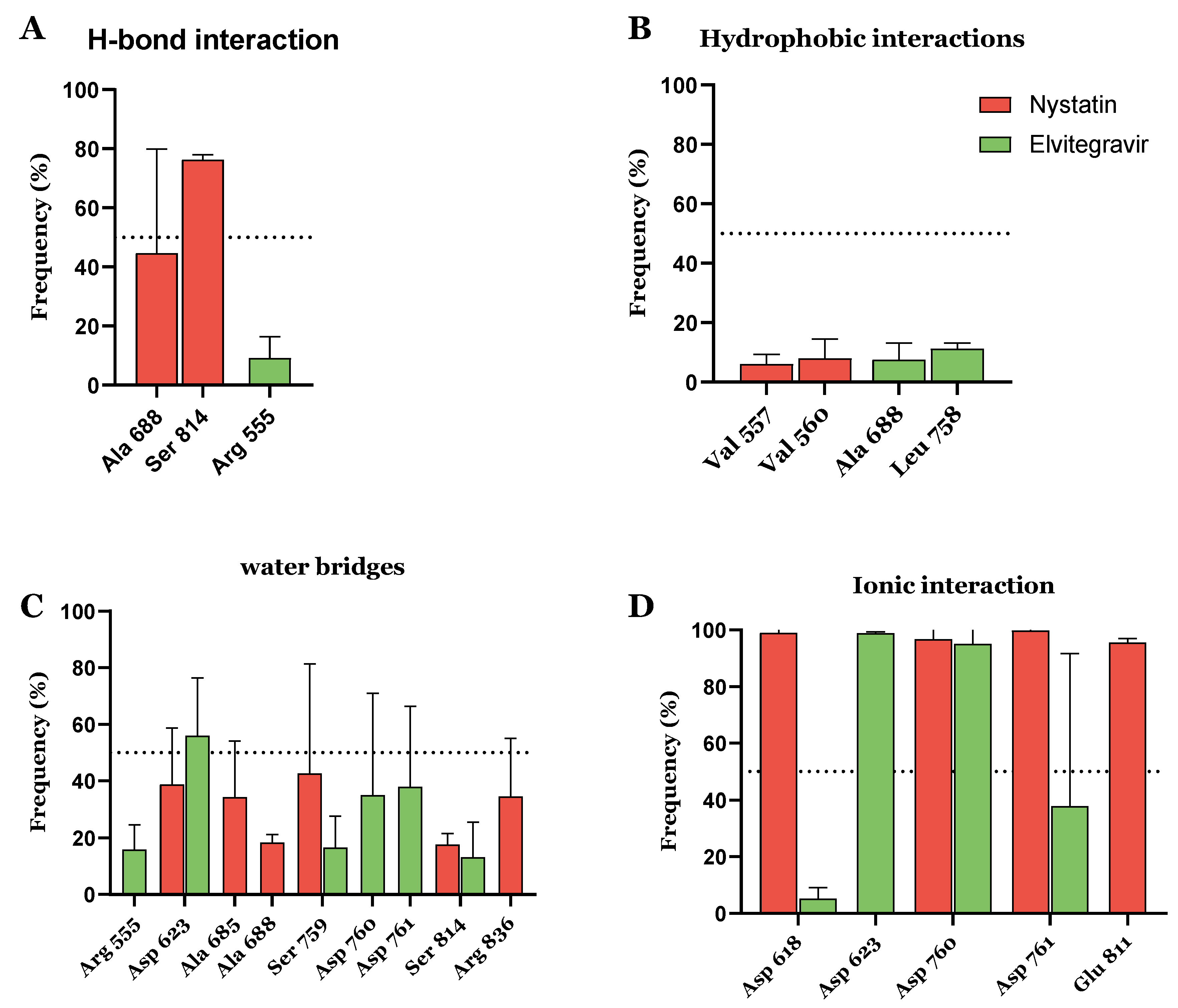
3.3. FDA-Approved Drug Candidates May Inhibit SARS-CoV-2 Replication
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salman, S.; Shah, F.H.; Idrees, J.; Idrees, F.; Velagala, S.; Ali, J.; Khan, A.A. Virtual screening of immunomodulatory medicinal compounds as promising anti-SARS-CoV-2 inhibitors. Future Virol. 2020, 15, 267–275. [Google Scholar] [CrossRef]
- Li, Y.C.; Bai, W.Z.; Hashikawa, T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J. Med. Virol. 2020, 92, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Han, B.; Wang, J. COVID-19: Gastrointestinal Manifestations and Potential Fecal-Oral Transmission. Gastroenterology 2020, 158, 1518–1519. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Li, K.S.M.; Tsang, A.K.L.; Lam, C.S.F.; Ahmed, S.; Chen, H.; Chan, K.-H.; Woo, P.C.Y.; Yuen, K.-Y. Genetic Characterization of Betacoronavirus Lineage C Viruses in Bats Reveals Marked Sequence Divergence in the Spike Protein of Pipistrellus Bat Coronavirus HKU5 in Japanese Pipistrelle: Implications for the Origin of the Novel Middle East Respiratory Sy. J. Virol. 2013, 87, 8638–8650. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, H.; Mashouri, L.; Okpechi, S.C.; Alahari, N. Repurposing existing drugs for the treatment of COVID-19/SARS-CoV-2 infection: A review describing drug mechanisms of action. Biochem. Pharmacol. 2021, 183, 114296. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Li, P.; Ju, X.; Rao, J.; Huang, W.; Ren, L.; Zhang, S.; Xiong, T.; Xu, K.; Zhou, X.; et al. In vivo structural characterization of the SARS-CoV-2 RNA genome identifies host proteins vulnerable to repurposed drugs. Cell 2021, 184, 1865–1883.e20. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Kanhed, A.M.; Patel, D.V.; Teli, D.M.; Patel, N.R.; Chhabria, M.T.; Yadav, M.R. Identification of potential Mpro inhibitors for the treatment of COVID-19 by using systematic virtual screening approach. Mol. Divers. 2021, 25, 383–401. [Google Scholar] [CrossRef]
- Tariq, A.; Mateen, R.M.; Afzal, M.S.; Saleem, M. Paromomycin: A potential dual targeted drug effectively inhibits both spike (S1) and main protease of COVID-19. Int. J. Infect. Dis. 2020, 98, 166–175. [Google Scholar] [CrossRef]
- Kumar, P.; Bhardwaj, T.; Kumar, A.; Gehi, B.R.; Kapuganti, S.K.; Garg, N.; Nath, G.; Giri, R. Reprofiling of approved drugs against SARS-CoV-2 main protease: An in-silico study. J. Biomol. Struct. Dyn. 2020, 40, 3170–3184. [Google Scholar] [CrossRef]
- Tsuji, M. Potential anti-SARS-CoV-2 drug candidates identified through virtual screening of the ChEMBL database for compounds that target the main coronavirus protease. FEBS Open Bio. 2020, 10, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Chakraborti, S.; Dimitrov, A.S.; Gramatikoff, K.; Dimitrov, D.S. The SARS-CoV S glycoprotein: Expression and functional characterization. Biochem. Biophys. Res. Commun. 2003, 312, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Unni, S.; Aouti, S.; Thiyagarajan, S.; Padmanabhan, B. Identification of a repurposed drug as an inhibitor of Spike protein of human coronavirus SARS-CoV-2 by computational methods. J. Biosci. 2020, 45, 130. [Google Scholar] [CrossRef]
- Yin, W.; Luan, X.; Li, Z.; Zhou, Z.; Wang, Q.; Gao, M.; Wang, X.; Zhou, F.; Shi, J.; You, E.; et al. Structural basis for inhibition of the SARS-CoV-2 RNA polymerase from SARS-CoV-2 by remdesivir. Nat. Struct. Mol. Biol. 2021, 28, 319–325. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S.; et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Mottin, M.; Ramos, P.R.P.S.; Sousa, B.K.P.; Neves, B.J.; Foil, D.H.; Zorn, K.M.; Braga, R.C.; Coffee, M.; Southan, C.; et al. Déjà vu: Stimulating open drug discovery for SARS-CoV-2. Drug Discov. Today 2020, 25, 928–941. [Google Scholar] [CrossRef] [PubMed]
- Tahir ul Qamar, M.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Liu, H.; Galasiti Kankanamalage, A.C.; Weerasekara, S.; Hua, D.H.; Groutas, W.C.; Chang, K.O.; Pedersen, N.C. Reversal of the Progression of Fatal Coronavirus Infection in Cats by a Broad-Spectrum Coronavirus Protease Inhibitor. PLoS Pathog. 2016, 12, e1005531. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Tortorici, M.A.; Veesler, D. Structural Insights into Coronavirus Entry. Adv. Virus Res. 2019, 105, 93–116. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Ahn, D.G.; Choi, J.K.; Taylor, D.R.; Oh, J.W. Biochemical characterization of a recombinant SARS coronavirus nsp12 RNA-dependent RNA polymerase capable of copying viral RNA templates. Arch. Virol. 2012, 157, 2095–2104. [Google Scholar] [CrossRef] [Green Version]
- Ziebuhr, J. The coronavirus replicase. Curr. Top. Microbiol. Immunol. 2005, 287, 57–94. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [Green Version]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019, 10, 2342. [Google Scholar] [CrossRef] [Green Version]
- Mcdonald, S.M. RNA synthetic mechanisms employed by diverse families of RNA viruses. Wiley Interdiscip. Rev. RNA 2013, 4, 351–367. [Google Scholar] [CrossRef]
- Van Hemert, M.J.; Van Den Worm, S.H.E.; Knoops, K.; Mommaas, A.M.; Gorbalenya, A.E.; Snijder, E.J. SARS-coronavirus replication/transcription complexes are membrane-protected and need a host factor for activity in vitro. PLoS Pathog. 2008, 4, e1000054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.C.; Chen, M.Y.; Lee, W.S.; Chang, Y.L. Potential therapeutic agents against COVID-19: What we know so far. J. Chin. Med. Assoc. 2020, 83, 534–536. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, T.P.; Sims, A.C.; Zhou, S.; Graham, R.L.; Pruijssers, A.J.; Agostini, M.L.; Leist, S.R.; Schafer, A.; Dinnon, K.H.; Stevens, L.J.; et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 2020, 12, eabb5883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, I.F.N.; Lung, K.C.; Tso, E.Y.K.; Liu, R.; Chung, T.W.H.; Chu, M.Y.; Ng, Y.Y.; Lo, J.; Chan, J.; Tam, A.R.; et al. Triple combination of interferon beta-1b, lopinavir–ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: An open-label, randomised, phase 2 trial. Lancet 2020, 395, 1695–1704. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput. Aided. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Adzhigirey, M.; Day, T.; Sherman, W.; Madhavi Sastry, G.; Annabhimoju, R. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins Struct. Funct. Bioinforma. 2004, 367, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, J.; Ikram, S.; Ahmad, F.; Rehman, I.U.; Mushtaq, M. SARS-CoV-2 RNA Dependent RNA polymerase (RdRp)—A drug repurposing study. Heliyon 2020, 6, e04502. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Sun, H.; Xi, L.; Li, J.; Yang, Y.; Liu, H.; Yao, X. Molecular modeling study of checkpoint kinase 1 inhibitors by multiple docking strategies and prime/MM-GBSA calculation. J. Comput. Chem. 2011, 32, 2800–2809. [Google Scholar] [CrossRef]
- Lyne, P.D.; Lamb, M.L.; Saeh, J.C. Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. J. Med. Chem. 2006, 49, 4805–4808. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins Struct. Funct. Bioinforma. 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [Green Version]
- Bowers, K.J.; Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the SC '06: 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Schrödinger, L. PyMol, The PyMOL Molecular Graphics System, Version 2.4; Schrödinger, LLC: New York, NY, USA, 2020. [Google Scholar]
- Thiruvengadam, R.; Awasthi, A.; Medigeshi, G.; Bhattacharya, S.; Mani, S.; Sivasubbu, S.; Shrivastava, T. Articles Effectiveness of ChAdOx1 nCoV-19 vaccine against SARS-CoV-2 infection during the delta ( B.1.617.2) variant surge in India: A test-negative, case-control study and a mechanistic study of post-vaccination immune responses. Lancet Infect. Dis. 2021, 3099, 1–10. [Google Scholar] [CrossRef]
- Public Health England. Technical Briefing 23. SARS-CoV-2 Variants of Concern and Variants under Investigation in England. 2021, pp. 1–61. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1018547/Technical_Briefing_23_21_09_16.pdf (accessed on 1 March 2022).
