The Potential Benefit of Beta-Blockers for the Management of COVID-19 Protocol Therapy-Induced QT Prolongation: A Literature Review

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
3. COVID-19 Treatment and Arrhythmia
3.1. Azithromycin
3.2. Chloroquine
3.3. Hydroxychloroquine–Azithromycin Combination Therapy
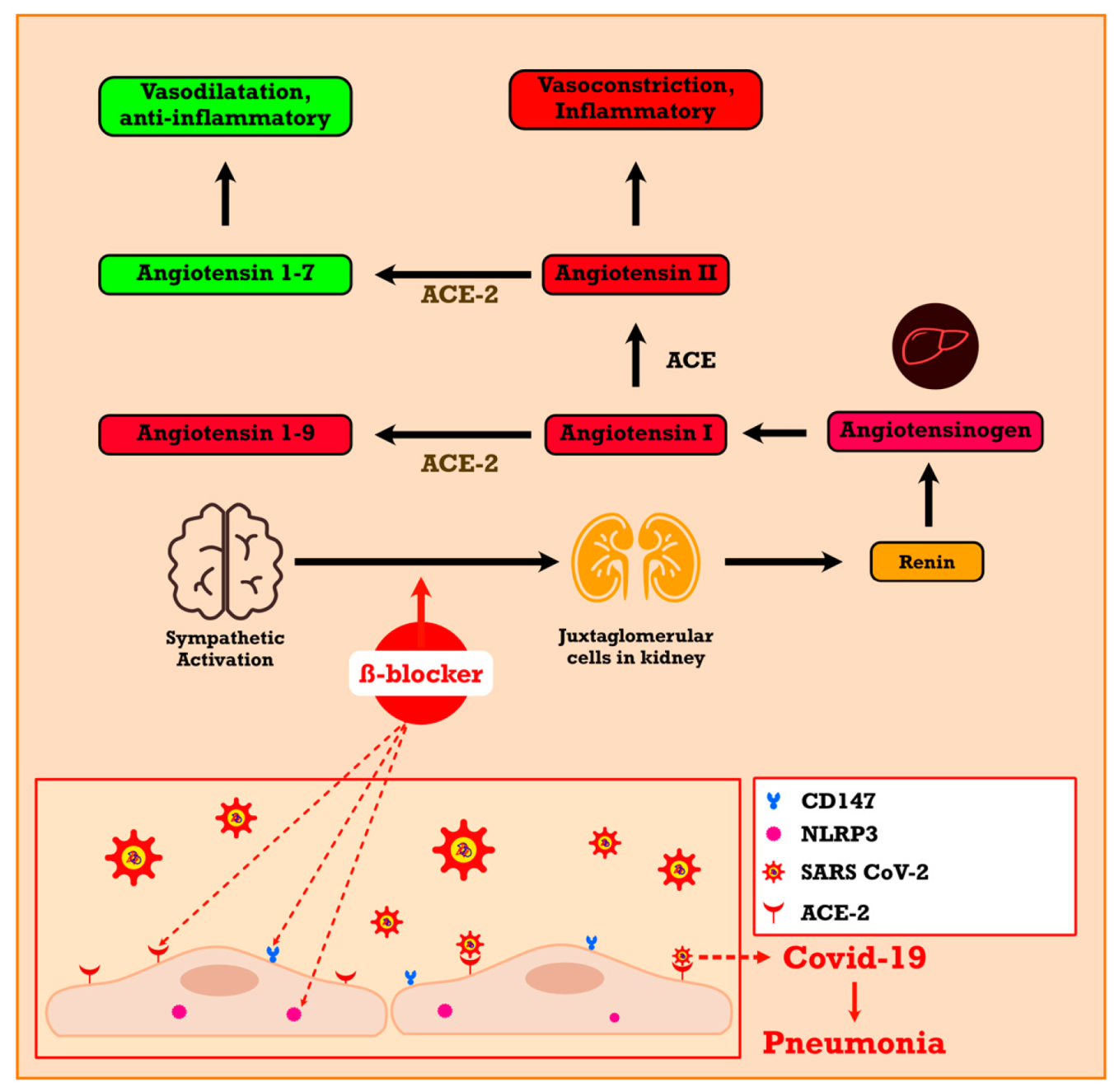
4. Beta-Blockers
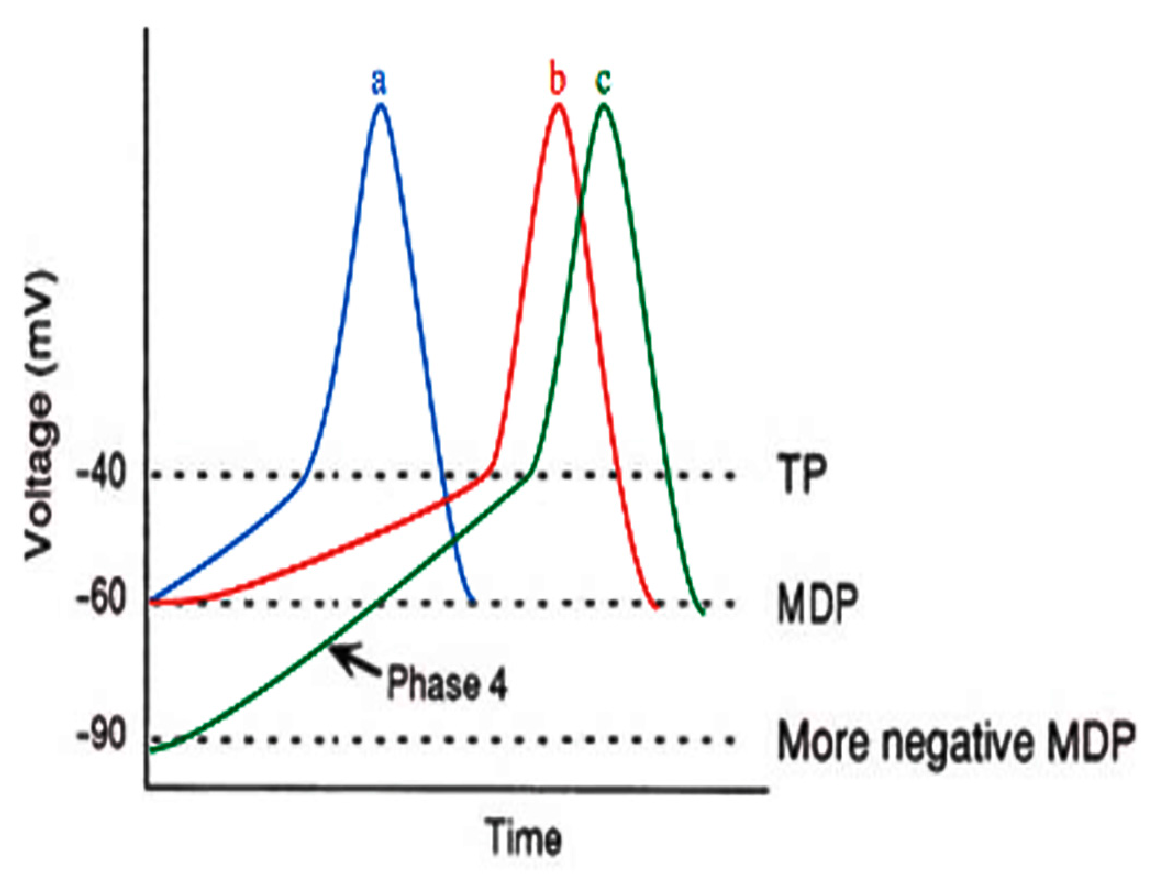
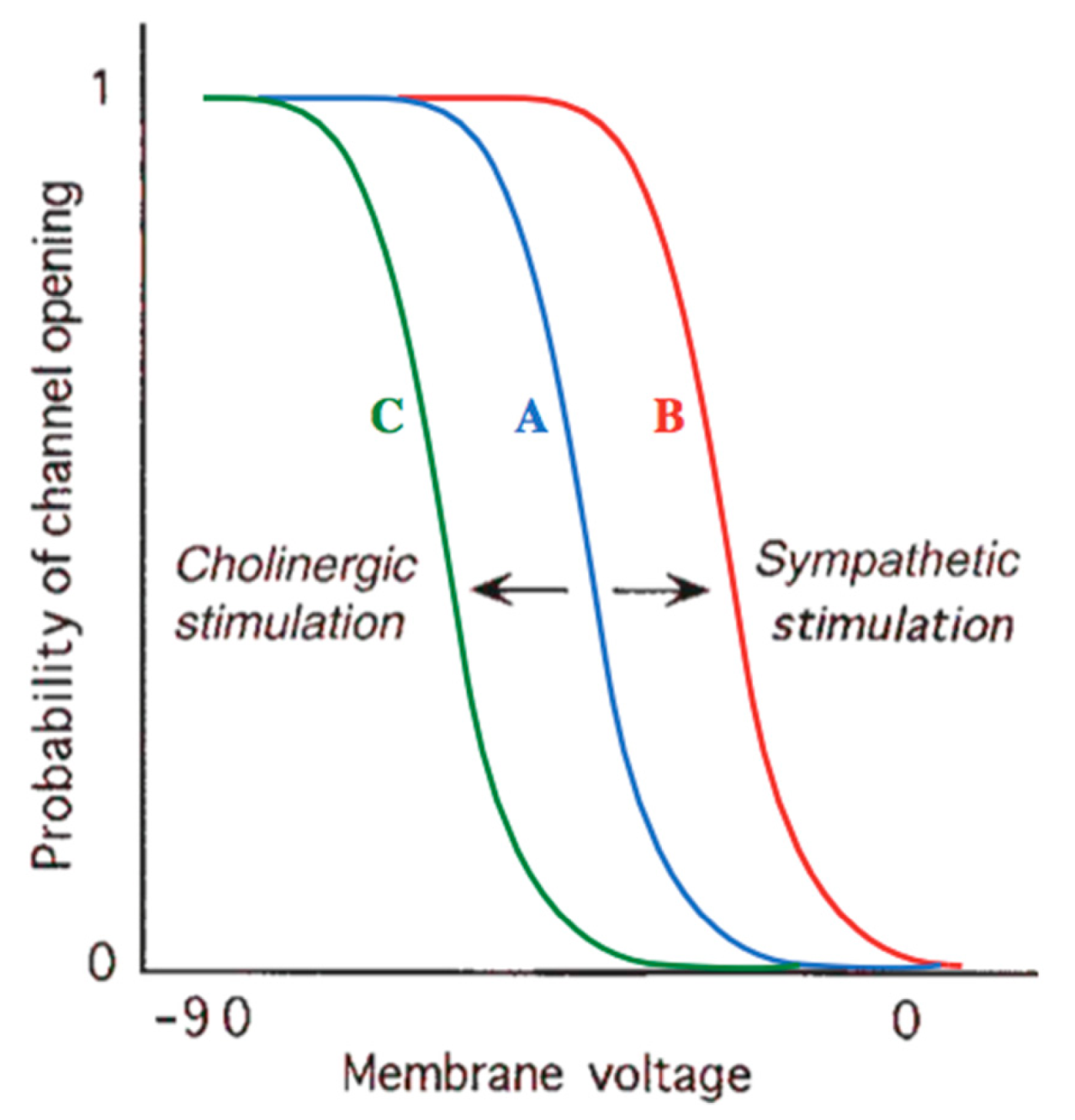
4.1. β-Adrenergic Physiology
4.2. Cardio-Selective Beta-Blockers Associated with Lung Function
4.3. Clinical Use of Beta-Blockers
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lu, H.C.; Stratton, W.; Tang, Y.W. Outbreak of pneumonia of unknown etiology in Wuhan, China: The mystery and the miracle. J. Med. Virol. 2020, 92, 401–402. [Google Scholar] [CrossRef] [PubMed]
- Hui, D.S. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 2020, 91, 264–266. [Google Scholar] [CrossRef] [PubMed]
- Burki, T. Outbreak of coronavirus disease 2019. Lancet Infect. Dis. 2020, 20, 292–293. [Google Scholar] [CrossRef]
- Harapan, H.; Itoh, N.; Yufika, A.; Winardi, W.; Keam, S.; Te, H.; Megawati, D.; Hayati, Z.; Wagner, A.L.; Mudatsir, M. Coronavirus disease 2019 (COVID-19): A literature review. J. Infect. Public Health 2020, 13, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhao, Y.; Zhang, F.; Wang, Q.; Li, T.; Liu, Z.; Wang, J.; Qin, Y.; Zhang, X.; Yan, X.; et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The experience of clinical immunologists from China. Clin. Immunol. 2020, 214, 108393. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tang, J.; Wei, F. Updated understanding of the outbreak of 2019 novel coronavirus (2019-nCoV) in Wuhan, China. J. Med. Virol. 2020, 92, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Elfiky, A.A. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sci. 2020, 248, 117477. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Patil, V.M.; Singhal, S.; Masand, A. A systematic review on use of aminoquinolines for the therapeutic management of COVID-19: Efficacy, safety and clinical trials. Life Sci. 2020, 254, 117775. [Google Scholar] [CrossRef]
- Saleh, M.; Gabriels, J.; Chang, D.; Kim, B.S.; Mansoor, A.; Mahmood, E.; Makker, P.; Ismail, H.; Goldner, B.; Willner, J.; et al. Effect of Chloroquine, Hydroxychloroquine, and Azithromycin on the Corrected QT Interval in Patients with SARS-CoV-2 Infection. Circ. Arrhythm. Electrophysiol. 2020, 13, 496–504. [Google Scholar] [CrossRef]
- Retallack, H.; Di Lullo, E.; Arias, C.; Knopp, K.A.; Laurie, M.T.; Sandoval-Espinosa, C.; Leon, W.R.M.; Krencik, R.; Ullian, E.M.; Spatazza, J.; et al. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl. Acad. Sci. USA 2016, 113, 14408–14413. [Google Scholar] [CrossRef] [PubMed]
- Jelić, D.; Antolović, R. From erythromycin to azithromycin and new potential ribosome-binding antimicrobials. Antibiotics 2016, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Sapp, J.L.; Alqarawi, W.; MacIntyre, C.J.; Tadros, R.; Steinberg, C.; Roberts, J.D.; Laksman, Z.; Healey, J.S.; Krahn, A.D. Guidance on Minimizing Risk of Drug-Induced Ventricular Arrhythmia During Treatment of COVID-19: A Statement from the Canadian Heart Rhythm Society. Can. J. Cardiol. 2020, 36, 948–951. [Google Scholar] [CrossRef]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Field, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS Guideline for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death. Circulation 2018, 138, e272–e391. [Google Scholar] [CrossRef]
- Grandi, E.; Ripplinger, C.M. Antiarrhythmic mechanisms of beta-blockers therapy. Pharmacol. Res. 2019, 146, 104274. [Google Scholar] [CrossRef] [PubMed]
- Lu, H. Drug treatment options for the 2019-new coronavirus (2019-nCoV). Biosci. Trends 2020, 14, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Gautret, P.; Lagier, J.-C.; Parola, P.; Hoang, V.T.; Meddeb, L.; Mailhe, M.; Doudier, B.; Courjon, J.; Giordanengo, V.; Vieira, V.E.; et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents 2020, 56, 105949. [Google Scholar] [CrossRef]
- Arshad, S.; Kilgore, P.; Chaudhry, Z.S.; Jacobsen, G.; Wang, D.D.; Huitsing, K.; Brar, I.; Alangaden, G.J.; Ramesh, M.S.; McKinnon, J.E.; et al. Treatment with hydroxychloroquine, azithromycin, and combination in patients hospitalized with COVID-19. Int. J. Infect. Dis. 2020, 97, 396–403. [Google Scholar] [CrossRef]
- Borba, M.G.S.; Val, F.F.A.; Sampaio, V.S.; Alexandre, M.A.A.; Melo, G.C.; Brito, M.; Mourão, M.P.G.; Brito-Sousa, J.D.; Baía-Da-Silva, D.; Guerra, M.V.F.; et al. Effect of High vs Low Doses of Chloroquine Diphosphate as Adjunctive Therapy for Patients Hospitalized With Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection: A Randomized Clinical Trial. JAMA Netw. Open 2020, 3, e208857. [Google Scholar] [CrossRef]
- Gandhi, R.T.; Lynch, J.B.; del Rio, C. Mild or Moderate Covid-19. N. Engl. J. Med. 2020, 1–9. [Google Scholar] [CrossRef]
- Ray, W.A.; Murray, K.T.; Hall, K.; Arbogast, P.G.; Stein, C.M. Azithromycin and the risk of cardiovascular death. N. Engl. J. Med. 2012, 366, 1881–1990. [Google Scholar] [CrossRef] [PubMed]
- Nachimuthu, S.; Assar, M.D.; Schussler, J.M. Drug-induced QT interval prolongation: Mechanisms and clinical management. Ther. Adv. Drug Saf. 2012, 3, 241–253. [Google Scholar] [CrossRef]
- Belardinelli, L.; Antzelevitch, C.; Vos, M.A. Assessing predictors of drug-induced torsade de pointes. Trends Pharmacol. Sci. 2003, 24, 619–625. [Google Scholar] [CrossRef]
- Damle, B.; Vourvahis, M.; Wang, E.; Leaney, J.; Corrigan, B. Clinical Pharmacology Perspectives on the Antiviral Activity of Azithromycin and Use in COVID-19. Clin. Pharmacol. Ther. 2020, 108, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Savarino, A.; Di Trani, L.; Donatelli, I.; Cauda, R.; Cassone, A. New insights into the antiviral effects of chloroquine. Lancet Infect. Dis. 2006, 6, 67–69. [Google Scholar] [CrossRef]
- Zhou, D.; Dai, S.M.; Tong, Q. COVID-19: A recommendation to examine the effect of hydroxychloroquine in preventing infection and progression. J. Antimicrob. Chemother. 2020, 75, 1667–1670. [Google Scholar] [CrossRef] [PubMed]
- White, N.J. Cardiotoxicity of antimalarial drugs. Lancet Infect. Dis. 2007, 7, 549–558. [Google Scholar] [CrossRef]
- Costedoat-Chalumeau, N.; Hulot, J.-S.; Amoura, Z.; Leroux, G.; Lechat, P.; Funck-Brentano, C.; Piette, J.-C. Heart conduction disorders related to antimalarials toxicity: An analysis of electrocardiograms in 85 patients treated with hydroxychloroquine for connective tissue diseases. Rheumatology 2007, 46, 808–810. [Google Scholar] [CrossRef]
- Wu, C.-I.; Postema, P.G.; Arbelo, E.; Behr, E.R.; Bezzina, C.R.; Napolitano, C.; Robyns, T.; Probst, V.; Schulze-Bahr, E.; Remme, C.A.; et al. SARS-CoV-2, COVID-19, and inherited arrhythmia syndromes. Hear. Rhythm. 2020, 17, 1456–1462. [Google Scholar] [CrossRef]
- Marquardt, K.; Albertson, T.E. Treatment of hydroxychloroquine overdose. Am. J. Emerg. Med. 2001, 19, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Mercuro, N.J.; Yen, C.F.; Shim, D.J.; Maher, T.R.; McCoy, C.M.; Zimetbaum, P.J.; Gold, H.S. Risk of QT Interval Prolongation Associated with Use of Hydroxychloroquine with or without Concomitant Azithromycin among Hospitalized Patients Testing Positive for Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 1036–1041. [Google Scholar] [CrossRef]
- Zhu, S.; Wang, J.; Wang, Y.; Chu, J.; Liu, Y.; Chen, X.; Chen, X. QTc prolongation during antiviral therapy in two COVID-19 patients. J. Clin. Pharm. Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Pandurangi, U.; Arora, V.; Gupta, A.; Jaswal, A.; Nabar, A.; Naik, A.; Naik, N.; Namboodiri, N.; Vora, A.; et al. Cardiovascular risks of hydroxychloroquine in treatment and prophylaxis of COVID-19 patients: A scientific statement from the Indian Heart Rhythm Society. Indian Pacing Electrophysiol. J. 2002, 20, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Eastman, R.T.; Roth, J.S.; Brimacombe, K.R.; Simeonov, A.; Shen, M.; Patnaik, S.; Hall, M.D. Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Cent. Sci. 2020, 6, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, J.E. Drug-induced QT interval prolongation and torsade de pointes: Role of the pharmacist in risk assessment, prevention and management. Can. Pharm. J. 2020, 149, 139–152. [Google Scholar] [CrossRef]
- Purwowiyoto, S.L.; Hermanto, D.Y.; Muhammad, I. Managing QT prolongation in the Era of Coronavirus Disease 2019 (COVID-19). Indones. J. Cardiol. 2020, 41, 108–111. [Google Scholar] [CrossRef]
- Cannon, W.B. Bodily Changes in Pain, Hunger, Fear and Rage: An Account of Recent Researches into the Function of Emotional Excitement; Cannon Press: Idaho, Moscow, 2008. [Google Scholar]
- DiFrancesco, D.; Tortora, P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature 1991, 351, 145–147. [Google Scholar] [CrossRef]
- Lakatta, E.G.; DiFrancesco, D. What keeps us ticking: A funny current, a calcium clock, or both? J. Mol. Cell. Cardiol. 2009, 47, 157–170. [Google Scholar] [CrossRef]
- Grandi, E.; Sanguinetti, M.C.; Bartos, D.C.; Bers, D.M.; Chen-Izu, Y.; Chiamvimonvat, N.; Colecraft, H.M.; Delisle, B.P.; Heijman, J.; Navedo, M.F.; et al. Potassium channels in the heart: Structure, function and regulation. J. Physiol. 2017, 595, 2209–2228. [Google Scholar] [CrossRef] [PubMed]
- Bartos, D.C.; Grandi, E.; Ripplinger, C.M. Ion channels in the heart. Compr. Physiol. 2015, 5, 1423–1464. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Leonard, M.L. Pathophysiology of the Heart Disease: A Collaborative Project of Medical Students and Faculty, 6th ed.; Wolters Kluwer: Philadelphia, PA, USA, 2016. [Google Scholar]
- Difrancesco, D. The role of the funny current in pacemaker activity. Circ. Res. 2010, 106, 434–446. [Google Scholar] [CrossRef]
- Vincent, G.M.; Schwartz, P.J.; Denjoy, I.; Swan, H.; Bithell, C.; Spazzolini, C.; Crotti, L.; Piippo, K.; Lupoglazoff, J.-M.; Villain, E.; et al. High efficacy of β-blockers in long-QT syndrome type 1: Contribution of noncompliance and QT-prolonging drugs to the occurrence of β-blocker treatment failures. Circulation 2009, 119, 215–221. [Google Scholar] [CrossRef]
- Ajijola, O.A.; Lux, R.L.; Khahera, A.; Kwon, O.; Aliotta, E.; Ennis, D.B.; Fishbein, M.C.; Ardell, J.L.; Shivkumar, K. Sympathetic modulation of electrical activation in normal and infracted myocardium: Implications for arrhythmogenesis. Am. J. Physiol. Hear. Circ. Physiol. 2017, 312, H608–H621. [Google Scholar] [CrossRef]
- Westfall, D.; Macarthur, H.; Westfall. The Autonomic and Somatic Motor Nervous Systems. In Goodman and Gilman’s the Pharmacological Basis of Therapeutics; Mc.Graw Hill: New York, NY, USA, 2018. [Google Scholar]
- Jamali, H.K.; Waqar, F.; Gerson, M.C. Cardiac autonomic innervation. J. Nucl. Cardiol. 2017, 24, 1558–1570. [Google Scholar] [CrossRef]
- Laitinen, P.J.; Brown, K.M.; Piippo, K.; Swan, H.; Devaney, J.M.; Brahmbhatt, B.; Donarum, E.A.; Marino, M.; Tiso, N.; Viitasalo, M.; et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 2001, 103, 185–190. [Google Scholar] [CrossRef]
- Myles, R.C.; Wang, L.; Kang, C.; Bers, D.M.; Ripplinger, C.M. Local β-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circ. Res. 2012, 110, 154–164. [Google Scholar] [CrossRef]
- Bundkirchen, A.; Brixius, K.; Bölck, B.; Nguyen, Q.; Schwinger, R.H.G. β1-adrenoceptor selectivity of nebivolol and bisoprolol. A comparison of [3H]CGP 12.177 and [125I]iodocyanopindolol binding studies. Eur. J. Pharmacol. 2003, 460, 19–26. [Google Scholar] [CrossRef]
- Lipworth, B.; Wedzicha, J.; Devereux, G.; Vestbo, J.; Dransfield, M.T. Beta-blockers in COPD: Time for reappraisal. Eur. Respir. J. 2016, 48, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Short, P.M.; Lipworth, S.I.; Elder, D.H.; Schembri, S.; Lipworth, B.J. Effect of β blockers in treatment of chronic obstructive pulmonary disease: A retrospective cohort study. BMJ 2011, 342, d2549. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.G. The selectivity of beta-adrenoceptor agonists at human beta1-, beta2- and beta3-adrenoceptors. Br. J. Pharmacol. 2010, 160, 1048–1061. [Google Scholar] [CrossRef] [PubMed]
- Kamp, O.; Metra, M.; