Inhibitors of 11β-Hydroxysteroid Dehydrogenase Type 1 as Potential Drugs for Type 2 Diabetes Mellitus—A Systematic Review of Clinical and In Vivo Preclinical Studies



Abstract
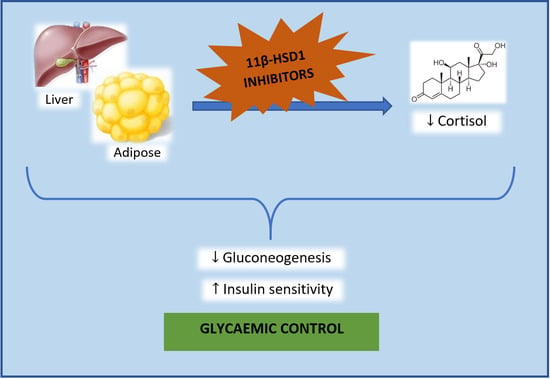
:
1. Introduction
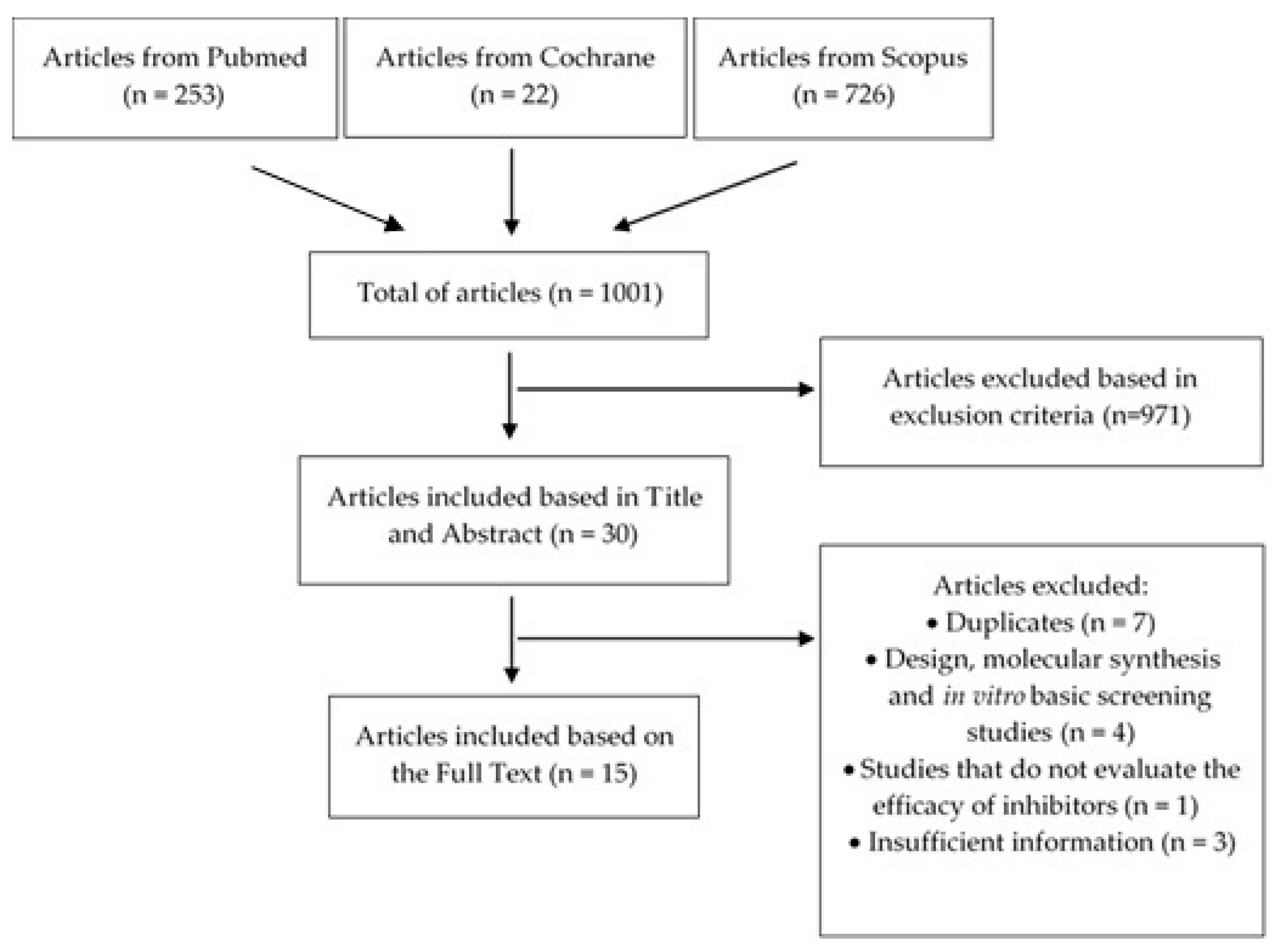
2. Materials and Methods
3. Results
3.1. Clinical Trials
3.2. In Vivo Preclinical Trials
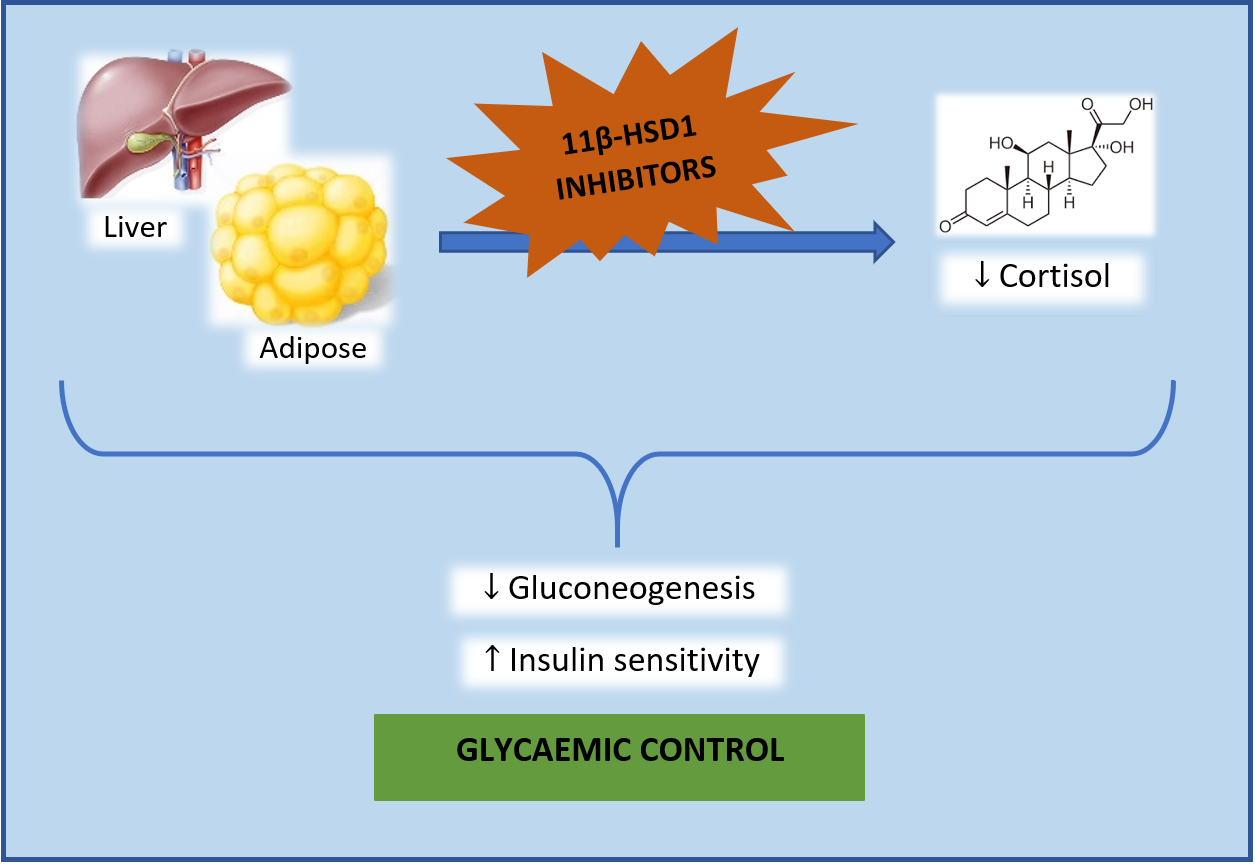
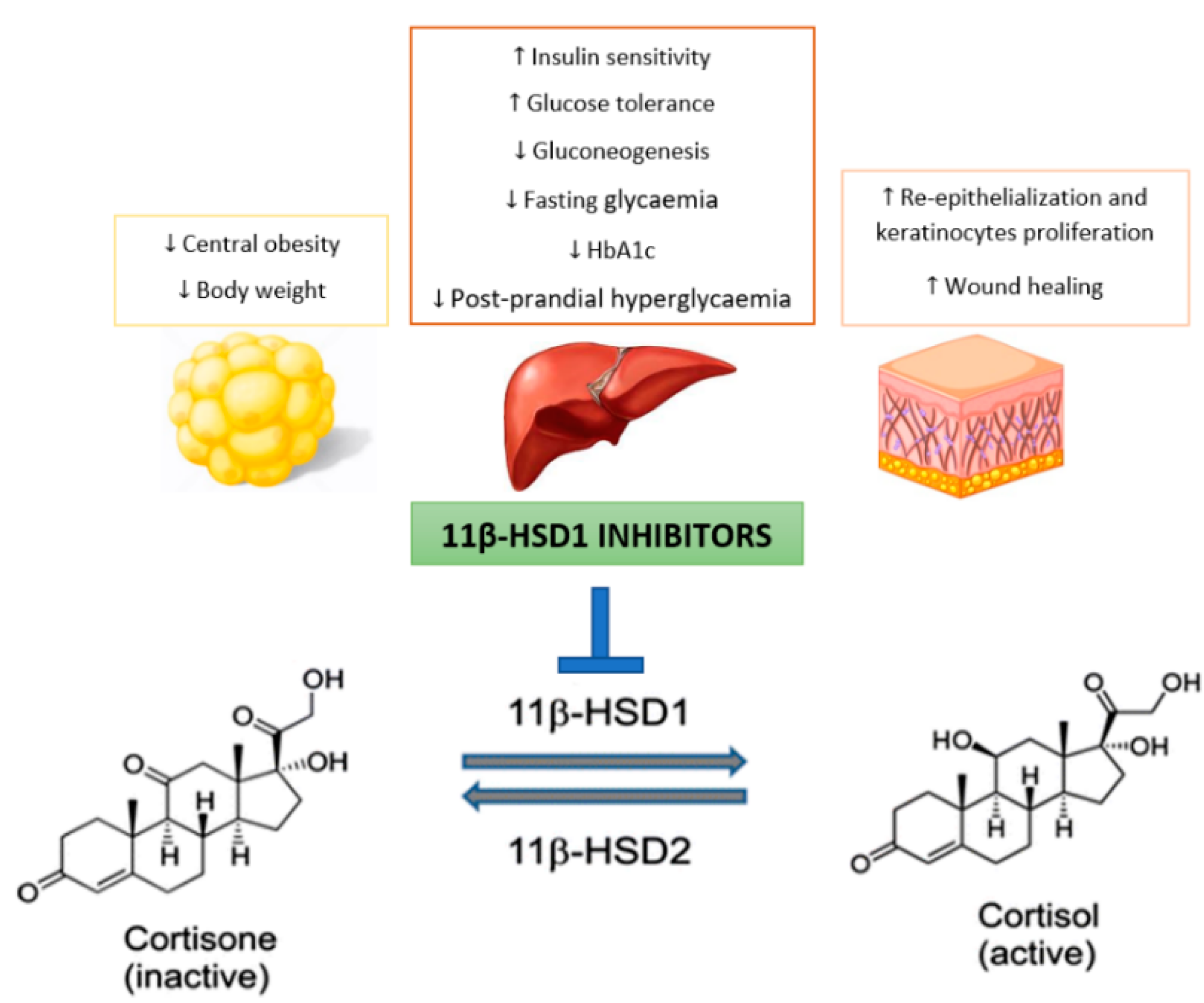
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
| PUBMED (Research Date: 18 November 2019) |
|
| COCHRANE (Research Date: 18 November 2019) |
|
| SCOPUS (Research Date: 14 January 2020) |
|
References
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Association, A.D. Classification and diagnosis of diabetes: Standards of Medical Care in Diabetes-2020. Diabetes Care 2020, 43, S14–S31. [Google Scholar] [CrossRef] [Green Version]
- Habtemariam, S.; Habtemariam, S. Pathophysiology of type 2 diabetes complications. Med. Foods Potential Ther. Type 2 Diabetes Assoc. Dis. 2019, 69–88. [Google Scholar] [CrossRef]
- Srinivasan, B.T.; Davies, M. Glycaemic management of type 2 diabetes. Medicine (Baltimore) 2019, 47, 32–39. [Google Scholar] [CrossRef]
- Byun, S.Y.; Shin, Y.J.; Nam, K.Y.; Hong, S.P.; Ahn, S.K. A novel highly potent and selective 11β-hydroxysteroid dehydrogenase type 1 inhibitor, UI-1499. Life Sci. 2015, 120, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Li, H.; Bai, H.; Su, Z.; Xiang, Q.; Wang, C.; Zhao, B.; Zhang, Y.; Zhang, Q.; Chu, Y.; et al. Synthesis of novel curcumin analogues for inhibition of 11β-hydroxysteroid dehydrogenase type 1 with anti-diabetic properties. Eur. J. Med. Chem. 2014, 77, 223–230. [Google Scholar] [CrossRef]