- Abdool, S.S.; Abdool, Q. Omicron SARS-CoV-2 Variant: A New Chapter in the COVID-19 Pandemic. Lancet 2021, 398, 2126–2128. [Google Scholar] [CrossRef]
- Cort, A.; Timur, M.; Ozdemir, E.; Kucuksayan, E.; Ozben, T. Synergistic anticancer activity of curcumin and bleomycin: An in vitro study using human malignant testicular germ cells. Mol. Med. Rep. 2012, 5, 1481–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgy, O.; Wettstein, G.; Bellaye, P.S.; Decologne, N.; Racoeur, C.; Goirand, F.; Beltramo, G.; Hernandez, J.F.; Kenani, A.; Camus, P.; et al. Deglycosylated bleomycin has the antitumor activity of bleomycin without pulmonary toxicity. Sci. Transl. Med. 2016, 8, 326ra20. [Google Scholar] [CrossRef] [PubMed]
- Che, R.; Zhu, Q.; Yu, J.; Li, J.; Yu, J.; Lu, W. Syntheses of two kinds of disaccharide subunits of antitumor antibiotic bleomycins. Tetrahedron 2017, 73, 6172–6180. [Google Scholar] [CrossRef]
- Cooper, I.; Atrakchi, D.; Walker, M.D.; Horovitz, A.; Fridkin, M.; Shechter, Y. Converting bleomycin into a prodrug that undergoes spontaneous reactivation under physiological conditions. Toxicol. Appl. Pharmacol. 2019, 384, 114782. [Google Scholar] [CrossRef]
- Zhang, Z.F.; Fan, S.H.; Zheng, Y.L.; Lu, J.; Wu, D.M.; Shan, Q.; Hu, B. Troxerutin improves hepatic lipid homeostasis by restoring NAD+-depletion-mediated dysfunction of lipin 1 signaling in high-fat diet-treated mice. Biochem. Pharmacol. 2014, 91, 74–86. [Google Scholar] [CrossRef]
- Saif, M.W.; Parikh, R.; Ray, D.; Kaye, J.A.; Kurosky, S.K.; Thomas, K.; Ramirez, R.A.; Halfdanarson, T.R.; Beveridge, T.J.R.; Mirakhur, B.; et al. Medical record review of transition to lanreotide following octreotide for neuroendocrine tumors. J. Gastrointest. Oncol. 2019, 10, 674–687. [Google Scholar] [CrossRef]
- Caron, P.J.; Bevan, J.S.; Petersenn, S.; Houchard, A.; Sert, C.; Webb, S.M. Effects of lanreotide Autogel primary therapy on symptoms and quality-of-life in acromegaly: Data from the PRIMARYS study. Pituitary 2016, 19, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Biermasz, N.R. New medical therapies on the horizon: Oral octreotide. Pituitary 2017, 20, 149–153. [Google Scholar] [CrossRef] [Green Version]
- Zafer, M.M.; El-Mahallawy, H.A.; Abdulhak, A.; Amin, M.A.; Al-Agamy, M.H.; Radwan, H.H. Emergence of colistin resistance in multidrug-resistant Klebsiella pneumoniae and Escherichia coli strains isolated from cancer patients. Ann. Clin. Microbiol. Antimicrob. 2019, 18, 40. [Google Scholar] [CrossRef]
- Nation, R.L.; Li, J. Colistin in the 21st century. Curr. Opin. Infect. Dis. 2009, 22, 535–543. [Google Scholar] [CrossRef]
- Stokowa-Sołtys, K.; Barbosa, N.A.; Kasprowicz, A.; Wieczorek, R.; Gaggelli, N.; Gaggelli, E.; Valensin, G.; Wrzesiński, J.; Ciesiołka, J.; Kuliński, T.; et al. Studies of viomycin, an anti-tuberculosis antibiotic: Copper(II) coordination, DNA degradation and the impact on delta ribozyme cleavage activity. Dalt. Trans. 2016, 45, 8645–8658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossman, R.I.; Modic, M.T. A randomized comparison of iodixanol and iohexol in adult intracranial computed tomography scanning. Acad. Radiol. 1996, 3 (Suppl S3), S488–S494. [Google Scholar] [CrossRef]
- Seong, J.M.; Choi, N.K.; Lee, J.; Chang, Y.; Kim, Y.J.; Yang, B.R.; Jin, X.M.; Kim, J.Y.; Park, B.J. Comparison of the safety of seven iodinated contrast media. J. Korean Med. Sci. 2013, 28, 1703–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manoharan, Y.; Haridas, V.; Vasanthakumar, K.C. Curcumin: A Wonder Drug as a Preventive Measure for COVID19 Management. Indian J. Clin. Biochem. 2020, 35, 373–375. [Google Scholar] [CrossRef]
- Gupta, S.C.; Patchva, S.; Aggarwal, B.B. Therapeutic roles of curcumin: Lessons learned from clinical trials. AAPS J. 2013, 15, 195–218. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, A.J.; Concepcion, R.; Kirchner, F.K.; McDougal, W.S.; Winfield, A.C. Ioversol for intravenous urography: A comparison study. Urol. Radiol. A J. Diagn. Imaging 1990, 12, 56–60. [Google Scholar] [CrossRef]
- Boyer, T.D. Advances in Hepatology. Gastroenterol. Hepatol. 2007, 3, 773–774. [Google Scholar]
