Development and Validation of a Rapid Analytical Method for the Simultaneous Quantification of Metabolic Syndrome Drugs by HPLC-DAD Chromatography

 ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Equipments
2.3. Chromatographic Conditions
2.4. Preparation of Standard Solutions
2.5. Working Standard Solution
2.6. Analytical Method Validation
2.6.1. Solution Stability
2.6.2. Linearity
2.6.3. Limit of Detection and Quantification
2.6.4. Precision and Accuracy
2.7. Sample Solutions of Commercially Available Drug Products
2.8. Working Sample Solutions of Commercially Available Drug Products
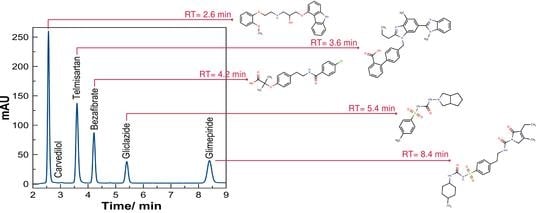
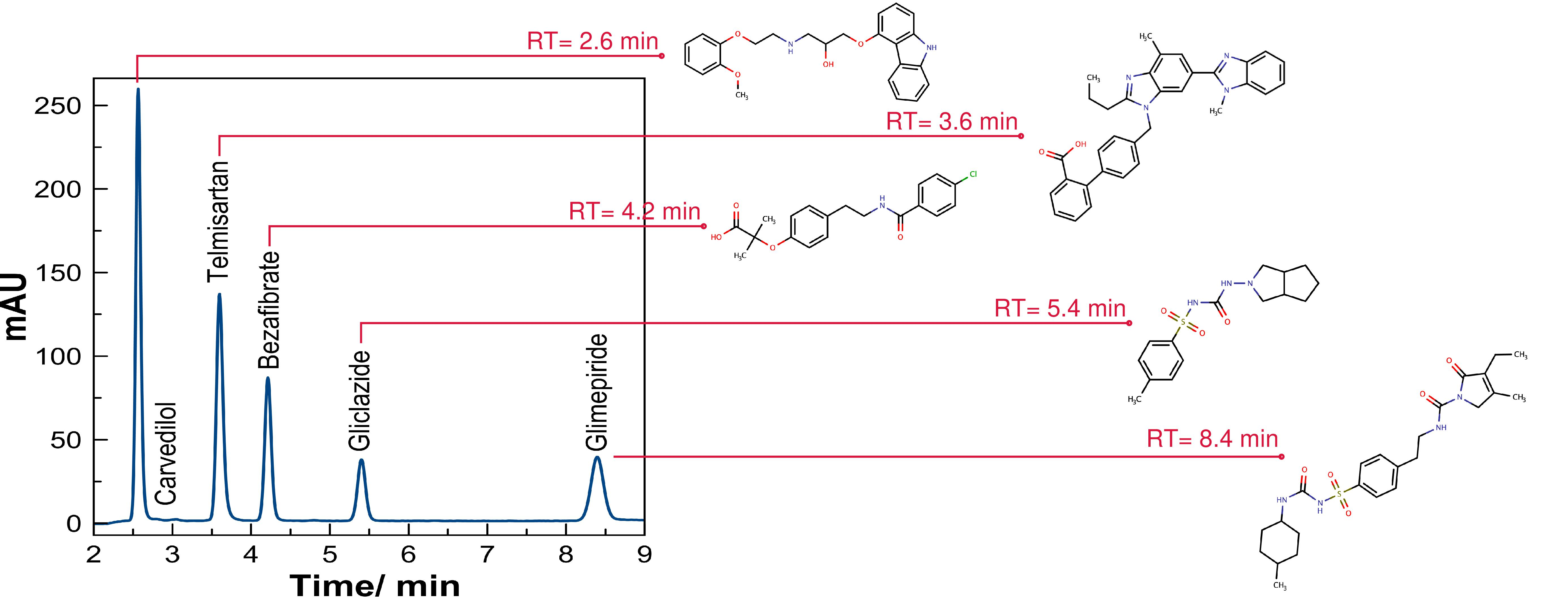
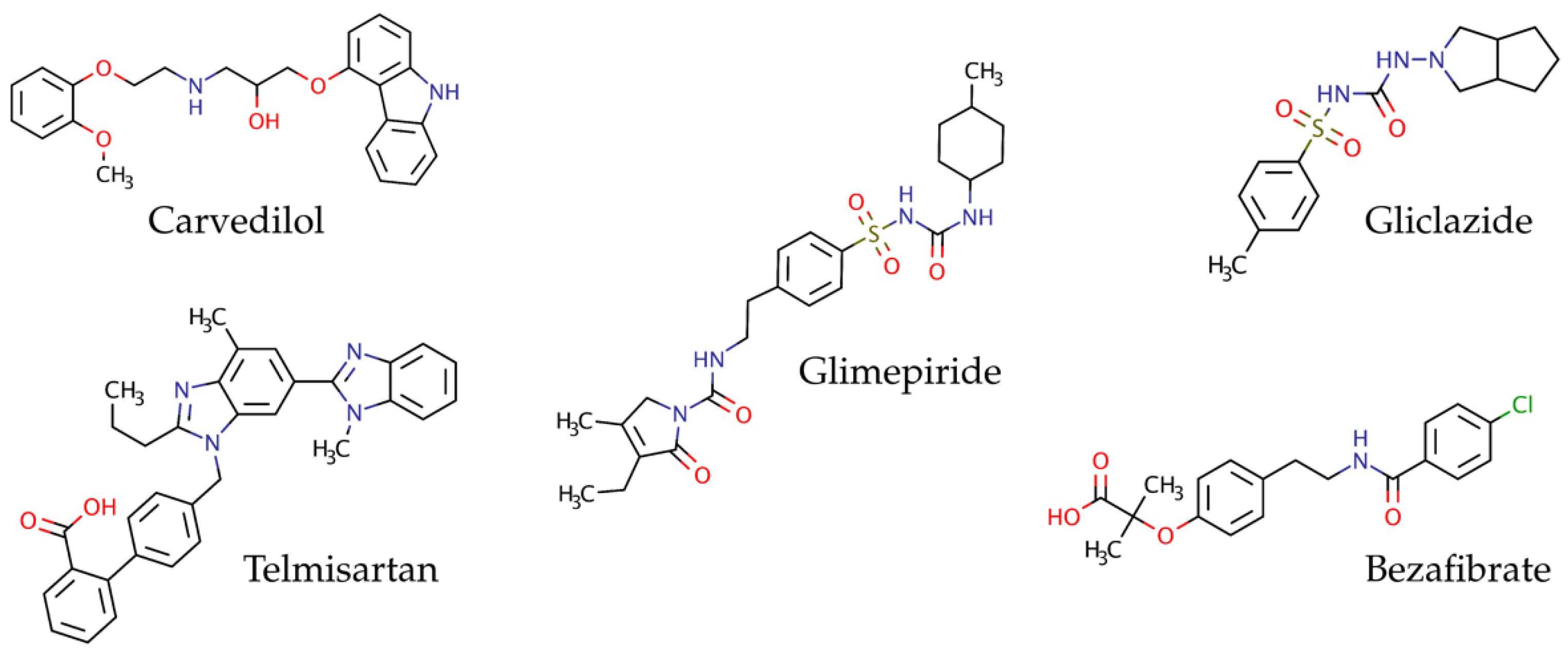
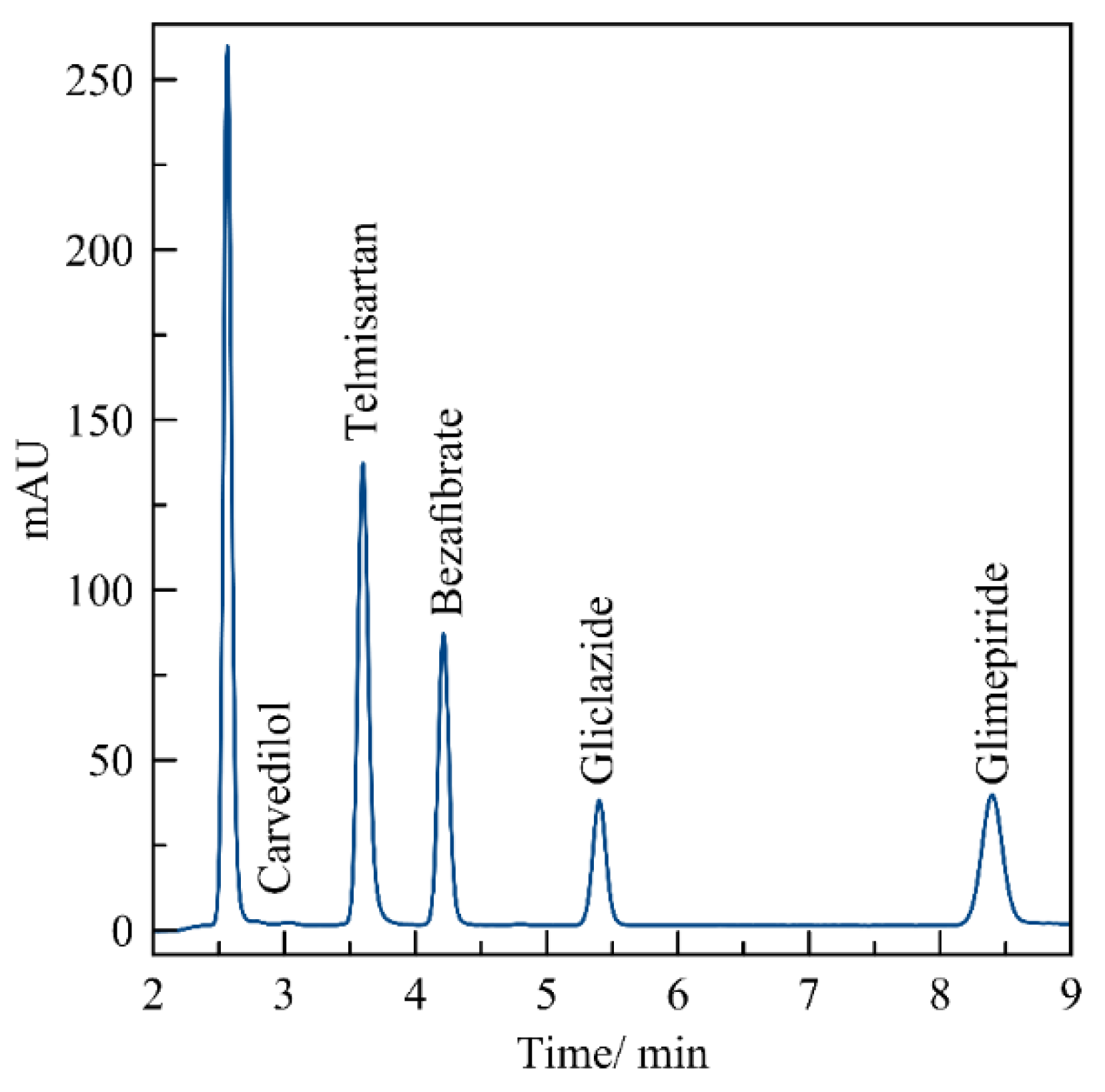
3. Results
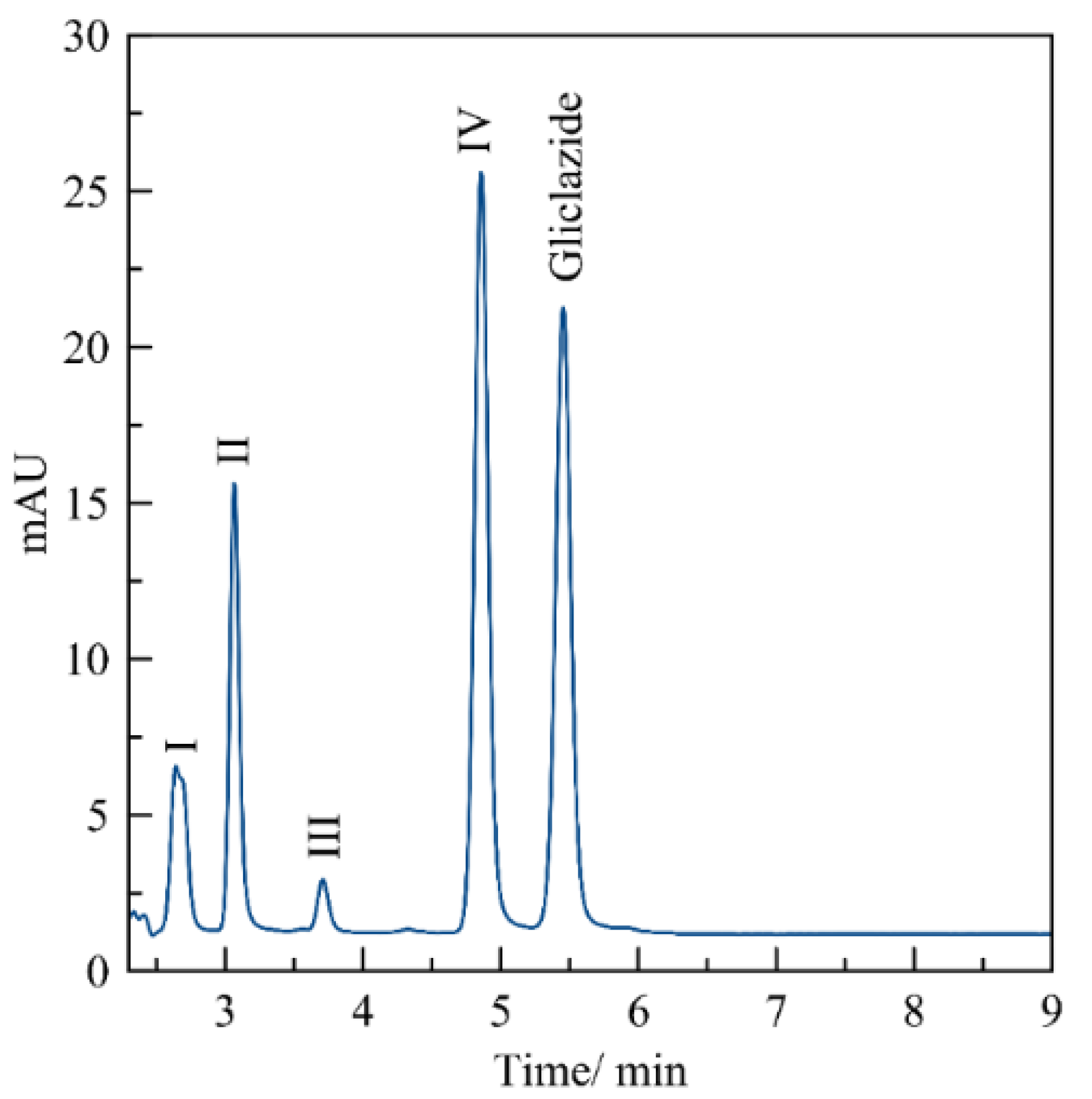
3.1. Standard Working Solution
3.2. Analytical Method Validation
3.2.1. Solution Stability
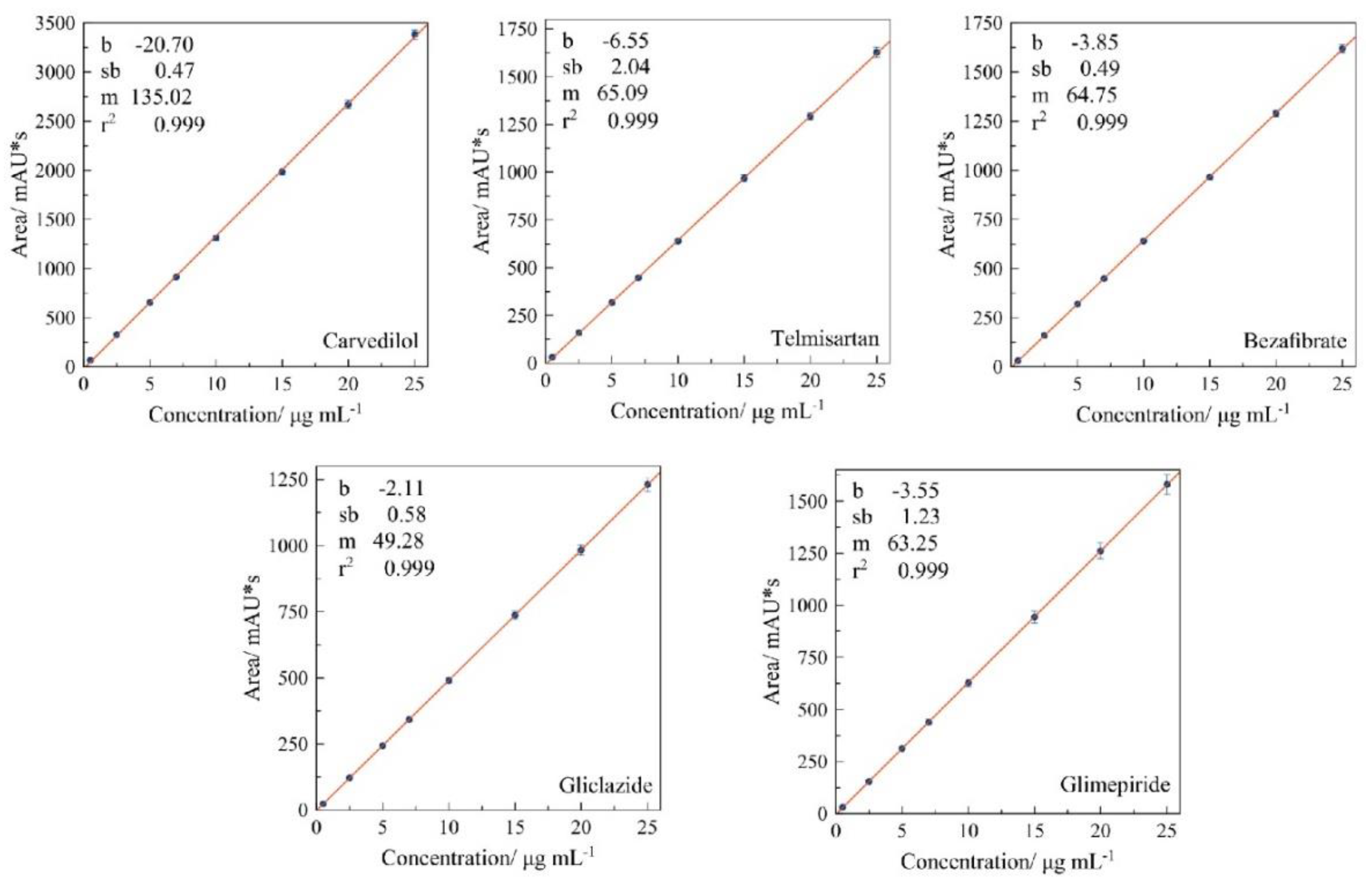
3.2.2. Linearity Range
3.2.3. Limit of Detection and Quantification
3.2.4. Precision and Accuracy
3.3. Evaluation of the Method by Working Sample Solutions
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MetS | Metabolic Syndrome |
| HPLC-DAD | High-Performance Liquid Chromatography with Diode-Array Detection |
| LOD | Limit of detection |
| LOQ | Limit of quantification |
| API | Active Pharmaceutical Ingredient |
| CVD | Carvedilol |
| TEL | Telmisartan |
| BZT | Bezafibrate |
| GZD | Gliclazide |
| GMP | Glimepiride |
| CV% | Coefficient of variation |
| RE% | Relative error |
References
- Nolan, P.B.; Carrick-Ranson, G.; Stinear, J.W.; Reading, S.A.; Dalleck, L.C. Prevalence of metabolic syndrome and metabolic syndrome components in young adults: A pooled analysis. Prev. Med. Rep. 2017, 7, 211–215. [Google Scholar] [CrossRef]
- O’Neill, S.; O’Driscoll, L. Metabolic syndrome: A closer look at the growing epidemic and its associated pathologies. Obes. Rev. 2015, 16, 1–12. [Google Scholar] [CrossRef]
