
Validation of an RP-HPLC Method for the Determination of Asenapine Maleate in Dissolution Media and Application to Study In Vitro Release from Co-Crystals


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation and Characterization of Co-Crystals
2.2.2. HPLC System and Chromatographic Conditions
2.2.3. Preparation of Solutions, Calibration Standards and Quality Control Samples
2.2.4. Validation
2.2.5. In Vitro Release Studies
3. Results
3.1. Method Validation
3.1.1. Linearity
3.1.2. Accuracy and Precision
3.1.3. Sensitivity
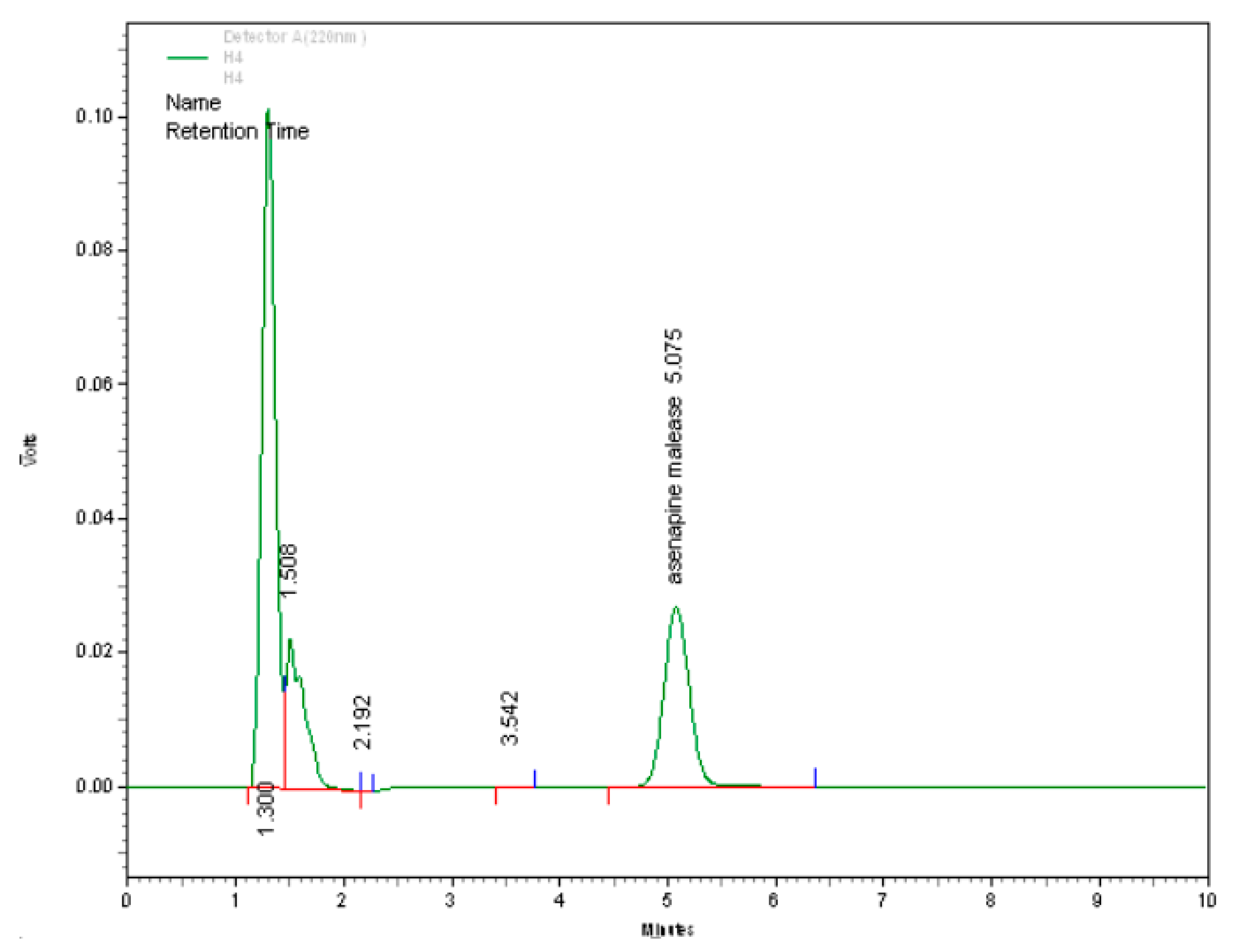
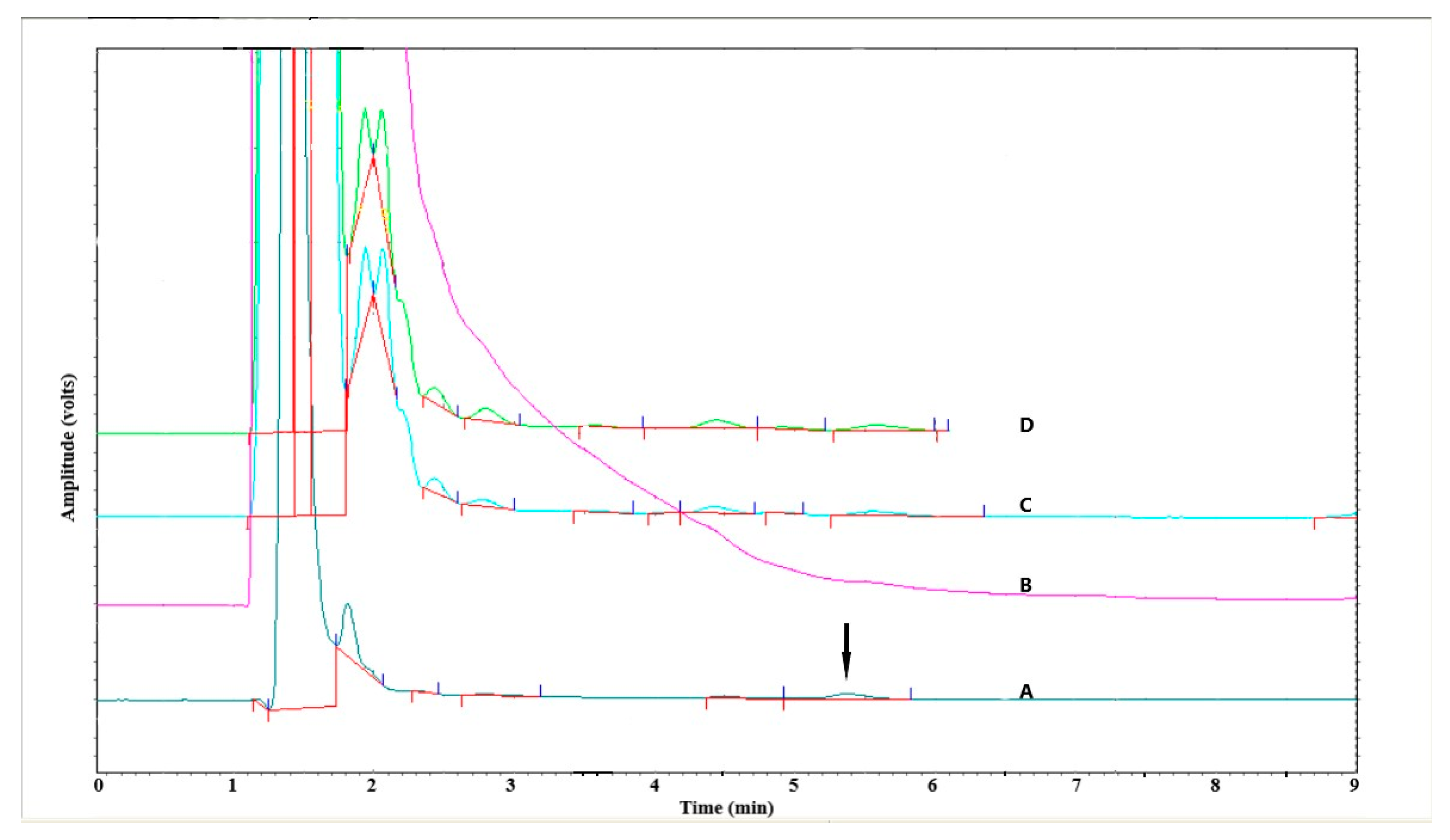
3.1.4. Specificity
3.1.5. Robustness
3.2. In Vitro Release Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Avachat, A.M.; Kapure, S.S. Asenapine maleate in situ forming biodegradable implant: An approach to enhance bioavailability. Int. J. Pharm. 2014, 477, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Avachat, A.M.; Avachat, C.M.; Pradhan, R.; Suryawanshi, T.S.; Khan, E.M.; Martis, E.A.; Coutinho, E.C.; Padhye, S. Preferential formulation of second generation antipsychotic asenapine as inclusion complex with sulphobutylether-βCD (captisol): In vitro and in vivo evaluation. Curr. Drug Deliv. 2018, 15, 520–531. [Google Scholar] [CrossRef]
- Gambhire, V.M.; Ranpise, N.S. Enhanced oral delivery of asenapine maleate from solid lipid nanoparticles: Pharmacokinetic and brain distribution evaluations. Asian J. Pharm. Sci. 2018, 12, 152–161. [Google Scholar]
- Managuli, R.S.; Gourishetti, K.; Shenoy, R.R.; Koteshwara, K.B.; Reddy, M.S.; Mutalik, S. Preclinical pharmacokinetics and biodistribution studies of asenapine maleate using novel and sensitive RP–HPLC method. Bioanalysis 2017, 9, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Shreya, A.B.; Managuli, R.S.; Menon, J.; Kondapalli, L.; Hegde, A.R.; Avadhani, K.; Menon, J.; Kondapalli, L.; Hegde, A.R.; Avadhani, K.; et al. Nano-transfersomal formulations for transdermal delivery of asenapine