- Bello, M.; Martínez-Muñoz, A.; Balbuena-Rebolledo, I. Identification of saquinavir as a potent inhibitor of dimeric SARS-CoV2 main protease through MM/GBSA. J. Mol. Model. 2020, 26, 340. [Google Scholar] [CrossRef]
- Noble, S.; Faulds, D. Saquinavir. Drug Eval. 1996, 52, 93–112. [Google Scholar] [CrossRef]
- Weidner, L.D.; Fung, K.L.; Kannan, P.; Moen, J.K.; Kumar, J.S.; Mulder, J.; Innis, R.B.; Gottesman, M.M.; Hall, M.D. Tariquidar is an inhibitor and not a substrate of human and mouse P-glycoprotein. Drug Metab. Dispos. 2016, 44, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Chung, B.H.; Horie, S.; Chiong, E. Clinical studies investigating the use of leuprorelin for prostate cancer in Asia. Prostate Int. 2020, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.T.; Grzeskowiak, C.L.; Fradette, J.J.; Gibson, L.A.; Rodriguez, L.B.; Creighton, C.J.; Scott, K.L.; Gibbons, D.L. TMEM106B drives lung cancer metastasis by inducing TFEB-dependent lysosome synthesis and secretion of cathepsins. Nat. Commun. 2018, 9, 2731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siklos, M.; BenAissa, M.; Thatcher, G.R.J. Cysteine proteases as therapeutic targets: Does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm. Sin. B 2015, 5, 506–519. [Google Scholar] [CrossRef] [Green Version]
- Heiser, K.; McLean, P.F.; Davis, C.; Fogelson, B.; Gordon, H.B.; Jacobson, P.; Hurst, B.; Miller, B.; Alfa, R.W.; Earnshaw, B.A.; et al. Identification of potential treatments for COVID-19 through artificial intelligence-enabled phenomic analysis of human cells infected with SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mahanta, S.; Chowdhury, P.; Gogoi, N.; Goswami, N.; Borah, D.; Kumar, R.; Chetia, D.; Borah, P.; Alak, K.; Gogoi, B. Potential anti-viral activity of approved repurposed drug against main protease of SARS- CoV-2: An in silico based approach. J. Biomol. Struct. Dyn. 2021, 39, 3802–3811. [Google Scholar] [CrossRef]
- Chakraborti, S.; Bheemireddy, S.; Srinivasan, N. Repurposing drugs against the main protease of SARS-CoV-2: Mechanism-based insights supported by available laboratory and clinical data. Mol. Omi. 2020, 16, 474–491. [Google Scholar] [CrossRef] [PubMed]
- George, P.M.; Wells, A.U.; Jenkins, R.G. Personal View Pulmonary fibrosis and COVID-19: The potential role for antifibrotic therapy. Lancet Respir. 2020, 8, 807–815. [Google Scholar] [CrossRef]
- Maffucci, I.; Contini, A. In Silico Drug Repurposing for SARS-CoV-2 Main Proteinase and Spike Proteins. J. Proteome Res. 2020, 19, 4637–4648. [Google Scholar] [CrossRef]
- Hajialyani, M.; Farzaei, M.H.; Echeverría, J.; Nabavi, S.M.; Uriarte, E.; Eduardo, S.S. Hesperidin as a neuroprotective agent: A review of animal and clinical evidence. Molecules 2019, 24, 648. [Google Scholar] [CrossRef] [Green Version]
- Lalani, S.; Ti, L.; Laa, C. Antiviral peptides against Enterovirus A71 causing hand, foot and mouth disease. Peptides 2021, 136, 170443. [Google Scholar] [CrossRef]
- O’Brien, J.J.; Campoli-Richards, D.M. Acyclovir: An Updated Review of its Antiviral Activity, Pharmacokinetic Properties and Therapeutic Efficacy. Drugs 1989, 37, 233–309. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L. Iron deficiency anemia: A common and curable disease. Cold Spring Harb. Perspect. Med. 2013, 3, a011866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, R.I.; Fillgrove, K.L.; Yee, K.L.; Liang, Y.; Lu, B.; Tatavarti, A.; Liu, R.; Anderson, M.S.; Behm, M.O.; Fan, L.; et al. Characterisation of the absorption, distribution, metabolism, excretion and mass balance of doravirine, a non-nucleoside reverse transcriptase inhibitor in humans. Xenobiotica 2019, 49, 422–432. [Google Scholar] [CrossRef]
- Van Gorkom, B.A.P.; Karrenbeld, A.; Van der Sluis, T.; Zwart, N.; De Vries, E.G.E.; Kleibeuker, J.H. Apoptosis induction by sennoside laxatives in man: Escape from a protective mechanism during chronic sennoside use? J. Pathol. 2001, 194, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; Carli, I.; Del Vecchio, C.; Xu, L.; Corona, A.; Grandi, N.; Piano, D.; Maccioni, E.; Distinto, S.; Parolin, C.; et al. Sennoside A, derived from the traditional chinese medicine plant Rheum L., is a new dual HIV-1 inhibitor effective on HIV-1 replication. Phytomedicine 2016, 23, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Zempleni, J.; Galloway, J.R.; McCormick, D.B. Pharmacokinetics of orally and intravenously administered riboflavin in healthy humans. Am. J. Clin. Nutr. 1996, 63, 54–66. [Google Scholar] [CrossRef] [Green Version]