- Ye, X.-Y.; Chen, S.Y.; Wu, S.; Yoon, D.S.; Wang, H.; Hong, Z.; O’Connor, S.P.; Li, J.; Li, J.J.; Kennedy, L.J.; et al. Discovery of Clinical Candidate 2-((2S,6S)-2-Phenyl-6-hydroxyadamantan-2-yl)-1-(3′-hydroxyazetidin-1-yl)ethanone [BMS-816336], an Orally Active Novel Selective 11β-Hydroxysteroid Dehydrogenase Type 1 Inhibitor. J. Med. Chem. 2017, 60, 4932–4948. [Google Scholar] [CrossRef]
- Freude, S.; Heise, T.; Woerle, H.-J.; Jungnik, A.; Rauch, T.; Hamilton, B.; Schölch, C.; Huang, F.; Graefe-Mody, U. Safety, pharmacokinetics and pharmacodynamics of BI 135585, a selective 11β-hydroxysteroid dehydrogenase-1 (HSD1) inhibitor in humans: Liver and adipose tissue 11β-HSD1 inhibition after acute and multiple administrations over 2 weeks. Diabetes. Obes. Metab. 2016, 18, 483–490. [Google Scholar] [CrossRef]
- Hollis, G.; Huber, R. 11β-Hydroxysteroid dehydrogenase type 1 inhibition in type 2 diabetes mellitus. Diabetes Obes. Metab. 2011, 13, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.C.I.; Epel, E.S.; White, M.L.; Standen, E.C.; Seckl, J.R.; Tomiyama, A.J. Hypothalamic-pituitary-adrenal axis dysregulation and cortisol activity in obesity: A systematic review. Psychoneuroendocrinology 2015, 62, 