maleate: In vitro and in vivo performance evaluations. J. Liposome Res. 2015, 26, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Dadhania, P.; Vuddanda, P.R.; Jain, A.; Velagac, S.; Singh, S. Intranasal delivery of asenapine loaded nanostructured lipid carriers: Formulation, characterization, pharmacokinetic and behavioural assessment. RSC Adv. 2016, 6, 2032–2045. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Avachat, A.M. Pharmacodynamic and pharmacokinetic investigation of cyclodextrin mediated asenapine maleate in situ nasal gel for improved bioavailability. Drug Dev. Ind. Pharm. 2017, 43, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Arelly, K.; Thimmaraju, M.K.; Nerella, R.; Allabotharam, S. Method development and validation of asenapine in bulk by RP-HPLC method. J. Chem. Pharm. Res. 2012, 4, 2580–2584. [Google Scholar]
- Halima, O.A.; Aneesh, T.P.; Reshma, G.; Thomas, N.R. Development and validation of UV spectrophotometric method for the estimation of asenapine maleate in bulk and pharmaceutical formulation. Der Pharma Chem. 2012, 4, 644–649. [Google Scholar]
- Parthasarathi, T.R.; Srinivas, T.S.; Sri, M.V.; Ram, S.S.; Mahaboob Basha, M.; Rajesh, P. Quantitative determination of asenapine maleate using reverse phase-high performance liquid chromatography. Int. J. Pharm. Bio. Sci. 2012, 3, 360–366. [Google Scholar]
- Gandhimathi, R.; Vijayaraj, S.; Jyothirmaie, M.P. Method development and validation of UV spectroscopic method for estimation of asenapine maleate in bulk and tablet formulation. IJMCA 2012, 12, 85–90. [Google Scholar]
- Mrudulesh, Y.; Sankar, P.R.; Devadasu, C.H.; Babu, P.S. Development of a validated UV spectrophotometric method for the quantitative estimation of asenapine maleate in bulk drug. J. Chem. Pharm. Sci. (JCPS) 2013, 6, 227–230. [Google Scholar]
- Yanamadala, G.; Ramamohan, G.V.; Praveen, S.P.; Rusyendra, G.V.; Lavanya, N. Development and validation of RP-HPLC method for the estimation of asenapine maleate in bulk and pharmaceutical dosage forms. AJPAMC 2013, 1, 132–139. [Google Scholar]