- Islam, R.; Parves, M.R.; Paul, A.S.; Uddin, N.; Rahman, M.S.; Mamun, A.A.; Hossain, M.N.; Ali, M.A.; Halim, M.A. A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 39, 3213–3224. [Google Scholar] [CrossRef]
- Chen, Y.W.; Yiu, C.P.B.; Wong, K.Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CLpro) structure: Virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research 2020, 9, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural Basis of Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Front. Chem. 2021, 9, 622898. [Google Scholar] [CrossRef]
- Jain, R.; Mujwar, S. Repurposing metocurine as main protease inhibitor to develop novel antiviral therapy for COVID-19. Struct. Chem. 2020, 31, 2487–2499. [Google Scholar] [CrossRef]
- Xue, X.; Yang, H.; Shen, W.; Zhao, Q.; Li, J.; Yang, K.; Chen, C.; Jin, Y.; Bartlam, M.; Rao, Z. Production of Authentic SARS-CoV Mpro with Enhanced Activity: Application as a Novel Tag-cleavage Endopeptidase for Protein Overproduction. J. Mol. Biol. 2007, 366, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.-J.; Huynh, T.-K.; Yang, C.-S.; Shen, Y.-C.; Tu, C.-Y.; Wu, Y.-C.; Tang, C.-H.; Huang, W.-C.; Chen, Y.; Ho, C.-Y. Hesperidin is a potential inhibitor against SARS-CoV-2 infection. Nutrients 2021, 13, 2800. [Google Scholar] [CrossRef] [PubMed]
- Santos, I.D.; Grosche, V.R.; Bergamini, F.R.G.; Sabino-Silva, R.; Jardim, A.C.G. Antivirals against Coronaviruses: Candidate Drugs for SARS-CoV-2 Treatment? Front. Microbiol. 2020, 11, 1818. [Google Scholar] [CrossRef] [PubMed]
- Peters, H.L.; Jochmans, D.; De Wilde, A.H.; Posthuma, C.C.; Snijder, E.J.; Neyts, J.; Seley-Radtke, K.L. Design, synthesis and evaluation of a series of acyclic fleximer nucleoside analogues with anti-coronavirus activity. Bioorg. Med. Chem. Lett. 2015, 25, 2923–2926. [Google Scholar] [CrossRef]
- Unal, M.A.; Bitirim, C.V.; Summak, G.Y.; Bereketoglu, S.; Zeytin, I.C.; Besbinar, O.; Gurcan, C.; Aydos, D.; Goksoy, E.; Kocakaya, E.; et al. Ribavirin shows antiviral activity against sars-cov-2 and downregulates the activity of tmprss2 and the expression of ace2 in vitro. Can. J. Physiol. Pharmacol. 2021, 99, 449–460. [Google Scholar] [CrossRef]
- DeJong, C.; Spinelli, M.A.; Okochi, H.; Gandhi, M. Tenofovir-based PrEP for COVID-19: An untapped opportunity? Aids 2021, 35, 1509–1511. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.; Gliwa, A.; et al. Transmission, infectivity, and antibody neutralization of an emerging SARS-CoV-2 variant in California carrying a L452R spike protein mutation. MedRxiv 2021. [Google Scholar] [CrossRef]
- Joshi, M.; Kumar, M.; Srivastava, V.; Kumar, D.; Pandit, R.; Joshi, C.G. First detection of SARS-CoV-2 Delta variant (B.1.617.2) in the wastewater of (Ahmedabad), India. MedRxiv 2021. [Google Scholar] [CrossRef]
- Yi, C.; Sun, X.; Ye, J.; Ding, L.; Liu, M.; Yang, Z.; Lu, X.; Zhang, Y.; Ma, L.; Gu, W.; et al. Key residues of the receptor binding motif in the spike protein of SARS-CoV-2 that interact with ACE2 and neutralizing antibodies. Cell. Mol. Immunol. 2020, 17, 621–630. [Google Scholar] [CrossRef]
- Ugur, I.; Marion, A. Molecular modelling reveals eight novel druggable binding sites in SARS-CoV-2’s spike protein. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Moore, J.P.; Offit, P.A. SARS-CoV-2 Vaccines and the Growing Threat of Viral Variants. JAMA-J. Am. Med. Assoc. 2021, 325, 821–822. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Schmidt, F.; Weisblum, Y.; Muecksch, F.; Barnes, C.O.; Finkin, S.; Schaefer-Babajew, D.; Cipolla, M.; Gaebler, C.; Lieberman, J.A.; et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature 2021, 592, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Zhou, S.; Tuttle, K.S.; Kim, A.; Li, W.; Dimitrov, D.S.; et al. Structural analysis of receptor binding domain mutations in SARS-CoV-2 variants of concern that modulate ACE2 and antibody binding. Cell Rep. 2021, 37, 110156. [Google Scholar] [CrossRef]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef]