301–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chourpiliadis, C.; Mohiuddin, S.S. Biochemistry, Gluconeogenesis; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Oh, K.; Han, H.; Kim, M.; Koo, S. CREB and FoxO1: Two transcription factors for the regulation of hepatic gluconeogenesis. BMB Rep. 2013, 46, 567–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M. Inhibitors of 11β-hydroxysteroid dehydrogenase type 1 in antidiabetic therapy. Handb. Exp. Pharmacol. 2011, 203, 127–146. [Google Scholar] [CrossRef]
- Liu, W.; Katz, D.A.; Locke, C.; Daszkowski, D.J.; Wang, Y.; Rieser, M.J.; Awni, W.M.; Marek, G.J.; Dutta, S. Clinical safety, pharmacokinetics, and pharmacodynamics of the 11β-hydroxysteroid dehydrogenase type 1 inhibitor ABT-384 in healthy volunteers and elderly adults. Clin. Pharmacol. Drug Dev. 2013, 2, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Hoshiro, M.; Ohno, Y.; Masaki, H.; Iwase, H.; Aoki, N. Comprehensive study of urinary cortisol metabolites in hyperthyroid and hypothyroid patients. Clin. Endocrinol. (Oxf.) 2006, 64, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Espinosa, J.J.; García-Jiménez, S.; Ríos, M.Y.; Medina-Franco, J.L.; López-Vallejo, F.; Webster, S.P.; Binnie, M.; Ibarra-Barajas, M.; Ortiz-Andrade, R.; Estrada-Soto, S. Antihyperglycemic and sub-chronic antidiabetic actions of morolic and moronic acids, in vitro and in silico inhibition of 11β-HSD 1. Phytomedicine 2013, 20, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.P.; Han, D.; Chang, K.H.; Ahn, S.K. A novel highly potent and selective 11β-hydroxysteroid dehydrogenase type 1 inhibitor, INU-101. Eur. J. Pharmacol. 2018, 835, 169–178. [Google Scholar] [CrossRef]