- Govindarajan, N.R.; Koulagari, S.; Methuku, A.; Podhuturi, S. Method development and validation of RP-HPLC method for determination of new antipsychotic agent asenapine maleate in bulk and pharmaceutical formulation. Eurasian J. Anal. Chem. 2014, 9, 58–65. [Google Scholar]
- Patel, P.S.; Patel, C.N.; Patel, M.M. Method development and validation of RP-HPLC for estimation of asenapine maleate in bulk drug and tablet dosage form. Int. J. Pharm. Res. Sci. (IJPRS) 2016, 5, 15–19. [Google Scholar]
- Borkar, A.A.; Gaikwad, N.J. UV Spectrophotometric and RP-HPLC estimation of drug asenapine in tablet dosage form. IJPSR 2016, 7, 3080–3084. [Google Scholar] [CrossRef]
- Patel, K.; Joshi, D.; Kumbhani, J.; Prajapati, V. HPLC Method Development for Estimation of Dissolution of Antipsychotic Drug as Sublingual Film Dosage Form. Chem. Sci. Trans. 2018, 7, 420–423. [Google Scholar] [CrossRef][Green Version]
- Ramadan, N.K.; Mohamed, T.A.; Fouad, R.M.; Moustafa, A.A. Potentiometric Determination of Asenapine Maleate Using PVC Membrane and Carbon Paste Ion-selective Electrodes. OMCIJ 2018, 8, 555726–555735. [Google Scholar] [CrossRef]
- Aneesh, T.P.; Rajasekaran, A. Stress Degradation studies and development and validation of RPHPLC method for the estimation of Asenapine maleate. Int. J. Pharm. Pharm. Sci. 2012, 4, 448–451. [Google Scholar]
- Chhalotiya, U.K.; Bhatt, K.K.; Shah, D.A.; Patel, J.R. Stability-indicating liquid chromatographic method for the quantification of the new antipsychotic agent asenapine in bulk and in pharmaceutical formulation. Sci. Pharm. 2012, 80, 407–417. [Google Scholar] [CrossRef]
- Managuli, R.S.; Kumar, L.; Chonkar, A.D.; Shirodkar, R.K.; Lewis, S.; Koteshwara, K.B.; Reddy, M.S.; Mutalik, S. Development and validation of a stability-indicating RP-HPLC method by a statistical optimization process for the quantification of asenapine maleate in lipidic nanoformulations. J. Chromatogr. Sci. 2016, 54, 1290–1300. [Google Scholar] [CrossRef]