- Miell, J.; Dhanjal, P.; Jamookeeah, C. Evidence for the use of demeclocycline in the treatment of hyponatraemia secondary to SIADH: A systematic review. Int. J. Clin. Pract. 2015, 69, 1396–1417. [Google Scholar] [CrossRef] [Green Version]
- Hasan, M.K.; Kamruzzaman, M.; Bin Manjur, O.H.; Mahmud, A.; Hussain, N.; Alam Mondal, M.S.; Hosen, M.I.; Bello, M.; Rahman, A. Structural analogues of existing anti-viral drugs inhibit SARS-CoV-2 RNA dependent RNA polymerase: A computational hierarchical investigation. Heliyon 2021, 7, e06435. [Google Scholar] [CrossRef]
- Gordon, C.J.; Tchesnokov, E.P.; Schinazi, R.F.; Götte, M. Molnupiravir promotes SARS-CoV-2 mutagenesis via the RNA template. J. Biol. Chem. 2021, 297, 100770. [Google Scholar] [CrossRef]
- Kabinger, F.; Stiller, C.; Schmitzová, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Höbartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef]
- Elfiky, A.A. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sci. 2020, 253, 117592. [Google Scholar] [CrossRef]
- Kim, S.H.; MacIntyre, D.A.; Hanyaloglu, A.C.; Blanks, A.M.; Thornton, S.; Bennett, P.R.; Terzidou, V. The Oxytocin Receptor Antagonist, Atosiban, Activates pro-Inflammatory Pathways in Human Amnion via G(αi) Signalling. Mol. Cell. Endocrinol. 2016, 420, 11–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, P.; Khan, F. A Mechanistic Review of the Anticancer Potential of Hesperidin, a Natural Flavonoid from Citrus Fruits. Nutr. Res. 2021, 92, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Radu, D.; Åhlin, A.; Svanborg, P.; Lindefors, N. Anxiogenic Effects of the CCK B Agonist Pentagastrin in Humans and Dose-Dependent Increase in Plasma C-Peptide Levels. Psychopharmacology 2002, 161, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Aggerbeck, M.; Guellaën, G.; Hanoune, J. N-Aralkyl Substitution Increases the Affinity of Adrenergic Drugs for the Alpha-Adrenoceptor in Rat Liver. Br. J. Pharmacol. 1979, 65, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Ackerson, N.O.B.; Liberatore, H.K.; Plewa, M.J.; Richardson, S.D.; Ternes, T.A.; Duirk, S.E. Disinfection Byproducts and Halogen-Specific Total Organic Halogen Speciation in Chlorinated Source Waters – The Impact of Iopamidol and Bromide. J. Environ. Sci. 2020, 89, 90–101. [Google Scholar] [CrossRef]
- Meijenhorst, G.C.; de Bruin, J.N. Hexabrix (ioxaglate), a New Low Osmolality Contrast Agent for Lumbar Epidural Double-Catheter Venography. Neuroradiology 1980, 20, 29–32. [Google Scholar] [CrossRef]
- Garriga, C.; Pérez-Elías, M.J.; Delgado, R.; Ruiz, L.; Nájera, R.; Pumarola, T.; del Mar Alonso-Socas, M.; García-Bujalance, S.; Menéndez-Arias, L. Mutational Patterns and Correlated Amino Acid Substitutions in the HIV-1 Protease after Virological Failure to Nelfinavir- and Lopinavir/ritonavir-Based Treatments. J. Med. Virol. 2007, 79, 1617–1628. [Google Scholar] [CrossRef]
- Karlgren, M.; Vildhede, A.; Norinder, U.; Wisniewski, J.R.; Kimoto, E.; Lai, Y.; Haglund, U.; Artursson, P. Classification of Inhibitors of Hepatic Organic Anion Transporting Polypeptides (OATPs): Influence of Protein Expression on Drug–Drug Interactions. J. Med. Chem. 2012, 55, 4740–4763. [Google Scholar] [CrossRef]
- Zwiener, C.; Glauner, T.; Sturm, J.; Wörner, M.; Frimmel, F.H. Electrochemical Reduction of the Iodinated Contrast Medium Iomeprol: Iodine Mass Balance and Identification of Transformation Products. Anal. Bioanal. Chem. 2009, 395, 1885–1892. [Google Scholar] [CrossRef]
- Chow, S.L.; Ng, T.M.H.; Litwinski, R.A.; Kangavari, S.; Weiss, M. Effect of Iodixanol and Ioxilan on QT Interval and Renal Function in Patients with Systolic Heart Failure. Int. J. Cardiol. 2012, 154, 17–21. [Google Scholar] [CrossRef]