- Slominski, A.T.; Zmijewski, M.A. Glucocorticoids Inhibit Wound Healing: Novel Mechanism of Action. J. Invest. Dermatol. 2017, 137, 1012–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NCT03313297. Glucocorticoids and Skin Healing in Diabetes (GC-SHealD). Available online: https://clinicaltrials.gov/show/nct03313297 (accessed on 13 January 2020).
- Hamilton, B.S.; Himmelsbach, F.; Nar, H.; Schuler-Metz, A.; Krosky, P.; Guo, J.; Guo, R.; Meng, S.; Zhao, Y.; Lala, D.S.; et al. Pharmacological characterization of the selective 11β-hydroxysteroid dehydrogenase 1 inhibitor, BI 135585, a clinical candidate for the treatment of type 2 diabetes. Eur. J. Pharmacol. 2015, 746, 50–55. [Google Scholar] [CrossRef]
- Anderson, A.; Walker, B.R. 11β-HSD1 Inhibitors for the Treatment of Type 2 Diabetes and Cardiovascular Disease. Drugs 2013, 73, 1385–1393. [Google Scholar] [CrossRef]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: Explanation and elaboration. PLoS Med. 2009, 6. [Google Scholar] [CrossRef]
- 2016 Annual Meeting of the American College of Clinical Pharmacology, September 25-27, 2016, Bethesda, MD. Clin. Pharmacol. Drug Dev. 2016, 5, 3–56. [CrossRef] [PubMed]
- Clinical Diabetes/Therapeutics. Diabetes. 65 (Suppl. 1), pp. A221–A360. Available online: http://diabetes.diabetesjournals.org/lookup/doi/10.2337/db16-861-1374 (accessed on 13 January 2020).
- Heise, T.; Morrow, L.; Hompesch, M.; Häring, H.-U.; Kapitza, C.; Abt, M.; Ramsauer, M.; Magnone, M.-C.; Fuerst-Recktenwald, S. Safety, efficacy and weight effect of two 11β-HSD1 inhibitors in metformin-treated patients with type 2 diabetes. Diabetes, Obes. Metab. 2014, 16, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.H.; Stone, J.A.; Crumley, T.M.; Wenning, L.; Zheng, W.; Yan, K.; Yang, A.Y.; Sun, L.; Cilissen, C.; Ramael, S.; et al. Pharmacokinetic-pharmacodynamic studies of the 11β-hydroxysteroid dehydrogenase type 1 inhibitor MK-0916 in healthy subjects. Br. J. Clin. Pharmacol. 2013, 76, 917–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anil, T.M.; Dandu, A.; Harsha, K.; Singh, J.; Shree, N.; Kumar, V.S.; Lakshmi, M.N.; Sunil, V.; Harish, C.; Balamurali, G.V.; et al. A novel 11β-hydroxysteroid dehydrogenase type1 inhibitor CNX-010-49 improves hyperglycemia, lipid profile and reduces body weight in diet induced obese C57B6/J mice with a potential to provide cardio protective benefits. BMC Pharmacol. Toxicol. 2014, 15, 43. [Google Scholar] [CrossRef] [Green Version]
- Oh, H.; Jeong, K.-H.; Han, H.Y.; Son, H.J.; Kim, S.S.; Lee, H.J.; Kim, S.; Sa, J.H.; Jun, H.-S.; Ryu, J.H.; et al. A potent and selective 11β-hydroxysteroid dehydrogenase type 1 inhibitor, SKI2852, ameliorates metabolic syndrome in diabetic mice models. Eur. J. Pharmacol. 2015, 768, 139–148. [Google Scholar] [CrossRef]
- Park, S.B.; Jung, W.H.; Kang, N.S.; Park, J.S.; Bae, G.H.; Kim, H.Y.; Rhee, S.D.; Kang, S.K.; Ahn, J.H.; Jeong, H.G.; et al. Anti-diabetic and anti-inflammatory effect of a novel selective 11β-HSD1 inhibitor in the diet-induced obese mice. Eur. J. Pharmacol. 2013, 721, 70–79. [Google Scholar] [CrossRef]
- Park, J.S.; Bae, S.J.; Choi, S.; Son, Y.H.; Park, S.B.; Rhee, S.D.; Kim, H.Y.; Jung, W.H.; Kang, S.K.; Ahn, J.H.; et al. A novel 11β-HSD1 inhibitor improves diabesity and osteoblast differentiation. J. Mol. Endocrinol. 2014, 52, 191–202. [Google Scholar] [CrossRef] [Green Version]
- Schnackenberg, C.G.; Costell, M.H.; Krosky, D.J.; Cui, J.; Wu, C.W.; Hong, V.S.; Harpel, M.R.; Willette, R.N.; Yue, T.-L. Chronic Inhibition of 11 β -Hydroxysteroid Dehydrogenase Type 1 Activity Decreases Hypertension, Insulin Resistance, and Hypertriglyceridemia in Metabolic Syndrome. BioMed Res. Int. 2013, 2013, 427640. [Google Scholar] [CrossRef] [Green Version]
- Winnick, J.J.; Ramnanan, C.J.; Saraswathi, V.; Roop, J.; Scott, M.; Jacobson, P.; Jung, P.; Basu, R.; Cherrington, A.D.; Edgerton, D.S. Effects of 11β-hydroxysteroid dehydrogenase-1 inhibition on hepatic glycogenolysis and gluconeogenesis. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E747–E756. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Li, H.; Bai, H.; Zhao, X.; Zhang, C.; Liu, H.; Zhang, Y.; Zhao, B.; Wu, Y.; Liu, J.; et al. The 11β-hydroxysteroid dehydrogenase type 1 inhibitor protects against the insulin resistance and hepatic steatosis in db/db mice. Eur. J. Pharmacol. 2016, 788, 140–151. [Google Scholar] [CrossRef]