- Shyamala; Swarupa, A.; Anitha, P. Validated stability-indicating RP-HPLC method for determination of asenapine. Indo Am. J. Pharm. Sci. (IAJPS) 2018, 5, 4107–4113. [Google Scholar] [CrossRef]
- Kalpana, G.L.; Devalarao, G.; Raju, M.B.; Praveenkumar, T. Validated stability indicating high performance liquid chromatographic method for the quantification of asenapine maleate. Int. J. Pharm. Pharm. Sci. 2015, 7, 61–65. [Google Scholar]
- Chowdary, A.K.; Suravarapu, N.L.R.; Meddala, S. Formulation and characterization of asenapine maleate nanoparticles. Int. J. Pharm. Sci. Rev. Res. 2016, 40, 1–5. [Google Scholar]
- Naik, B.; Gandhi, J.; Shah, P.; Naik, H.; Sarolia, J. Asenapine maleate loaded solid lipid nanoparticles for oral delivery. Int. Res. J. Pharm. 2017, 8, 45–53. [Google Scholar] [CrossRef]
- Supriya, A.; Sundaraseelan, J.; Murthy, B.R.S.; Priya, M.B. Formulation and evaluation of capsules of asenapine maleate loaded chitosan nanoparticles. ASPS 2018, 2, 29–37. [Google Scholar]
- Patel, M.H.; Mundada, V.P.; Sawant, K.K. Novel drug delivery approach via self-microemulsifying drug delivery system for enhancing oral bioavailability of asenapine maleate: Optimization, characterization, cell uptake, and in vivo pharmacokinetic studies. AAPS PharmSciTech 2019, 20, 44. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Malviya, K.; Kumar, S.; Golwala, V.D.K.; Mohapatra, P.K. Formulation and evaluation of sublingual tablets of asenapine maleate by 32 full factorial design. Aegaeum J. (AJ) 2020, 8, 1236–1250. [Google Scholar]
- Blatter, F.; Reichenbacher, K. Novel Crystalline Salts of Asenapine with Organic di-Acids and Tri-Acids. U.S. Patent WO 2012/156383 Al, 21 November 2012. [Google Scholar]
- Managuli, R.S.; Wang, J.T.; Faruqu, F.N.; Kushwah, V.; Raut, S.Y.; Shreya, A.B.; Al-Jamal, K.T.; Jain, S.; Mutalik, S. Asenapine maleate-loaded nanostructured lipid carriers: Optimization and in vitro, ex vivo and in vivo evaluations. Nanomedicine 2018, 14, 889–910. [Google Scholar] [CrossRef]