- Grundy, S.M.; Cleeman, J.I.; Daniels, S.R.; Donato, K.A.; Eckel, R.H.; Franklin, B.A.; Gordon, D.J.; Krauss, R.M.; Savage, P.J.; Smith, S.C.; et al. Diagnosis and Management of the Metabolic Syndrome: An American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 2005, 112, 2735–2752. [Google Scholar] [CrossRef]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Bahiru, E.; de Cates, A.N.; Farr, M.R.; Jarvis, M.C.; Palla, M.; Rees, K.; Ebrahim, S.; Huffman, M.D. Fixed-dose combination therapy for the prevention of atherosclerotic cardiovascular diseases. Cochrane Database Syst. Rev. 2017, 3, CD009868. [Google Scholar] [CrossRef]
- Hirano, T.; Kazumi, T.; Yoshino, G. Long-Term Efficacy of Bezafibrate in Reduction of Small, Dense Low-Density Lipoprotein by Hypotriglyceridemic Action. Curr. Ther. Res. 2000, 61, 127–136. [Google Scholar] [CrossRef]
- El-Kommos, M.E.; Mohamed, N.A.; Ali, H.R.H.; Abdel Hakiem, A.F. Simultaneous Estimation of Metformin Hydrochloride and Certain Cardiovascular Drugs with a Pharmacokinetic Study. J. Liq. Chromatogr. Relat. Technol. 2015, 38, 1759–1766. [Google Scholar] [CrossRef]
- Rouini, M.-R.; Mohajer, A.; Tahami, M.-H. A simple and sensitive HPLC method for determination of gliclazide in human serum. J. Chromatogr. B 2003, 785, 383–386. [Google Scholar] [CrossRef]
- Foroutan, S.M.; Zarghi, A.; Shafaati, A.; Khoddam, A. Application of monolithic column in quantification of gliclazide in human plasma by liquid chromatography. J. Pharm. Biomed. Anal. 2006, 42, 513–516. [Google Scholar] [CrossRef]
- Jondhale, S.; Bhise, S.; Pore, Y. Physicochemical Investigations and Stability Studies of Amorphous Gliclazide. AAPS PharmSciTech 2012, 13, 448–459. [Google Scholar] [CrossRef]
- Sandhya, S.M.; Kumar, P.S. Simple and Rapid Simultaneous RP-HPLC Method for Determination of Glimepiride and Metformin in Tablet Dosage Form. Res. J. Pharm. Technol. 2014, 7, 906–909. [Google Scholar]
- Vaingankar, P.N.; Amin, P.D. Development and Validation of Stability-Indicating RP-HPLC Method for Simultaneous Determination of Metformin HCI and Glimepiride in Fixed-Dose Combination. Anal. Chem. Insights 2016, 11, 13–20. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Singh, O.P.B.; Biswal, S.; Sahoo, J.; Murthy, P.N. Physicochemical Properties of Glimepiride in Solid Dispersions with Polyethylene Glycol 20,000. Int. J. Pharm. Sci. Nanotechnol. 2009, 2, 537–543. [Google Scholar]
- Babu, K.R.; Sarma, E.S.R.S.; Kumari, N.A.; Raju, G.M.J.; Sarma, G.V.S. Simple and stability indicating RP-HPLC assay method development and validation lisinopril dihydrate by RP-HPLC in bulk and dosage form. Der. Pharm. Lett. 2015, 7, 274–280. [Google Scholar]