- Yao, F.; Chen, L.; Fan, Z.; Teng, F.; Zhao, Y.; Guan, F.; Zhang, M.; Liu, Y. Interplay between H6PDH and 11β-HSD1 implicated in the pathogenesis of type 2 diabetes mellitus. Bioorg. Med. Chem. Lett. 2017, 27, 4107–4113. [Google Scholar] [CrossRef] [PubMed]
- Stimson, R.H.; Walker, B.R. The role and regulation of 11β-hydroxysteroid dehydrogenase type 1 in obesity and the metabolic syndrome. Horm. Mol. Biol. Clin. Investig. 2013, 15, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.C.; Rooyackers, O.; Walker, B.R. Effects of the 11β-hydroxysteroid dehydrogenase inhibitor carbenoxolone on insulin sensitivity in men with type 2 diabetes. J. Clin. Endocrinol. Metab. 2003, 88, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Chatzigeorgiou, A.; Halapas, A.; Kalafatakis, K.; Kamper, E. The Use of Animal Models in the Study of Diabetes Mellitus. In Vivo 2009, 23, 245–258. [Google Scholar] [PubMed]
- King, A.J.F. The use of animal models in diabetes research. Br. J. Pharmacol. 2012, 166, 877–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, C.J.; Day, C. The future of new drugs for diabetes management. Diabetes Res. Clin. Pract. 2019, 155, 107785. [Google Scholar] [CrossRef]
- Gutierrez, P.M.; Gyte, A.; Deschoolmeester, J.; Ceuppens, P.; Swales, J.; Stacey, C.; Eriksson, J.W.; Sjostrand, M.; Nilsson, C.; Leighton, B. Continuous inhibition of 11β-hydroxysteroid dehydrogenase type i in adipose tissue leads to tachyphylaxis in humans and rats but not in mice. Br. J. Pharmacol. 2015, 172, 4806–4816. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.S.; Stewart, P.M. 11Β-Hydroxysteroid Dehydrogenase Type 1 and Its Role in the Hypothalamus-Pituitary-Adrenal Axis, Metabolic Syndrome, and Inflammation. J. Clin. Endocrinol. Metab. 2009, 94, 4645–4654. [Google Scholar] [CrossRef] [Green Version]
- Anderson, A.J.; Andrew, R.; Homer, N.Z.; Jones, G.C.; Smith, K.; Livingstone, D.E.; Walker, B.R.; Stimson, R.H. Metformin Increases Cortisol Regeneration by 11βHSD1 in Obese Men With and Without Type 2 Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2016, 101, 3787–3793. [Google Scholar] [CrossRef]
- Terao, M.; Murota, H.; Kimura, A.; Kato, A.; Ishikawa, A.; Igawa, K.; Miyoshi, E.; Katayama, I. 11Β-Hydroxysteroid Dehydrogenase-1 Is a Novel Regulator of Skin Homeostasis and a Candidate Target for Promoting Tissue Repair. PLoS ONE 2011, 6, e25039. [Google Scholar] [CrossRef]
- Tiganescu, A.; Tahrani, A.; Morgan, S.A.; Otranto, M.; Desmoulière, A.; Abrahams, L.; Hassan-Smith, Z.; Walker, E.A.; Rabbitt, E.H.; Cooper, M.S.; et al. 11β-Hydroxysteroid dehydrogenase blockade prevents age-induced skin structure and function defects. J. Clin. Investig. 2013, 123, 3051–3060. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, J.; Yang, Q.; Shao, S. 11β-Hydroxysteroid Dehydrogenase Type 1 in Obese Subjects With Type 2 Diabetes Mellitus. Am. J. Med. Sci. 2017, 354, 408–414. [Google Scholar] [CrossRef] [PubMed]
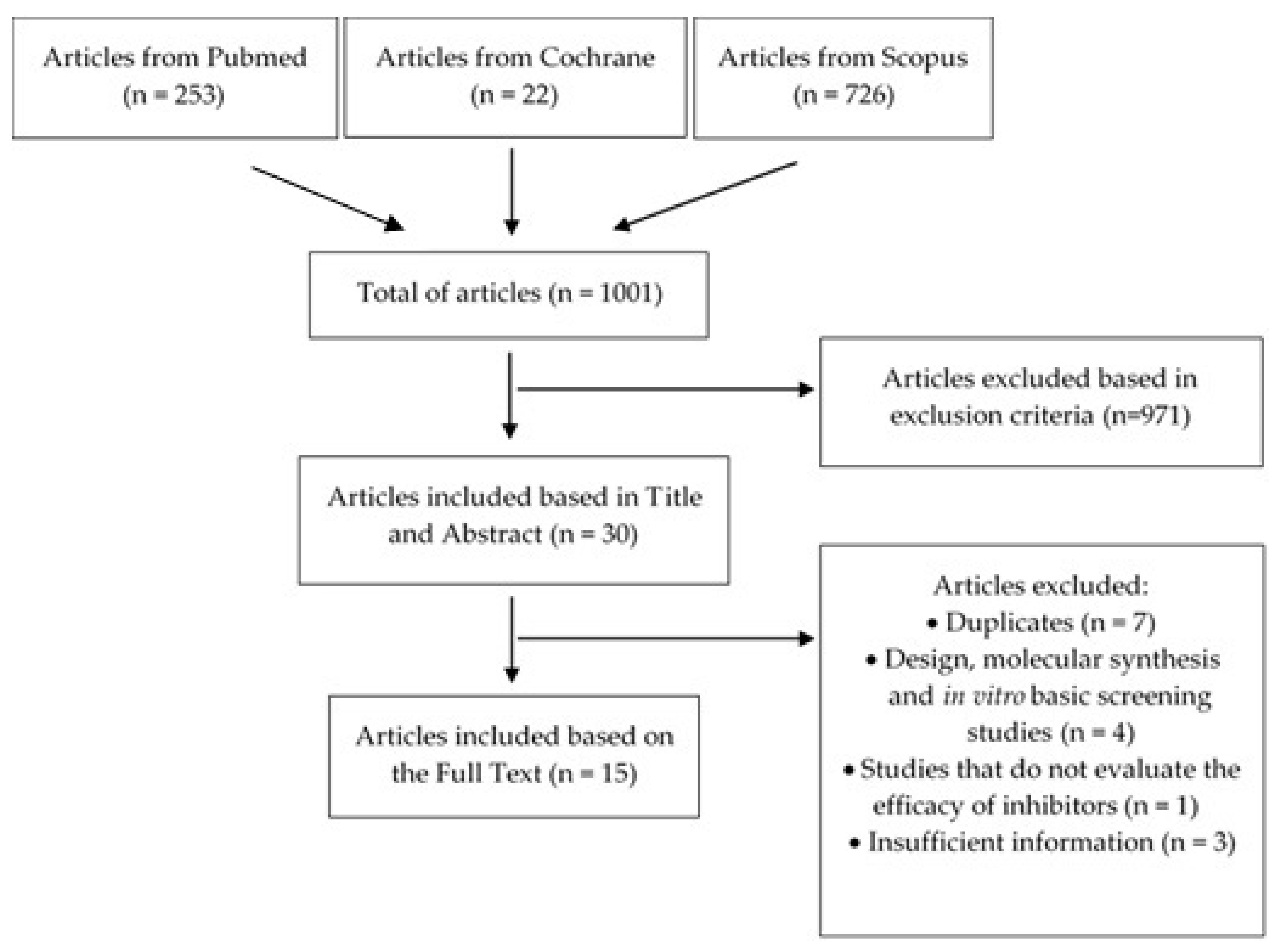
| Inclusion Criteria: |
|
| Exclusion Criteria: |
|
| Experimental Drug | Healthy Patients (without T2DM) | Dosage Used (Oral) | Patients with T2DM | Dosage Used (Oral) | Control Group | Duration | Inhibition of 11β-HSD1 | Reduction Ratio (Allo-THF + THF)/THE | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Liver | Adipose Tissue | |||||||||
| Wu et al., 2016 [23] | BI 187004 | Obesus or overweight: 72 | Single doses, between 2.5–360 mg, once daily (1id) | 71 | Multiple doses, between 10–360 mg, 1id, in monotherapy | Placebo | 14 days | 95% | 90% at doses 160 mg/day | 75% |
| Freude et al., 2016 [24] | BI 187004 | - | - | Obesus or overweight: 103 | Group 1: 20, 80, 240 mg, 1id, in monotherapy | Placebo | 28 days | - | - | 75% |
| Group 2: 240 mg, 1id, in combination with metformin | ||||||||||
| Freude et al., 2016 [8] | BI 135585 | 9 | Single doses, 200 mg, 1id | 72 | Multiple doses, between 5, 12.5, 25, 50, 100, 200 mg, 1id, in monotherapy | Placebo | 14 days | - | 90% | 65–75% after single and 75% after multiple dosing in the lowest dose |
| Heise et al., 2014 [25] | RO 5093151/RO-151 | - | - | 110 | 5 or 200 mg, twice daily (2id), in combination with metformin | Placebo | 28 days | 86–88% in low doses and 92% in high doses | - | - |
| RO 5027383/RO-838 | 50 or 200 mg, 1id, in combination with metformin | 54–55% in low doses and 62–69% in high doses | ||||||||
| Liu et al., 2013 [14] | ABT-384 | 103 | Adults: Single doses, between 1–240 mg, and multiple doses, between 1–100 mg, 1id, in monotherapy | - | - | Placebo | Adults: 7–14 days | - | - | 87–97% |
| Elderly: Multiple doses, between 10–100 mg, 1id, in monotherapy | Elderlies: 21 days | |||||||||
| Wright et al., 2013 [26] | MK-0916 | 80 | Single doses, between 0.2 and 225 mg, followed by multiple doses, between 0.2 and 100 mg, 1id, in monotherapy | - | - | Placebo | 14 days | 84% | - | 31–42% |
| Experimental Drug | Animal Model | Number of Animals | Treatment (Oral) | Control Group | Duration | Inhibition of 11β-HSD1 | Reduction in Fasting Glucose | Reduction of HbA1c Levels | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Liver | Adipose Tissue | |||||||||
| Anil et al., 2014 [27] | CNX-010-49 | Mice DIO-C57B6/J a | 8 | 30 mg/Kg, 2id | vehicle | 10 weeks | 58% at 1 h and 24% at 7 h | 41% | 15% at week 5 | - |
| Byun et al., 2015 [5] | UI-1499 | Mice C57BL/6J | 34 | 45 mg/Kg | vehicle | 2–6 h | 88.8% at 2 h and 37.2% at 6 h | 40.6% at 2 h and 28% at 6 h | - | - |
| Mice KKAy b,c | 10 or 30 mg/Kg | 3 weeks | - | - | 40.1% in lower dose and 30.6% in higher dose | 1.4% to 30 mg/Kg | ||||
| Hong et al., 2018 [17] | INU-101 | Mice C57BL/6J | 38 | 45 mg/Kg | vehicle | 2–6 h | 56.8% at 2 h and 40.3% at 6 h | 38.3% at 2 h and 23.9% at 6 h | - | - |