- Managuli, R.S.; Wang, J.T.-Z.; Faruqu, F.M.; Pandey, A.; Jain, S.; Al-Jamal, K.T.; Mutalik, S. Surface engineered nanoliposomal platform for selective lymphatic uptake of asenapine maleate: In vitro and in vivo studies. Mater. Sci. Eng. C 2019, 109. [Google Scholar] [CrossRef]
- Tzanavaras, P.D. A green HPLC method for the determination of n-acetylcysteine using post-column derivatization with methyl-propiolate. Instrum. Sci. Technol. 2012, 40, 150–160. [Google Scholar] [CrossRef]
- Yabré, M.; Ferey, L.; Somé, T.I.; Sivadier, G.; Gaudin, K. Development of a green HPLC method for the analysis of artesunateand amodiaquine impurities using quality by design. J. Pharm. Biomed. Anal. 2020, 190, 113507–113516. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Liu, X.; Dong, Y.; Yang, J.; Zhang, J.; He, S.; Yang, F.; Wang, Z.; Dong, Y. A green HPLC method for determination of nine sulfonamides in milk and beef, and its greenness assessment with analytical eco-scale and greenness profile. J. AOAC Int. 2020, 103, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.-Z.; Ragan, F.A., Jr.; Prasad, C. Use of Triethylamine as an Ion-Pairing Reagent. J. Liq. Chromatogr. 1994, 17, 2383–2394. [Google Scholar] [CrossRef]
- Park, J.H.; Ryu, Y.K.; Lim, H.J.; Lee, H.S.; Park, J.K.; Lee, Y.K.; Jang, M.D.; Suh, J.K.; Carr, P.W. Effect of triethylamine in the mobile phase on the retention properties of conventional polymeric and horizontally polymerized octadecylsilica in RPLC. Chromatographia 1999, 49, 635–642. [Google Scholar] [CrossRef]
- United States Pharmacopeia and National Formulary (USP30-NF25); 1225, Validation of Compendial Procedures; United States Pharmacopeial Convention: Rockville, MD, USA, 2007.
- Validation of Analytical Procedures. Text and Methodology. In Proceedings of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, Q2(R1), Geneva, Switzerland, 1–13 November 2005.