- Patel, J.M.; Dhingani, A.P.; Garala, K.C.; Raval, M.K.; Sheth, N.R. Development and Validation of Bioanalytical HPLC Method For Estimation of Telmisartan In Rat Plasma: Application To Pharmacokinetic Studies. Dhaka Univ. J. Pharm. Sci. 2012, 11, 121–127. [Google Scholar] [CrossRef]
- Bajaj, A.; Rao, M.R.P.; Pardeshi, A.; Sali, D. Nanocrystallization by Evaporative Antisolvent Technique for Solubility and Bioavailability Enhancement of Telmisartan. AAPS PharmSciTech 2012, 13, 1331–1340. [Google Scholar] [CrossRef]
- Naidu, K.B.; Reddy, M.R.M.; Naidu, N.V. Development and validation of RP-HPLC method for determination of carvedilol in bulk and pharmaceutical dosage forms. Der. Pharm. Lett. 2014, 6, 198–206. [Google Scholar]
- Swetha, E.; Vijitha, C.; Veeresham, C. HPLC Method Development and Validation of S (-)-Carvedilol from API and Formulations. Am. J. Anal. Chem. 2015, 6, 437–445. [Google Scholar] [CrossRef]
- Jouyban, A.; Sorouraddin, M.H.; Farajzadeh, M.A.; Somi, M.H.; Fazeli-Bakhtiyari, R. Determination of five antiarrhythmic drugs in human plasma by dispersive liquid–liquid microextraction and high-performance liquid chromatography. Talanta 2015, 134, 681–689. [Google Scholar] [CrossRef]
- De Melo, J.; Hurtado, F.K.; Poitevin, F.S.; Flores, F.C.; Sonego, E.; Dalmora, S.L.; Madalena, C.; Rolim, B. HPLC Determination of Bezafibrate in Human Plasma and its Application to Pharmacokinetics Studies. J. Chromatogr. Sci. 2010, 48, 362–366. [Google Scholar] [CrossRef]
- Zhang, W.; Xiang, B.-R.; Zhan, Y.; Yu, L.-Y.; Wang, T.; Wang, C.-Y. HPLC Method for the Determination of Bezafibrate in Human Plasma and Application to a Pharmacokinetic Study of Bezafibrate Dispersible Tablet. J. Chromatogr. Sci. 2008, 46, 844–847. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhou, Z.; Jiang, J.-Q. Simultaneous Detection of Sulfamethoxazole, Diclofenac, Carbamazepine, and Bezafibrate by Solid Phase Extraction and High Performance Liquid Chromatography with Diode Array Detection. J. Appl. Spectrosc. 2014, 81, 273–278. [Google Scholar] [CrossRef]