- Diaz-Montero, C.M.; Wang, Y.; Shao, L.; Feng, W.; Zidan, A.-A.; Pazoles, C.J.; Montero, A.J.; Zhou, D. The Glutathione Disulfide Mimetic NOV-002 Inhibits Cyclophosphamide-Induced Hematopoietic and Immune Suppression by Reducing Oxidative Stress. Free Radic. Biol. Med. 2012, 52, 1560–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stover, P.J.; Field, M.S. Trafficking of Intracellular Folates. Adv. Nutr. 2011, 2, 325–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, V.T.G.; Suno, M. Levoleucovorin as Replacement for Leucovorin in Cancer Treatment. Ann. Pharmacother. 2012, 46, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Adolph, J.M.G.; Engelkamp, H.; Herbig, W.; Peters, P.E.; Wenzel-Hora, B.I. Iotrolan in Urography: Efficacy and Tolerance in Comparison with Iohexol and Iopamidol. Eur. Radiol. 1995, 5, S63–S68. [Google Scholar] [CrossRef]
- Gan, X.-D.; Wei, B.-Z.; Fang, D.; Fang, Q.; Li, K.-Y.; Ding, S.-L.-Y.; Peng, S.; Wan, J. Efficacy and Safety Analysis of New P2Y12 Inhibitors versus Clopidogrel in Patients with Percutaneous Coronary Intervention: A Meta-Analysis. Curr. Med. Res. Opin. 2015, 31, 2313–2323. [Google Scholar] [CrossRef]
- Holleran, J.L.; Parise, R.A.; Guo, J.; Kiesel, B.F.; Taylor, S.E.; Percy Ivy, S.; Chu, E.; Beumer, J.H. Quantitation of Iohexol, a Glomerular Filtration Marker, in Human Plasma by LC–MS/MS. J. Pharm. Biomed. Anal. 2020, 189, 113464. [Google Scholar] [CrossRef]
- Dhondt, L.; Croubels, S.; De Cock, P.; Dhont, E.; De Baere, S.; De Paepe, P.; Devreese, M. Volumetric Absorptive Microsampling as Alternative Sampling Technique for Renal Function Assessment in the Paediatric Population Using Iohexol. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2021, 1171, 122623. [Google Scholar] [CrossRef]
- Hofmann, M.; Maecke, H.; Börner, A.; Weckesser, E.; Schöffski, P.; Oei, M.; Schumacher, J.; Henze, M.; Heppeler, A.; Meyer, G.; et al. Biokinetics and Imaging with the Somatostatin Receptor PET Radioligand 68Ga-DOTATOC: Preliminary Data. Eur. J. Nucl. Med. 2001, 28, 1751–1757. [Google Scholar] [CrossRef]
- Carlquist, M.; Frejd, T.; Gorwa-Grauslund, M.F. Flavonoids as Inhibitors of Human Carbonyl Reductase 1. Chem. Biol. Interact. 2008, 174, 98–108. [Google Scholar] [CrossRef]
- Zoghbi, G.J.; Iskandrian, A.E. Selective Adenosine Agonists and Myocardial Perfusion Imaging. J. Nucl. Cardiol. 2012, 19, 126–141. [Google Scholar] [CrossRef]
- Hirshfeld, J.W., Jr.; Wieland, J.; Davis, C.A.; Giles, B.D.; Passione, D.; Ray, M.B.; Ripley, N.S. Hemodynamic and Electrocardiographic Effects of Ioversol during Cardiac Angiography. Investig. Radiol. 1989, 24, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Peters, F.; Ellermann, I.; Steinbicker, A.U. Intravenous Iron for Treatment of Anemia in the 3 Perisurgical Phases: A Review and Analysis of the Current Literature. Anesth. Analg. 2018, 126, 1268–1282. [Google Scholar] [CrossRef] [PubMed]
- Geisser, P.; Burckhardt, S. The Pharmacokinetics and Pharmacodynamics of Iron Preparations. Pharmaceutics 2011, 3, 12–33. [Google Scholar] [CrossRef]
- Charytan, C.; Levin, N.; Al-Saloum, M.; Hafeez, T.; Gagnon, S.; Van Wyck, D.B. Efficacy and Safety of Iron Sucrose for Iron Deficiency in Patients with Dialysis-Associated Anemia: North American Clinical Trial. Am. J. Kidney Dis. 2001, 37, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Leonard, R.; Yellowlees, A.; Mansi, J.; Fallowfield, L.; Jenkins, V. The Affect of Goserelin on the QoL of Women Having Chemotherapy for EBC: Results from the OPTION Trial. Breast 2020, 52, 122–131. [Google Scholar]
- Aljabri, B.; Lilleby, W.; Switlyk, M.D.; Tafjord, G. Restart of Androgen Deprivation Therapy after Goserelin Induced Pituitary Apoplexy in a Patient with Disseminated Prostate Cancer a Case Report and Five-Years Follow-Up. Urol. Case Rep. 2021, 37, 101648. [Google Scholar] [CrossRef]
- Tuyen, D.T.; Yew, G.Y.; Cuong, N.T.; Hoang, L.T.; Yen, H.T.; Thao, P.T.H.; Thao, N.T.; le Thanh, N.S.; Trang, N.T.H.; Trung, N.T.; et al. Selection, Purification, and Evaluation of Acarbose−an α-Glucosidase Inhibitor from Actinoplanes Sp. Chemosphere 2021, 265, 129167. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, J.; Hu, J.; Chen, Y.; Dong, B.; Wang, Y. A Comparative Study of Acarbose, Vildagliptin and Saxagliptin Intended for Better Efficacy and Safety on Type 2 Diabetes Mellitus Treatment. Life Sci. 2021, 274, 119069. [Google Scholar] [CrossRef]