- Karim, S.; Hay, K.Y.; Baie, S.H.; Bukhari, N.I.; Murtaza, G. Study of comparative bioavailability of omeprazole pellets. Acta Pol. Pharm. 2014, 71, 463–468. [Google Scholar] [PubMed]
- Suresh, R.; Anarthanan, S.; Manavalan, R.; Valliappan, K. Aspects of validation in HPLC method development for pharmaceutical analysis-comparison of validation requirements by FDA, USA and ICH. Int. J. Pharm. Sci. Res. 2010, 12, 123–133. [Google Scholar]
- Rao, R.; Talluri, M.V.; Raju, A.N.; Shinde, D.D.; Ramanjaneyulu, G.S. Development of a validated RP-LC/ESI-MS–MS method for separation, identification and determination of related substances of tamsulosin in bulk drugs and formulations. J. Pharm. Biomed. Anal. 2008, 46, 94–103. [Google Scholar]
- Reddy, L.S.; Reddy, S.L.N.P.; Reddy, G.S. Development and validation of a stability indicating liquid chromatographic method for simultaneous estimation of dutasteride and tamsulosin in combined dosage form. Orient J. Chem. 2013, 29, 1665–1673. [Google Scholar] [CrossRef][Green Version]
- Tiwari, G.; Tiwari, R. Bioanalytical method validation: An updated review. Pharm. Methods 2010, 1, 25–38. [Google Scholar] [CrossRef]
- Lindholm, J. Development and Validation of HPLC Methods for Analytical and Preparative Purposes. Master Thesis, Acta Universitatis Upsaliensis, Uppsala, Sweden, 2004. [Google Scholar]
- US Food and Drug Administration. Guidance for Industry-Bioanalytical Method Validation; US Food and Drug Administration: Rockville, MD, USA, 2001.
- US Food and Drug Administration. Reviewer Guidance: Validation of Chromatographic Method; US Food and Drug Administration: Rockville, MD, USA, 1994.
- CHROMacademy. The Theory of HPLC Chromatographic Parameters. Available online: http://www.Chromacademy.com (accessed on 24 February 2019).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Calib. Curve # | Conc. (µg/mL) | Slope | Intercept | R2 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.1 | 0.5 | 1 | 2 | 4 | 8 | 10 | 14 | ||||
| Area under the Peak | |||||||||||
| 1 | 3768 | 17,276 | 36,890 | 76,744 | 154,435 | 310,322 | 377,191 | 536,969 | 38,312 | 444.35 | 0.9998 |
| 2 | 3539 | 16,413 | 34,897 | 68,696 | 145,550 | 285,936 | 361,038 | 498,709 | 35,844 | 579.21 | 0.9999 |
| 3 | 2846 | 15,543 | 34,995 | 63,027 | 121,823 | 267,408 | 329,266 | 449,850 | 32,534 | 450.44 | 0.9991 |
| 4 | 3080 | 18,022 | 38,372 | 73,429 | 138,106 | 295,102 | 369,782 | 508,066 | 36,552 | 438.69 | 0.9996 |
| 5 | 3215 | 16,194 | 35,921 | 75,730 | 151107 | 300,606 | 368,445 | 514,839 | 36,927 | 470.3 | 0.9998 |
| 6 | 4551 | 16,004 | 35,087 | 71,747 | 147,177 | 287,246 | 365,097 | 508,659 | 36,384 | 656.84 | 0.9999 |
| Mean | 3500 | 16,575 | 36,027 | 71,562 | 143,033 | 291,103 | 361,803 | 502,849 | |||
| SD | 610 | 911 | 1377 | 5076 | 11,772 | 14,688 | 16,821 | 28,965 | |||
| RSD | 17 | 5 | 4 | 7 | 8 | 5 | 5 | 6 | |||
| Source of Variation | DF | SS | MS | F Ratio | Prop > F |