- Reichal, C.R.; Rao, M.G. Development and validation of PR-HPLC method for simultaneous estimation of gliclazide and sitagliptin phosphate monohydrate in bulk and tablet dosage form. Int. J. Pharma Sci. 2015, 6, 361–369. [Google Scholar]
- Chakradhar, L.; Kallem, R.; Karthik, A.; Sundari, B.T.; Ramesh, S.; Mullangi, R.; Srinivas, N.R. A rapid and highly sensitive method for the determination of glimepiride in human plasma by liquid chromatography–electrospray ionization tandem mass spectrometry: Application to a pre-clinical pharmacokinetic study. Biomed. Chromatogr. 2008, 22, 58–63. [Google Scholar] [CrossRef]
- Surekha, M.L.; Kumara Swamy, G.; Ashwini, G.L. Development and Validation of RP—HPLC method for the estimation of Telmisartan in bulk and tablet dosage Form. Int. J. Drug Dev. Res. 2012, 4, 200–205. [Google Scholar]
- Sree Janardhanan, V.; Manavalan, R.; Valliappan, K. Chemometric technique for the optimization of chromatographic system: Simultaneous HPLC determination of Rosuvastatin, Telmisartan, Ezetimibe and Atorvastatin used in combined cardiovascular therapy. Arab. J. Chem. 2016, 9, S1378–S1387. [Google Scholar] [CrossRef]
- Sultana, N.; Arayne, M.S.; Shafi, N.; Siddiqui, F.A.; Hussain, A. Development of a RP-HPLC method for the simultaneous analysis of diltiazem and statin: Application in pharmaceuticals and human serum. Anal. Methods 2010, 2, 1571. [Google Scholar] [CrossRef]
- Pasha, M.K.; Muzeeb, S.; Basha, S.J.S.; Shashikumar, D.; Mullangi, R.; Srinivas, N.R. Analysis of five HMG-CoA reductase inhibitors—Atorvastatin, lovastatin, pravastatin, rosuvastatin and simvastatin: Pharmacological, pharmacokinetic and analytical overview and development of a new method for use in pharmaceutical formulations analysis an. Biomed. Chromatogr. 2006, 20, 282–293. [Google Scholar] [CrossRef]
- Alhazmi, H.; Alnami, A.; Arishi, M.; Alameer, R.; Al Bratty, M.; Rehman, Z.; Javed, S.; Arbab, I. A Fast and Validated Reversed-Phase HPLC Method for Simultaneous Determination of Simvastatin, Atorvastatin, Telmisartan and Irbesartan in Bulk Drugs and Tablet Formulations. Sci. Pharm. 2018, 86, 1. [Google Scholar] [CrossRef]
- Vishnuvardhan, C.; Radhakrishnanand, P.; Navalgund, S.G.; Atcha, K.R.; Satheeshkumar, N. RP-HPLC Method for the Simultaneous Estimation of Eight Cardiovascular Drugs. Chromatographia 2014, 77, 265–275. [Google Scholar] [CrossRef]
- Soltani, S.; Jouyban, A. Optimization and validation of an isocratic HPLC-UV method for the simultaneous determination of five drugs used in combined cardiovascular therapy in human plasma. Asian J. Chem. 2011, 23, 1728–1734. [Google Scholar]
- Gonzalez, O.; Iriarte, G.; Ferreirós, N.; Maguregui, M.I.; Alonso, R.M.; Jiménez, R.M. Optimization and validation of a SPE-HPLC-PDA-fluorescence method for the simultaneous determination of drugs used in combined cardiovascular therapy in human plasma. J. Pharm. Biomed. Anal. 2009, 50, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, P.; Harisudhan, T.; Choudhury, H.; Mullangi, R.; Srinivas, N.R. Simultaneous estimation of six anti-diabetic drugs—glibenclamide, gliclazide, glipizide, pioglitazone, repaglinide and rosiglitazone: Development of a novel HPLC method for use in the analysis of pharmaceutical formulations and its application to human pl. Biomed. Chromatogr. 2006, 20, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.K.; Hassan, A.A. Development of a RP-HPLC Method for Simultaneous Determination of Some Antidiabetic Sulfonylurea Drugs in Bulk and Pharmaceutical Dosage Forms. Int. J. Pharm. Sci. Rev. Res. 2014, 25, 207–210. [Google Scholar]
- Pednekar, S.; Lokhande, R.; Sutar, R.; Kolhal, S.; Surve, S.; Gudekar, S. Simultaneous Determination of Metformin, Sitagliptin, Saxagliptin, Linagliptin and Vildagliptin in Multicomponent Pharmaceutical Preparations by RP-HPLC. Int. J. Pharm. Sci. Rev. Res. 2014, 28, 128–133. [Google Scholar]
- AbuRuz, S.; Millership, J.; McElnay, J. The development and validation of liquid chromatography method for the simultaneous determination of metformin and glipizide, gliclazide, glibenclamide or glimperide in plasma. J. Chromatogr. B 2005, 817, 277–286. [Google Scholar] [CrossRef]
- Sultana, N.; Arayne, M.S.; Iftikhar, B. Simultaneous Determination of Atenolol, Rosuvastatin, Spironolactone, Glibenclamide and Naproxen Sodium in Pharmaceutical Formulations and Human Plasma by RP-HPLC. J. Chin. Chem. Soc. 2008, 55, 1022–1029. [Google Scholar] [CrossRef]
- Eldin, A.B.; Ismaiel, O.A.; Hassan, W.E.; Shalaby, A.A. Green analytical chemistry: Opportunities for pharmaceutical quality control. J. Anal. Chem. 2016, 71, 861–871. [Google Scholar] [CrossRef]
- International Conference on Harmonization (ICH). Topic Q2 (R1): Validation of Analytical Procedures: Text and Methodology; ICH: Geneva, Switzerland, 1994. [Google Scholar]
- Shah, V.P.; Midha, K.K.; Dighe, S.; McGilveray, I.J.; Skelly, J.P.; Yacobi, A.; Layloff, T.; Viswanathan, C.T.; Edgar Cook, C.; Mcdowall, R.D.; et al. Analytical Methods Validation: Bioavailability, Bioequivalence, and Pharmacokinetic Studies. J. Pharm. Sci. 1992, 81, 309–312. [Google Scholar] [CrossRef]
- Karnes, H.T.; Shiu, G.; Shah, V.P. Validation of Bioanalytical Methods. Pharm. Res. 1991, 08, 421–426. [Google Scholar] [CrossRef]
- Bansal, G.; Singh, M.; Jindal, K.C. Forced Degradation Study on Gliclazide and Application of Validated Stability-Indicating HPLC-UV Method in Stability Testing of Gliclazide Tablets. Chromatographia 2007, 66, 751–755. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| API | Parameter | Area/mUA·s, after 72 h in Storage at | |||
|---|---|---|---|---|---|
| −20 °C | 4 °C | 25 °C | 80 °C a | ||
| Carvedilol | 1327.90 ± 3.72 | 1330.19 ± 4.46 | 1329.21 ± 5.00 | 1338.70 ± 5.60 | |
| CV% | 0.28 | 0.34 | 0.38 | 0.42 | |
| RE% | 1.33 | 1.5 | 1.43 | 2.13 | |
| % Recovery | 101.35 | 101.52 | 101.45 | 102.17 | |
| Telmisartan | 645.66 ± 3.22 | 644.15 ± 2.19 | 643.89 ± 5.74 | 642.92 ± 1.36 | |
| CV% | 0.50 | 0.34 | 0.89 | 0.21 | |
| RE% | 0.92 | 0.69 | 0.64 | 0.49 | |
| % Recovery | 100.93 | 100.69 | 100.65 | 100.50 | |
| Bezafibrate | 649.27 ± 1.85 | 651.01 ± 2.85 | 649.02 ± 0.72 | 649.28 ± 1.73 | |
| CV% | 0.29 | 0.44 | 0.11 | 0.27 | |
| RE% | 1.36 | 1.62 | 1.32 | 1.36 | |
| % Recovery | 101.38 | 101.65 | 101.34 | 101.38 | |
| Gliclazide | 498.30 ± 1.05 | 497.07 ± 1.82 | 439.70 ± 8.74 | 109.23 ± 49.98 | |
| CV% | 0.21 | 0.37 | 1.99 | 45.76 | |
| RE% | 1.69 | 1.44 | −11.42 | −348.52 | |
| % Recovery | 101.71 | 101.46 | 89.75 | 22.30 | |
| Glimepiride | 639.32 ± 3.61 | 641.20 ± 1.74 | 639.36 ± 3.00 | 595.82 ± 2.54 | |
| CV% | 0.56 | 0.27 | 0.47 | 0.43 | |
| RE% | 1.85 | 2.14 | 1.86 | −5.31 | |
| % Recovery | 101.89 | 102.19 | 101.90 | 94.96 | |
| API | Parameter | Concentration/μg mL−1 | ||
|---|---|---|---|---|
| Below LOQ | LOQ | Above LOQ | ||
| Target concentration | 0.005 | 0.035 | 0.065 | |
| Carvedilol | Avg. of experimental concentration | - a | 0.035 ± 0.001 | 0.062 ± 0.02 |
| CV% | - a | 2.518 | 3.71 | |
| RE% | - a | 1.373 | −5.32 | |
| Target concentration | 0.283 | 0.313 | 0.343 | |
| Telmisartan | Avg. of experimental concentration | 0.34 ± 0.07 | 0.35 ± 0.04 | 0.38 ± 0.04 |
| CV% | 20.97 | 11.58 | 11.36 | |
| RE% | 16.62 | 10.34 | 10.62 | |
| Target concentration | 0.046 | 0.076 | 0.106 | |
| Bezafibrate | Avg. of experimental concentration | 0.06 ± 0.01 | 0.08 ± 0.01 | 0.11 ± 0.02 |
| CV% | 9.70 | 13.69 | 13.89 | |
| RE% | 23.79 | 8.67 | 5.79 | |
| Target concentration | 0.087 | 0.117 | 0.147 | |
| Gliclazide | Avg. of experimental concentration | 0.09 ± 0.01 | 0.10 ± 0.01 | 0.13 ± 0.01 |
| CV% | 16.22 | 10.08 | 1.43 | |
| RE% | 4.12 | −12.60 | −11.8 | |
| Target concentration | 0.097 | 0.127 | 0.157 | |
| Glimepiride | Avg. of experimental concentration | 0.12 ± 0.01 | 0.13 ± 0.01 | 0.17 ± 0.01 |
| CV% | 8.31 | 6.03 | 6.83 | |
| RE% | 21.16 | 2.32 | 8.87 | |
| API | Parameter | Repeatability Concentration Intra-Day/μg mL−1 | Reproducibility Concentration Inter-Day/μg mL−1 | ||||
|---|---|---|---|---|---|---|---|
| 2.5 | 10 | 25 | 2.5 | 10 | 25 | ||
| Carvedilol | Avg. of experimental concentration | 2.56 ± 0.04 | 9.88 ± 0.01 | 25.25 ± 0.34 | 2.57 ± 0.01 | 9.92 ± 0.05 | 25.38 ± 0.18 |
| CV% | 1.57 | 1.31 | 1.34 | 0.49 | 0.52 | 0.71 | |
| RE% | 2.27 | 1.22 | 0.99 | 2.59 | 0.77 | 1.52 | |
| Telmisartan | Avg. of experimental concentration | 2.55 ± 0.03 | 10.03 ± 0.17 | 25.12 ± 0.39 | 2.57 ± 0.04 | 9.99 ± 0.08 | 25.16 ± 0.11 |
| CV% | 1.40 | 1.72 | 1.56 | 1.59 | 0.78 | 0.42 | |
| RE% | 2.25 | 0.35 | 0.49 | 2.62 | -0.07 | 0.63 | |
| Bezafibrate | Avg. of experimental concentration | 2.53 ± 0.03 | 9.97 ± 0.13 | 25.10 ± 0.33 | 2.52 ± 0.01 | 10.00 ± 0.03 | 25.18 ± 0.08 |
| CV% | 1.06 | 1.29 | 1.31 | 0.35 | 0.30 | 0.33 | |
| RE% | 1.02 | 0.31 | 0.39 | 0.66 | 0.02 | 0.73 | |
| Gliclazide | Avg. of experimental concentration | 2.50 ± 0.05 | 9.97 ± 0.22 | 25.01 ± 0.55 | 2.48 ± 0.02 | 9.94 ± 0.03 | 24.97 ± 0.05 |
| CV% | 2.08 | 2.21 | 2.19 | 0.80 | 0.29 | 0.19 | |
| RE% | 0.14 | 0.28 | 0.05 | −0.67 | −0.58 | −0.12 | |
| Glimepiride | Avg. of experimental concentration | 2.49 ± 0.07 | 9.97 ± 0.29 | 25.06 ± 0.76 | 2.50 ± 0.02 | 10.01 ± 0.03 | 25.12 ± 0.05 |
| CV% | 2.86 | 2.95 | 3.02 | 0.60 | 0.29 | 0.22 | |
| RE% | −0.58 | −0.26 | 0.23 | 0.02 | 0.07 | 0.48 | |
| API | Parameter | Experimental Concentration a /μg mL−1 | Mass of a Single Tablet of Commercial Drug/mg |
|---|---|---|---|
| Carvedilol | 7.70 ± 0.03 | 95.77 ± 0.07 | |
| CV% | 0.43 | 0.07 | |
| RE% | −29.79 | −4.42 | |
| % Recovery | 77.05 | 95.77 | |
| Telmisartan | 9.46 ± 0.06 | 153.13 ± 0.31 | |
| CV% | 0.61 | 0.20 | |
| RE% | −3.73 | −4.49 | |
| % Recovery | 96.46 | 95.70 | |
| Bezafibrate | 10.72 ± 0.04 | 803.81 ± 8.00 | |
| CV% | 0.33 | 1.00 | |
| RE% | 6.76 | 0.47 | |
| % Recovery | 107.25 | 100.48 | |
| Gliclazide | 6.97 ± 0.08 | 220.10 ± 0.23 | |
| CV% | 1.13 | 0.11 | |
| RE% | −43.48 | −9.04 | |
| % Recovery | 69.70 | 91.71 | |
| Glimepiride | 9.94 ± 1.42 | 19.22 ± 0.03 | |
| CV% | 14.27 | 0.14 | |
| RE% | −0.61 | −4.04 | |
| % Recovery | 99.40 | 96.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz-Angeles, J.; Martínez, L.M.; Videa, M.; Rodríguez-Rodríguez, J.; Martínez-Jiménez, C. Development and Validation of a Rapid Analytical Method for the Simultaneous Quantification of Metabolic Syndrome Drugs by HPLC-DAD Chromatography. Sci. Pharm. 2021, 89, 8. https://doi.org/10.3390/scipharm89010008
Cruz-Angeles J, Martínez LM, Videa M, Rodríguez-Rodríguez J, Martínez-Jiménez C. Development and Validation of a Rapid Analytical Method for the Simultaneous Quantification of Metabolic Syndrome Drugs by HPLC-DAD Chromatography. Scientia Pharmaceutica. 2021; 89(1):8. https://doi.org/10.3390/scipharm89010008
Chicago/Turabian StyleCruz-Angeles, Jorge, Luz María Martínez, Marcelo Videa, José Rodríguez-Rodríguez, and Cecilia Martínez-Jiménez. 2021. "Development and Validation of a Rapid Analytical Method for the Simultaneous Quantification of Metabolic Syndrome Drugs by HPLC-DAD Chromatography" Scientia Pharmaceutica 89, no. 1: 8. https://doi.org/10.3390/scipharm89010008
APA StyleCruz-Angeles, J., Martínez, L. M., Videa, M., Rodríguez-Rodríguez, J., & Martínez-Jiménez, C. (2021). Development and Validation of a Rapid Analytical Method for the Simultaneous Quantification of Metabolic Syndrome Drugs by HPLC-DAD Chromatography. Scientia Pharmaceutica, 89(1), 8. https://doi.org/10.3390/scipharm89010008