| Monkey Cynomolgus | 20 mg/Kg | 2 h | 41.7% | 49.4% | - | - | ||||
| Mice KKAy c | 10 or 30 mg/Kg | 3 weeks | 36% to 30 mg/Kg | 65% to 30 mg/Kg | 41.3% in higher dose and 26.7% in lower dose | 14.1% in higher dose and 8.0% in lower dose | ||||
| Mice ob/ob b | 10 or 30 mg/Kg | 3 weeks | - | - | 31.1% to 30 mg/Kg | 12.2% to 30 mg/Kg | ||||
| Rats ZDF c | 60 or 120 mg/Kg | 8 weeks | - | - | 11.4% to 60 mg/Kg and 32.7% to 120 mg/kg | - | ||||
| Oh et al., 2015 [28] | SKI2852 | Mice DIO-C57BL/6 a,c | 30 | 10 or 20 mg/Kg, 1id | vehicle | 8.5 weeks | 80% | 80% | - | - |
| Mice ob/ob b,c | 12.5 mg/Kg, 2id | 18 days | - | - | 21% | |||||
| Park et al., 2013 [29] | KR-67105 | Mice C57BL/6 | ND | 1–60 mg/Kg | vehicle | 2–24 h | 65–70% to 60 mg/Kg and 50% to 40 mg/Kg at 2 h | 75–80% to 40 mg/Kg and 60–65% to 60 mg/Kg at 2 h | - | - |
| Mice DIO-C57BL/6 a | 100 mg/Kg | 28 days | 55–60% | 40–45% | ||||||
| Park et al., 2014 [30] | KR-67500 | Mice DIO-C57BL/6 a | ND | 50 mg/Kg | vehicle | 28 days | 80–90% | 80% | - | - |
| C57BL/6 mice | 16 | 0.5–10 mg/Kg | 2–6 h | 85–90% to 10 mg/Kg at 2 h | 90–95% to 10 mg/Kg at 2 h | |||||
| Schnackenberg et al., 2013 [31] | Compound 11 | Rats SHR-cp | 44 | 10 mg/Kg d | vehicle | 4 weeks | Lean rats: 96% | Lean rats: 92% | - | - |
| Obese rats: 90% | Obese rats: 97% | |||||||||
| Winnick et al., 2013 [32] | HSD1-I | 42 h Fasted Dogs | 12 | 75 mg d | Placebo | 7 days | 100% | - | - | - |
| Yuan et al., 2016 [33] | H8 | Mice db/db c | 40 | 5 or 10 mg/kg | vehicle | 6 weeks | 70% to 10 mg/Kg and 50% to 5 mg/Kg | 60–70% to 10 mg/Kg and 50–60% to 5 mg/Kg | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida, C.; Monteiro, C.; Silvestre, S. Inhibitors of 11β-Hydroxysteroid Dehydrogenase Type 1 as Potential Drugs for Type 2 Diabetes Mellitus—A Systematic Review of Clinical and In Vivo Preclinical Studies. Sci. Pharm. 2021, 89, 5. https://doi.org/10.3390/scipharm89010005
Almeida C, Monteiro C, Silvestre S. Inhibitors of 11β-Hydroxysteroid Dehydrogenase Type 1 as Potential Drugs for Type 2 Diabetes Mellitus—A Systematic Review of Clinical and In Vivo Preclinical Studies. Scientia Pharmaceutica. 2021; 89(1):5. https://doi.org/10.3390/scipharm89010005
Chicago/Turabian StyleAlmeida, Cristiana, Cristina Monteiro, and Samuel Silvestre. 2021. "Inhibitors of 11β-Hydroxysteroid Dehydrogenase Type 1 as Potential Drugs for Type 2 Diabetes Mellitus—A Systematic Review of Clinical and In Vivo Preclinical Studies" Scientia Pharmaceutica 89, no. 1: 5. https://doi.org/10.3390/scipharm89010005
APA StyleAlmeida, C., Monteiro, C., & Silvestre, S. (2021). Inhibitors of 11β-Hydroxysteroid Dehydrogenase Type 1 as Potential Drugs for Type 2 Diabetes Mellitus—A Systematic Review of Clinical and In Vivo Preclinical Studies. Scientia Pharmaceutica, 89(1), 5. https://doi.org/10.3390/scipharm89010005