|---|---|---|---|---|---|
| Between groups | 7 | 1,447,908,839,267.33 | 206,844,119,895.33 | 1099.44 | 0 |
| Within group | 40 | 7,525,440,295.57 | 188,136,007.39 | ||
| Total | 47 | 1,455,434,279,562.89 |
| QC Samples (n = 6) | Conc. (μg/mL) | Inter-Day Assay | Inter-Day Assay | ||||
|---|---|---|---|---|---|---|---|
| Mean | Accuracy | Precision (CV %) | Mean | Accuracy | Precision (CV %) | ||
| LLOQ | 0.10 | 0.10 | 101.00 | 3.71 | 0.10 | 104.62 | 6.62 |
| QC low | 0.30 | 0.29 | 97.85 | 5.50 | 0.30 | 99.67 | 3.43 |
| QC mid | 7.00 | 6.71 | 95.87 | 3.26 | 7.02 | 100.23 | 4.45 |
| QC high | 12.00 | 11.66 | 97.17 | 1.80 | 12.36 | 103.02 | 5.28 |
| Concentration Level | Source of Variation | DF | SS | MS | F Ratio | Prop > F |
|---|---|---|---|---|---|---|
| LLOQ | between groups | 2 | 0.0004 | 0.0002 | 6.82 | 0.0078 |
| within group | 15 | 0.0004 | 0 | |||
| Total | 17 | 0.0008 | ||||
| QC Low | between groups | 2 | 0.0003 | 0.0002 | 1.837 | 0.2014 |
| within group | 12 | 0.0011 | 0.0001 | |||
| Total | 14 | 0.0015 | ||||
| QC Mid | between groups | 2 | 1.0562 | 0.5281 | 20.6702 | 0.0001 |
| within group | 12 | 0.3066 | 0.0255 | |||
| Total | 14 | 1.3628 | ||||
| QC High | between groups | 2 | 4.8364 | 2..4182 | 25.5275 | 0 |
| within group | 12 | 1.1368 | 0.0947 | |||
| Total | 14 | 5.9732 |
| Parameters Studied | Rs | k | N | As | |
|---|---|---|---|---|---|
| Flow rate (mL/min) | 0.80 | 7.88 | 4.00 | 1644.68 | 1.85 |
| 1.20 | 4.53 | 2.23 | 1819.72 | 1.76 | |
| pH of the buffer | 3.30 | 10.89 | 2.86 | 2436.00 | 0.98 |
| 3.70 | 8.40 | 2.92 | 1187.55 | 1.41 | |
| Buffer:acetonitrile | 68:32 | 8.27 | 2.04 | 2171.01 | 1.24 |
| 72:28 | 12.17 | 3.80 | 2380.00 | 1.06 | |
| Nominal condition | 10.89 | 2.86 | 2178.07 | 1.20 | |
| Target | >2 | 1 < k’ < 10 | >2000 | ≤2 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Nimry, S.S.; Khanfar, M.S. Validation of an RP-HPLC Method for the Determination of Asenapine Maleate in Dissolution Media and Application to Study In Vitro Release from Co-Crystals. Sci. Pharm. 2021, 89, 14. https://doi.org/10.3390/scipharm89010014
Al-Nimry SS, Khanfar MS. Validation of an RP-HPLC Method for the Determination of Asenapine Maleate in Dissolution Media and Application to Study In Vitro Release from Co-Crystals. Scientia Pharmaceutica. 2021; 89(1):14. https://doi.org/10.3390/scipharm89010014
Chicago/Turabian StyleAl-Nimry, Suhair S., and Mai S. Khanfar. 2021. "Validation of an RP-HPLC Method for the Determination of Asenapine Maleate in Dissolution Media and Application to Study In Vitro Release from Co-Crystals" Scientia Pharmaceutica 89, no. 1: 14. https://doi.org/10.3390/scipharm89010014
APA StyleAl-Nimry, S. S., & Khanfar, M. S. (2021). Validation of an RP-HPLC Method for the Determination of Asenapine Maleate in Dissolution Media and Application to Study In Vitro Release from Co-Crystals. Scientia Pharmaceutica, 89(1), 14. https://doi.org/10.3390/scipharm89010014