- Loho, T.; Dharmayanti, A. Colistin: An Antibiotic and Its Role in Multiresistant Gram-Negative Infections. Acta Med. Indones. 2015, 47, 157–168. [Google Scholar] [PubMed]
- Weiner, C.P.; Buhimschi, C. Drugs for Pregnant and Lactating Women E-Book; Elsevier Health Sciences: Hoboken, NJ, USA, 2009; ISBN 9781437721362. [Google Scholar]
- Barza, M. Imipenem: First of a New Class of Beta-Lactam Antibiotics. Ann. Intern. Med. 1985, 103, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D.; Perry, C.M. Zoledronic Acid. Drugs Aging 2008, 25, 963–986. [Google Scholar] [CrossRef] [PubMed]
- Wellington, K.; Goa, K.L. Zoledronic Acid. Drugs 2012, 63, 417–437. [Google Scholar] [CrossRef]
- Zhong, X.; Ma, Z.; Su, Y.; Li, Z.; Liao, Y.; Pan, X.; Zang, L.; Zhou, S. Flavin Adenine Dinucleotide Ameliorates Hypertensive Vascular Remodeling via Activating Short Chain Acyl-CoA Dehydrogenase. Life Sci. 2020, 258, 118156. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Oft, J.; Dorff, T.; Pal, S.; Agarwal, N.; Figlin, R.A.; Posadas, E.M.; Freedland, S.J.; Gong, J. COVID-19 and Androgen-Targeted Therapy for Prostate Cancer Patients. Endocr. Relat. Cancer 2020, 27, R281–R292. [Google Scholar] [CrossRef]
- Müller, J.; Jewell, K.S.; Schulz, M.; Hermes, N.; Ternes, T.A.; Drewes, J.E.; Hübner, U. Capturing the Oxic Transformation of Iopromide – A Useful Tool for an Improved Characterization of Predominant Redox Conditions and the Removal of Trace Organic Compounds in Biofiltration Systems? Water Res. 2019, 152, 274–284. [Google Scholar] [CrossRef]
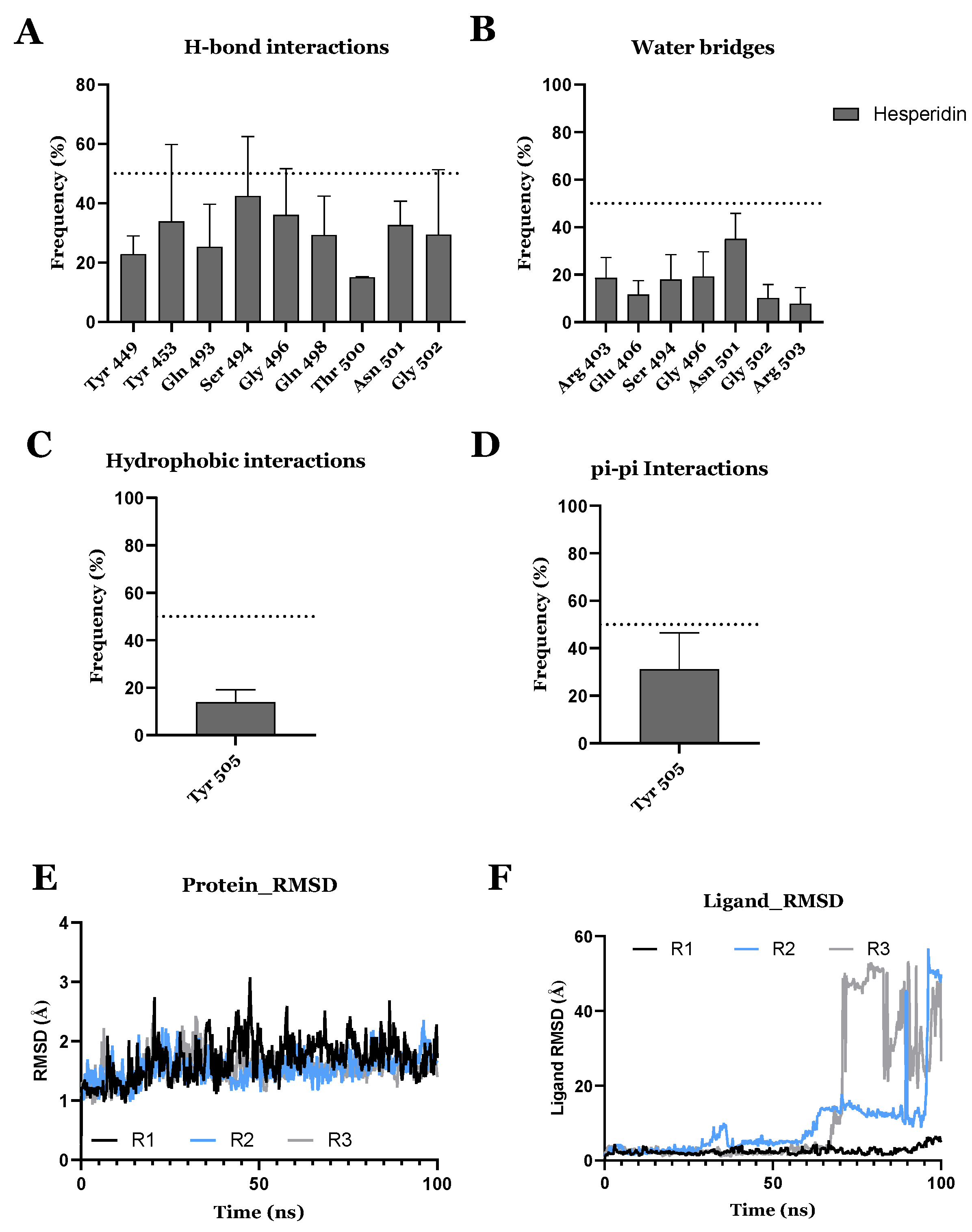
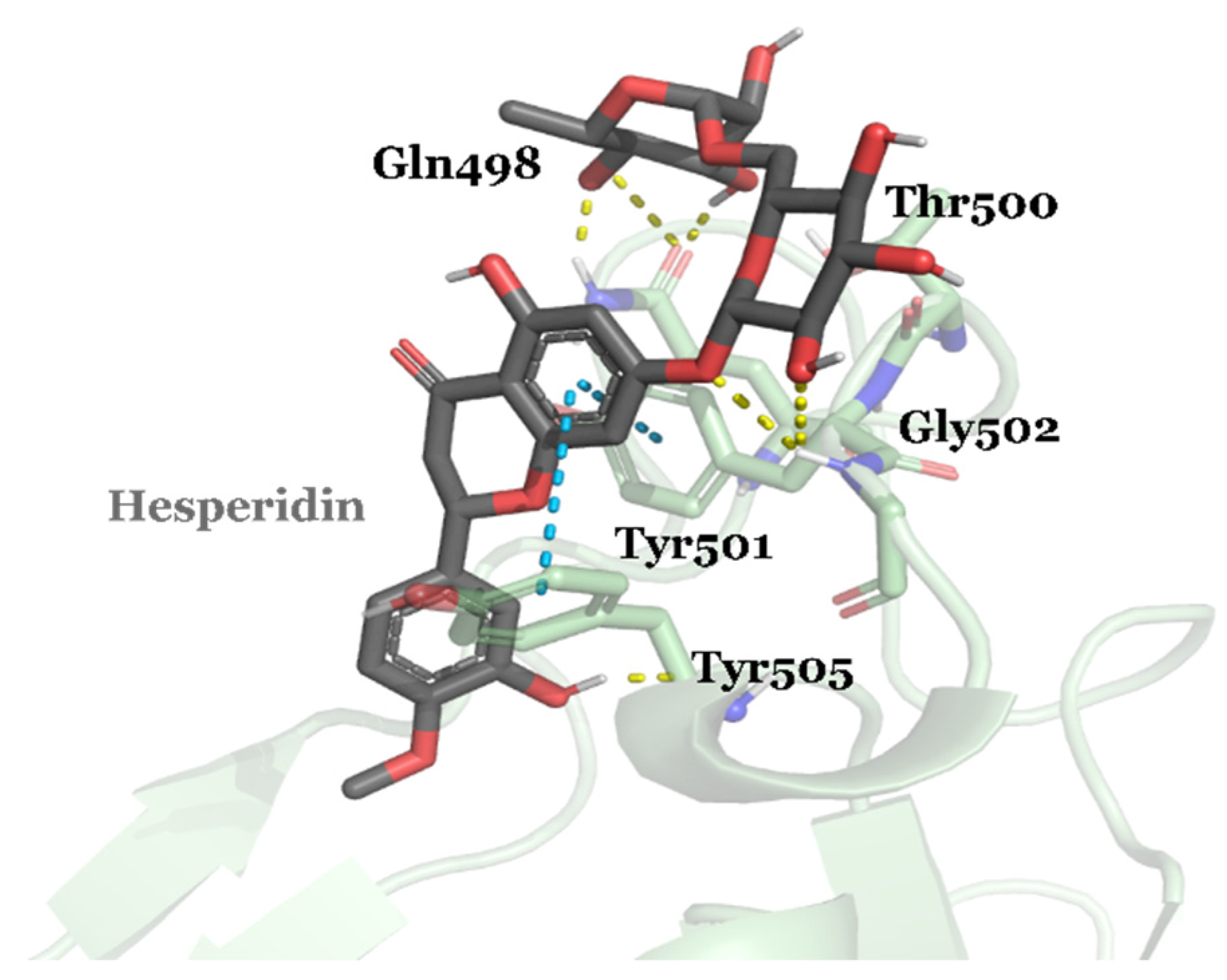
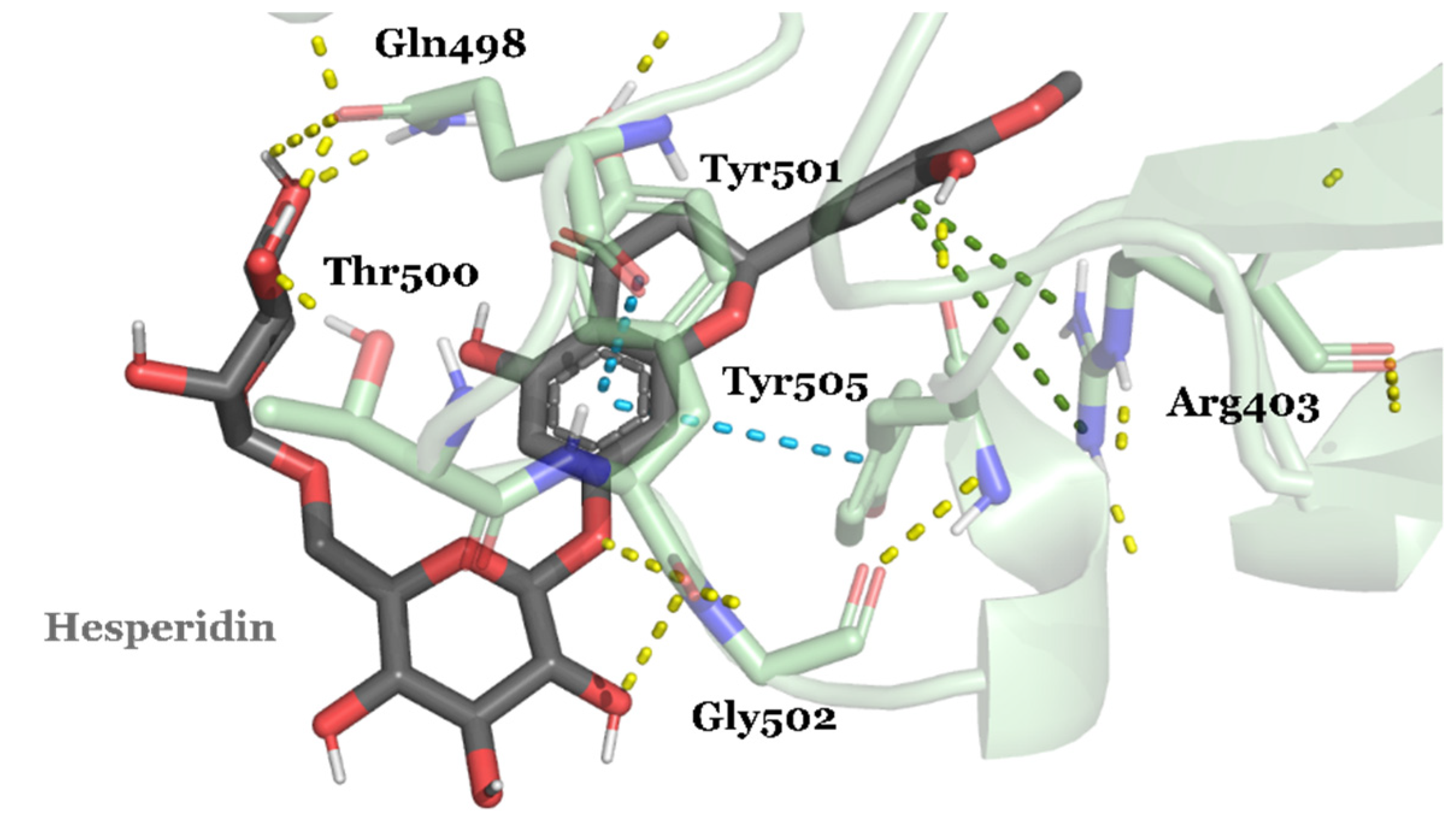
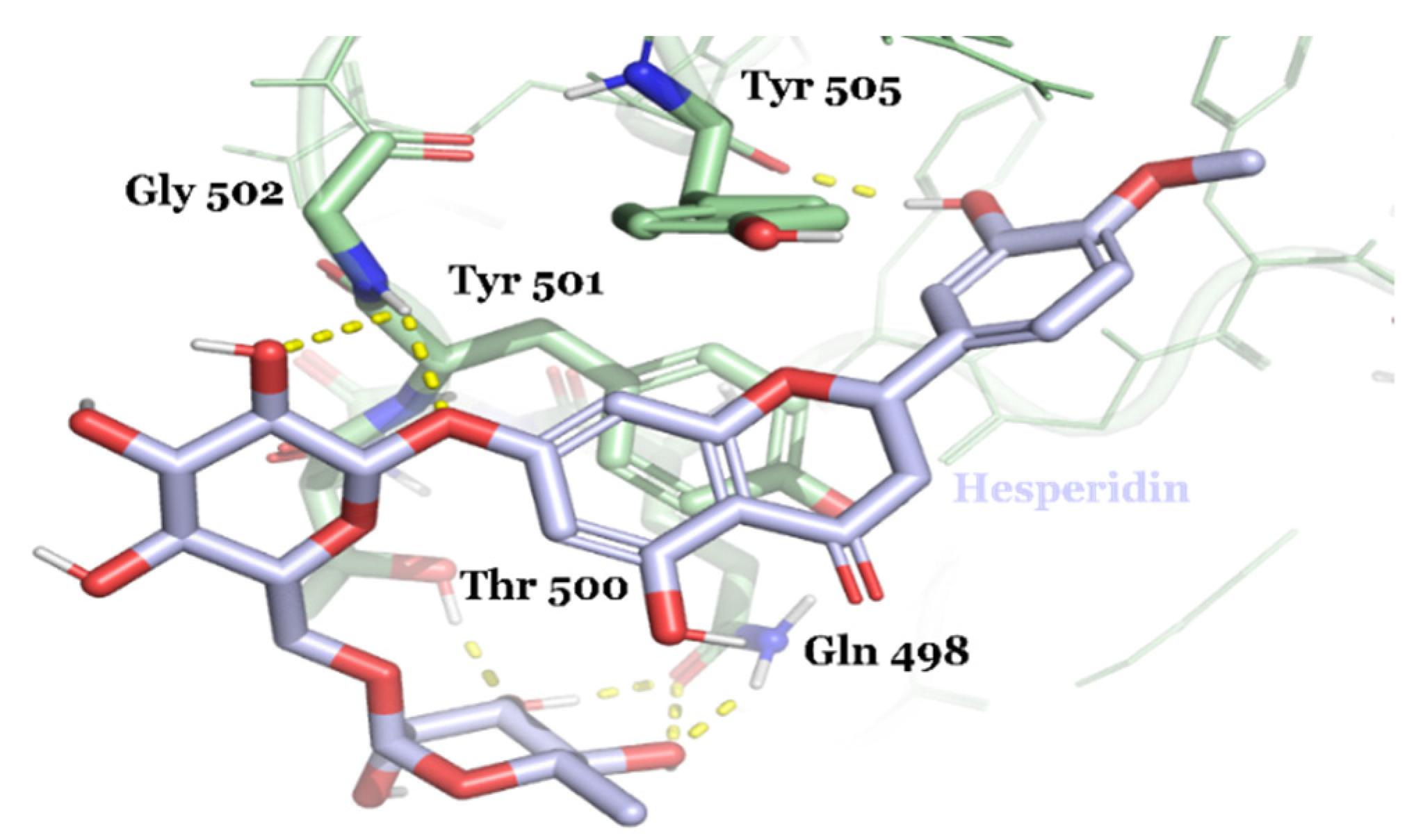
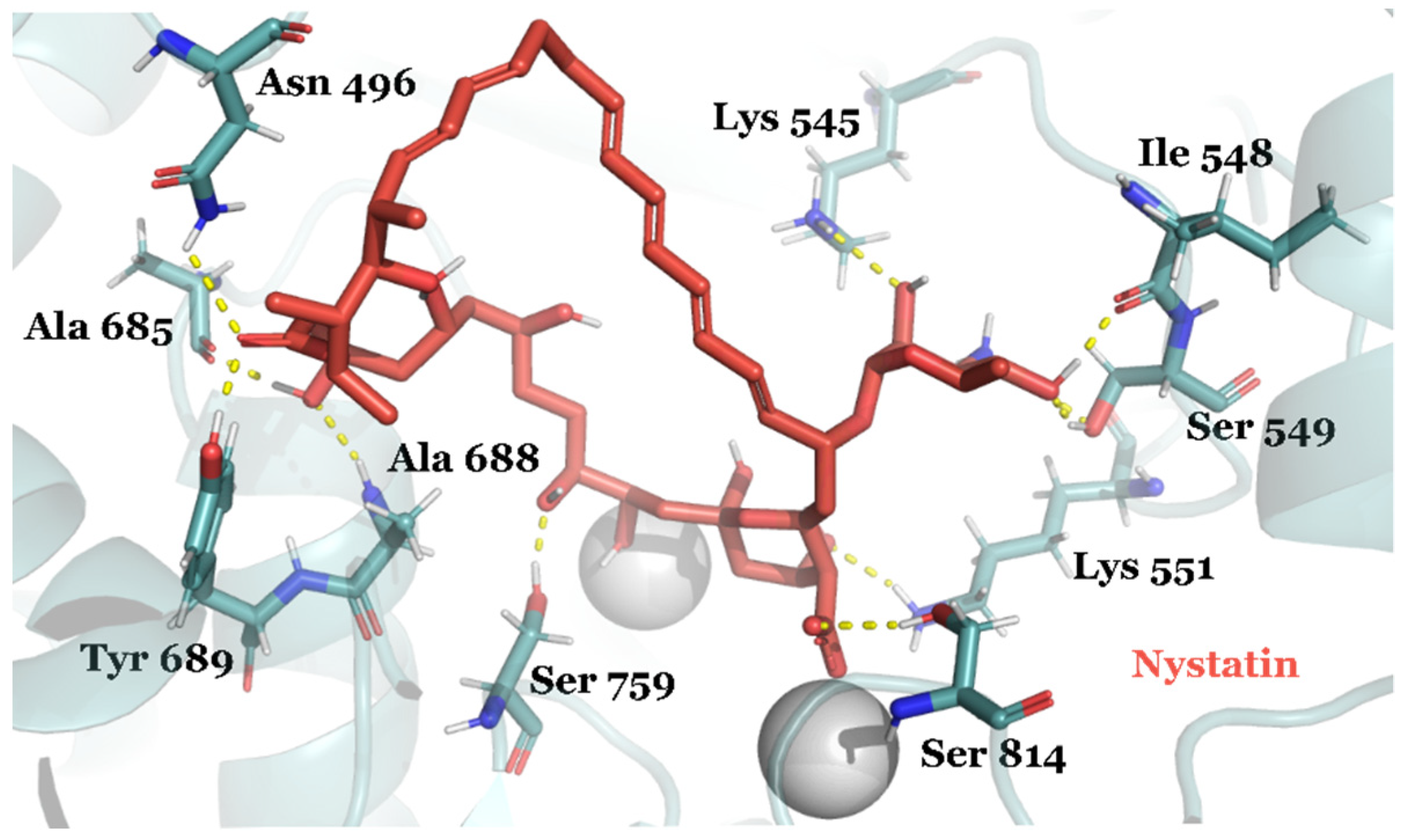
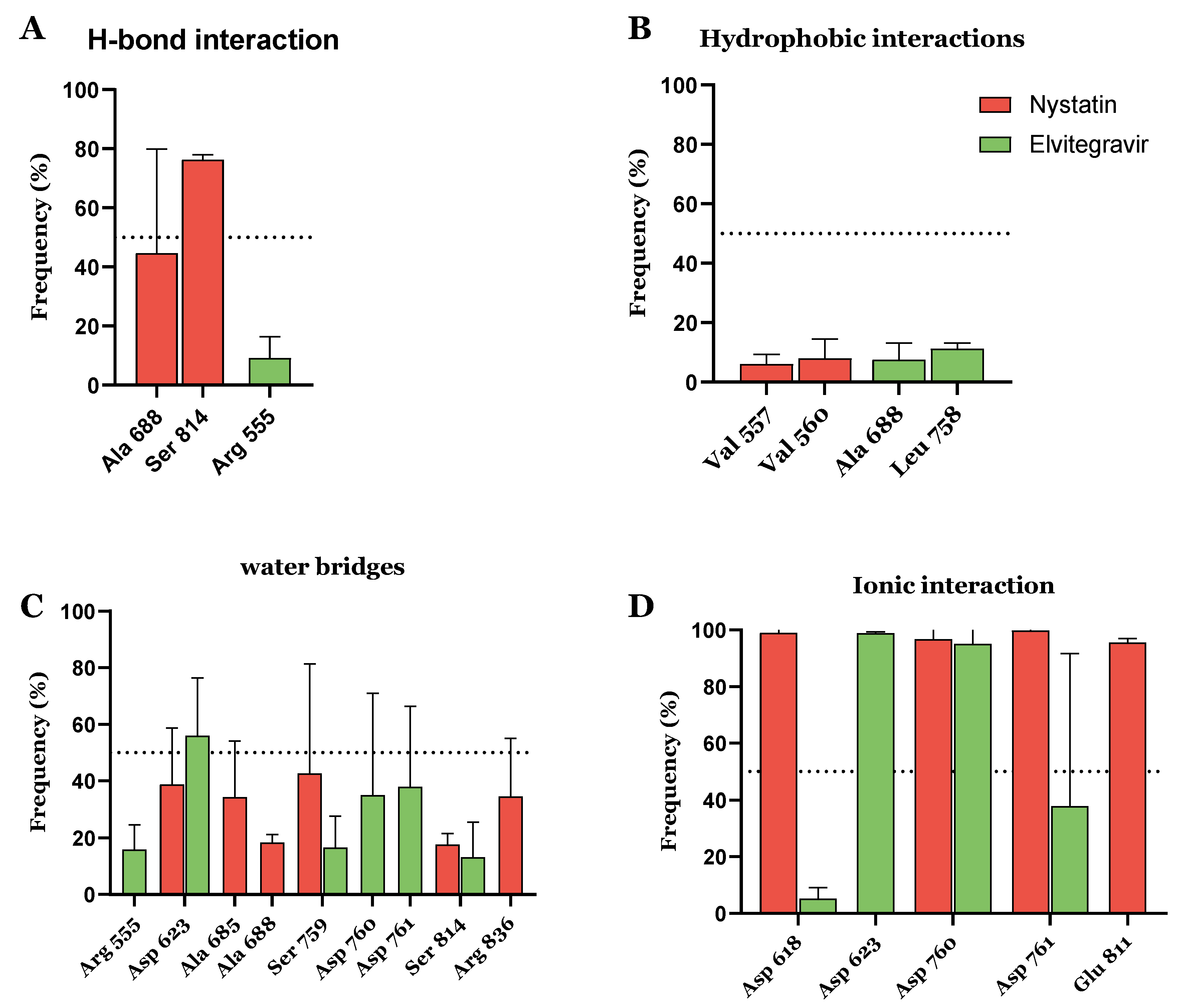
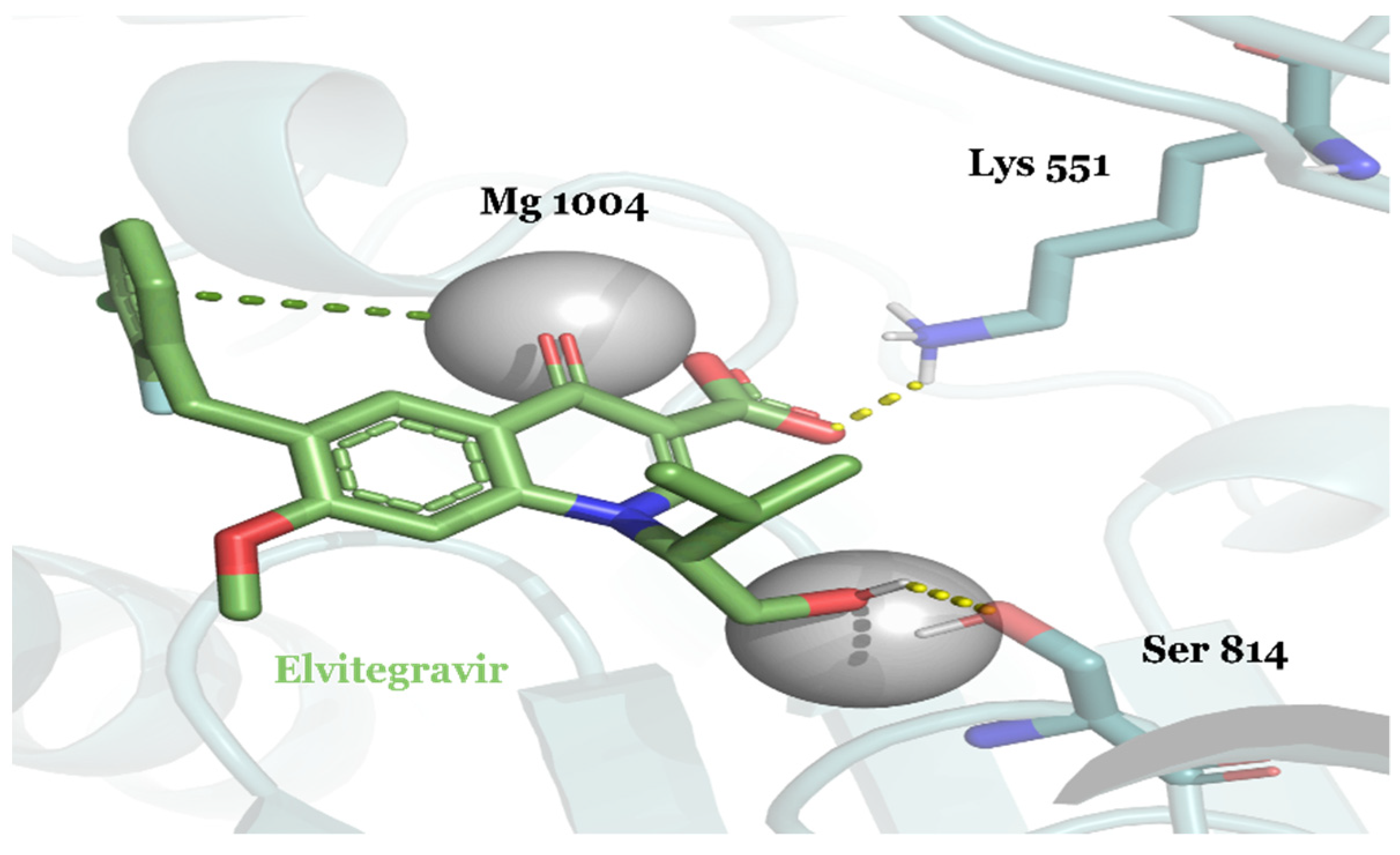

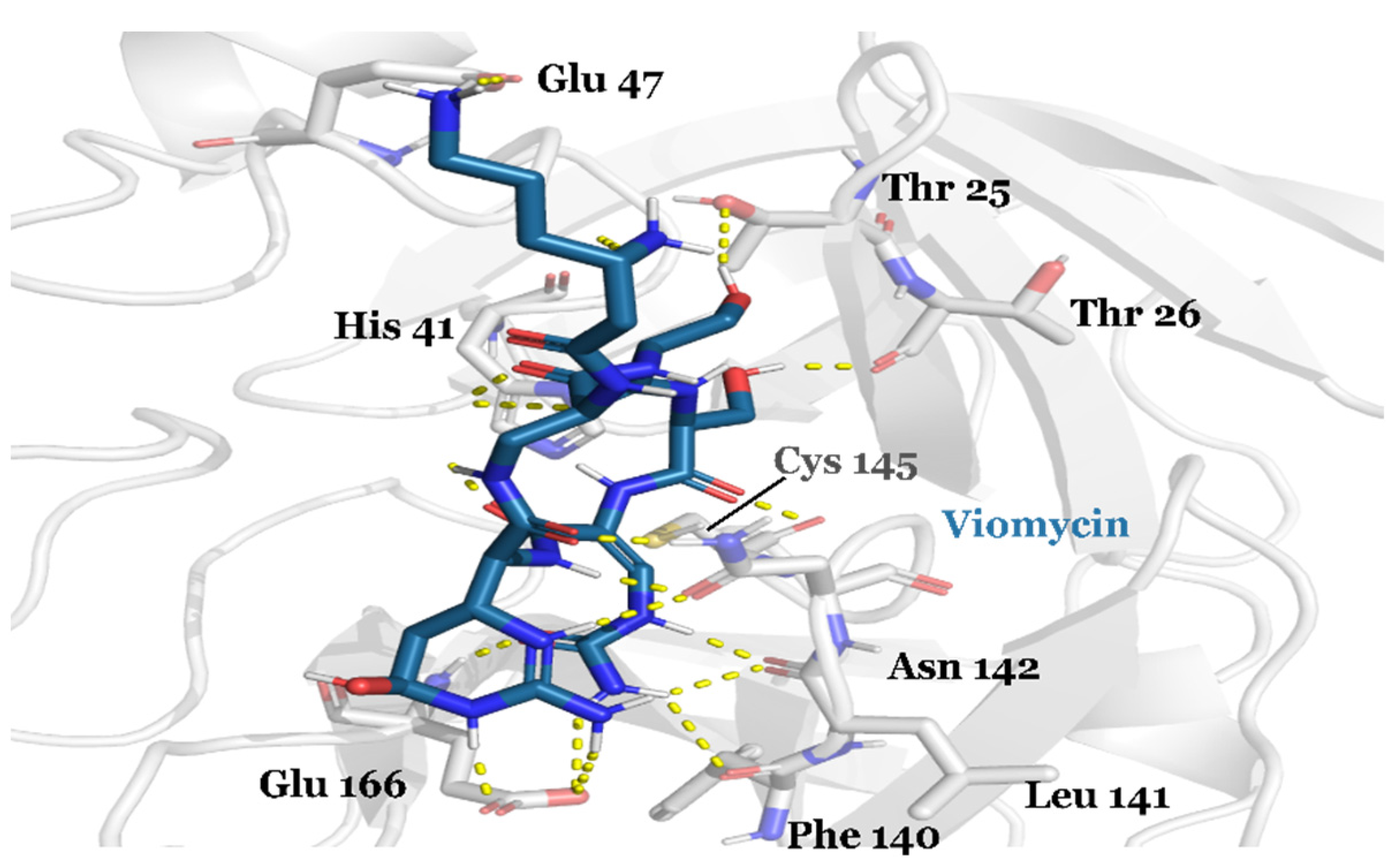{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound ID | Traditional Name | FDA Status | Docking Score | ΔG(bind) Kcal/mol | Biological Activity |
|---|---|---|---|---|---|
| DB00290 | Bleomycin | Approved | −12.119 | −96.63 | Anticancer, antibiotic [54,55,56,57] |
| DB09487 | Iotrolan | Approved | −13.411 | −78.80 | Radiocontrast agent [8] |
| DB01698 | Troxerutin | Approved | −11.707 | −83.08 | Antioxidant, vasoprotective agent [58] SARS-CoV-2 Mpro [10] |
| DB06791 | Lanreotide | Approved | −9.932 | −65.15 | Anticancer [59,60,61] |
| DB00803 | Colistin | Approved | −11.315 | −94.83 | Antibiotic [62,63] |
| CHEMBL3989823 | Viomycin Sulfate | Approved | −10.319 | −74.63 | Anti-tuberculosis, antibiotic [64] |
| DB01249 | Iodixanol | Approved | −9.88 | −91.33 | Radiocontrast agent [8,65,66] |
| DB11672 | Curcumin | Approved | −9.831 | −70.24 | Anti-inflammatory, hypoglycemic, antioxidant, and antimicrobial [54,67,68] |
| DB09134 | Ioversol | Approved | −9.149 | −44.95 | Radiocontrast agent [66,69] |
| DB02638 | Terlipressin | Approved | −8.513 | −89.37 | Hepatorenal syndrome, vasoactive drug in the management of hypotension [70] |
| DB00104 | Octreotide | Approved | −8.21 | −82.34 | Anticancer [59,61] |
| DB01232 | Saquinavir | Approved | −7.978 | −64.49 | HIV-1 protease inhibitor, inhibitor SARS-CoV-2 Mpro [71,72] |
| DB06240 | Tariquidar | Approved | −7.224 | −79.24 | P-gp transporter inhibitor [73] |
| DB00007 | Leuprolide | Approved | −6.406 | 125.56 | Anticancer [74] |
| Compound ID | Traditional Name | FDA Status | Docking Score | ΔG(bind) Kcal/mol | Activity |
|---|---|---|---|---|---|
| Site 1 | |||||
| DB01362 | Iohexol | Approved | −9.175 | −58.99 | Contrast agent [65] |
| DB04703 | Hesperidin | Approved | −8.947 | −66.09 | Neuroprotective agent [82] |
| DB09487 | Iotrolan | Approved | −8.313 | −76.17 | Radiocontrast agent [8] |
| DB09135 | Ioxilan | Approved | −8.082 | −62.37 | Contrast agent [8] |
| DB14126 | Tenofovir | Approved | −4.658 | −46.10 | Used to treat constipation 24, anti-ribonuclease H activity in HIV [83] |
| DB00811 | Ribavirin | Approved | −5.117 | −35.21 | Antiviral agent [34] |
| DB00787 | Acyclovir | Approved | −5.088 | −46.10 | Nucleotide analog antiviral used to treat herpes simplex [84] |
| Site 2 | |||||
| DB15617 | Ferric derisomaltose | Approved | −10.192 | −30.76 | Used to treat iron-deficiency anemia [85] |
| DB09487 | Iotrolan | Approved | −10.047 | −60.03 | Radiocontrast agent [8] |
| DB01362 | Iohexol | Approved | −8.431 | −57.64 | Contrast agent [8] |
| DB12301 | Doravirine | Approved | −8.199 | −49.40 | Used to treat HIV-1 infection [86] |
| CHEMBL2368547 | Senosside A | Approved | −8.677 | −39.58 | Used to treat constipation [87], anti-ribonuclease H activity in HIV [88] |
| CHEMBL1534 | Riboflavin | Approved | −8.027 | −59.09 | Used to treat vitamin B2 deficiency [89] |
| CHEMBL1200455 | Iohexol | Approved | −7.997 | −43.54 | Contrast agent [66] |
| Site 3 | |||||
| DB01249 | Iodixanol | Approved | −10.49 | −61.92 | Radiocontrast agent [66] |
| DB15617 | Ferric derisomaltosa | Approved | −9.852 | −55.90 | Used to treat iron deficiency anemia [85] |
| Compound ID | Traditional Name | FDA Status | Docking Score | ΔG(bind) Kcal/mol | Activity |
|---|---|---|---|---|---|
| DB00618 | Demeclocycline | Approved | −14.225 | −2.73 | Antibiotic, also is used to treat hyponatremia [109] |
| DB11365 | Sennoside | Research | −14.040 | −47.94 | Used to treat constipation 24 |
| DB06796 | Mangafodipir | Approved | −13.433 | −31.72 | Contrast agent |
| DB00650 | Leucovorin | Approved | −13.424 | −0.07 | Used to prevent toxic effects after high-dose methotrexate therapy |
| DB11596 | Levoleucovorin | Approved | −13.322 | −7.54 | Used to prevent toxic effects after high-dose methotrexate therapy |
| DB00923 | Ceforanide | Approved | −12.969 | 63.36 | Antibacterial activity |
| DB09487 | Iotrolan | Approved | −12.951 | 6.25 | Radiocontrast agent 5 |
| DB15062 | Inarigivir | Research | −11.882 | −1.37 | Used to treat hepatitis B virus |
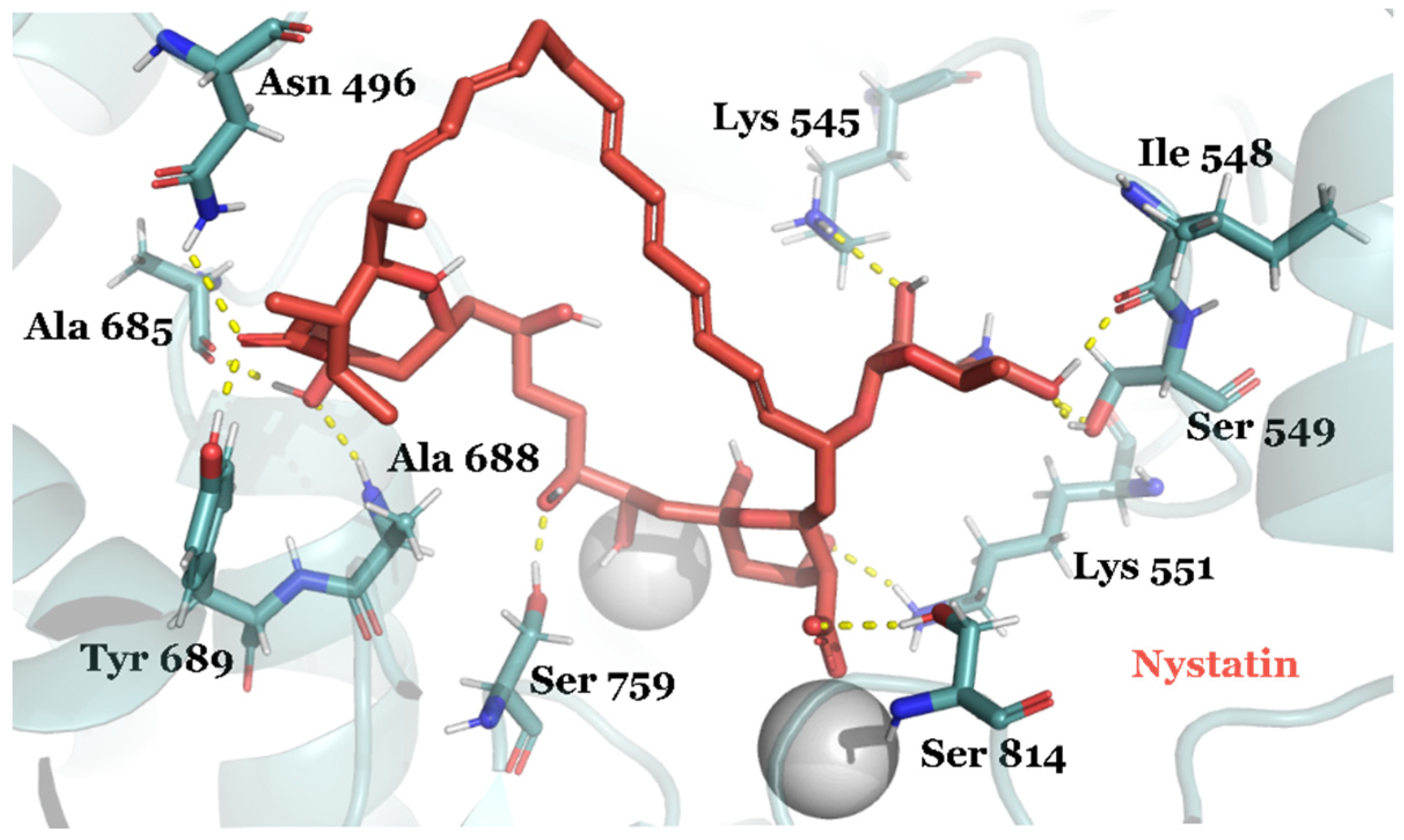
| DB00646 | Nystatin | Approved | −11.669 | −60.13 | Antifungal activity |
| DB01076 | Atorvastatin | Approved | −11.117 | 39.19 | Antilipemic agent |
| DB01607 | Ticarcillin | Approved | −11.095 | −34.52 | Antibiotic |
| DB00369 | Cidofovir | Approved | −10.972 | 32.78 | Antiviral activity against human cytomegalovirus |
| DB06794 | Lodoxamide | Approved | −10.967 | 7.96 | Ophthalmic agent |
| DB00558 | Zanamivir | Approved | −10.869 | 90.24 | Used to treat influenza viruses A and B |
| DB04570 | Latamoxef | Approved | −10.843 | 24.25 | Antibiotic used against Gram-positive and Gram-negative bacteria |
| DB11808 | Faldaprevir | Research | −10.750 | 45.70 | Antiviral agent against hepatitis C virus |
| DB09292 | Sacubitril | Approved | −10.650 | 7.38 | Used to treat cardiovascular diseases |
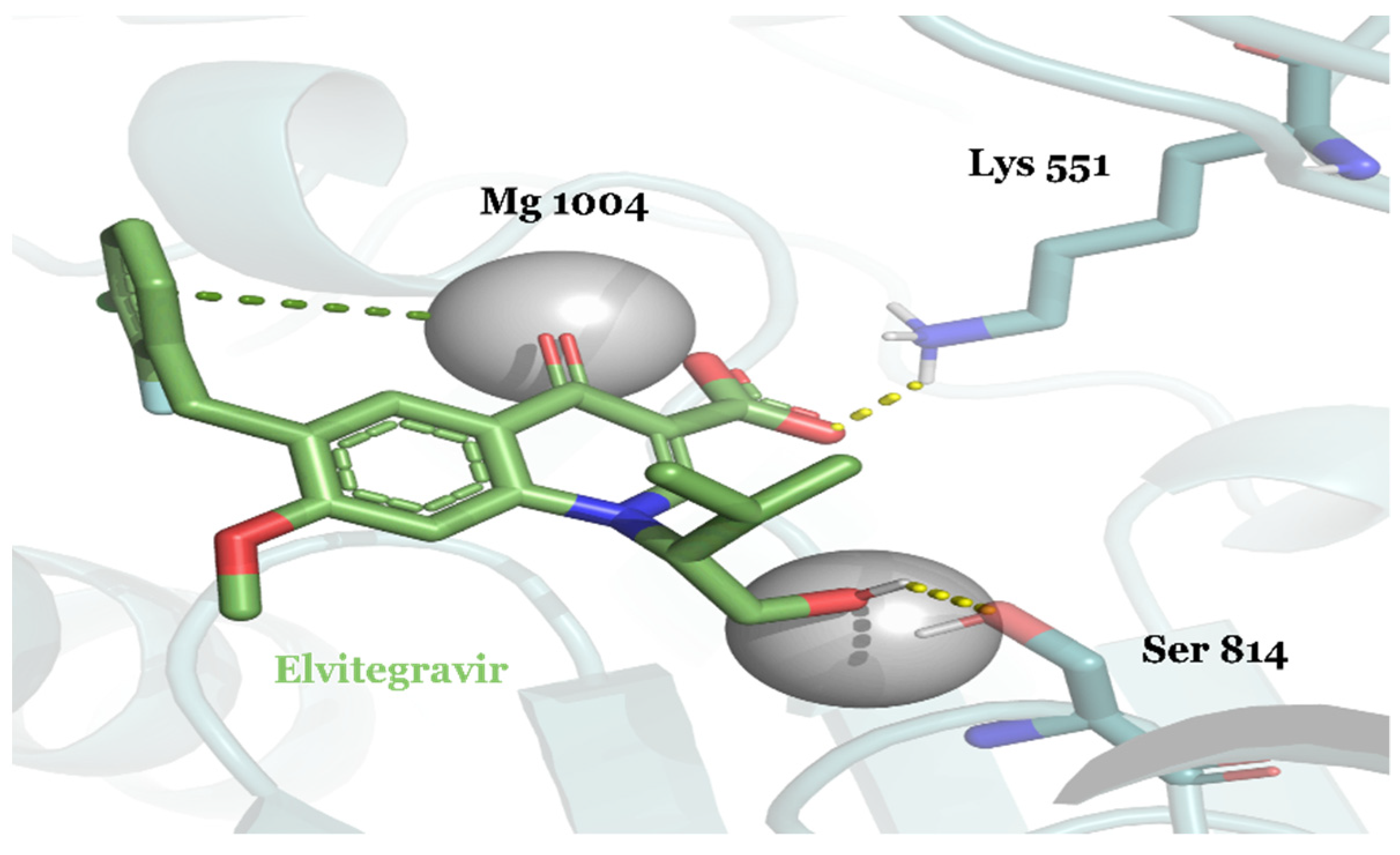
| DB09101 | Elvitegravir | Approved | −10.432 | −34.56 | HIV-1 treatment |
| DB14663 | Ribavirin monophosphate | Approved | −9.075 | −19.72 | Antiviral agent against hepatitis C virus |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vesga, L.C.; Ruiz-Hernández, C.A.; Alvarez-Jacome, J.J.; Duque, J.E.; Rincon-Orozco, B.; Mendez-Sanchez, S.C. Repurposing of Four Drugs as Anti-SARS-CoV-2 Agents and Their Interactions with Protein Targets. Sci. Pharm. 2022, 90, 24. https://doi.org/10.3390/scipharm90020024
Vesga LC, Ruiz-Hernández CA, Alvarez-Jacome JJ, Duque JE, Rincon-Orozco B, Mendez-Sanchez SC. Repurposing of Four Drugs as Anti-SARS-CoV-2 Agents and Their Interactions with Protein Targets. Scientia Pharmaceutica. 2022; 90(2):24. https://doi.org/10.3390/scipharm90020024
Chicago/Turabian StyleVesga, Luis C., Camilo A. Ruiz-Hernández, Jeimmy J. Alvarez-Jacome, Jonny E. Duque, Bladimiro Rincon-Orozco, and Stelia C. Mendez-Sanchez. 2022. "Repurposing of Four Drugs as Anti-SARS-CoV-2 Agents and Their Interactions with Protein Targets" Scientia Pharmaceutica 90, no. 2: 24. https://doi.org/10.3390/scipharm90020024
APA StyleVesga, L. C., Ruiz-Hernández, C. A., Alvarez-Jacome, J. J., Duque, J. E., Rincon-Orozco, B., & Mendez-Sanchez, S. C. (2022). Repurposing of Four Drugs as Anti-SARS-CoV-2 Agents and Their Interactions with Protein Targets. Scientia Pharmaceutica, 90(2), 24. https://doi.org/10.3390/scipharm90020024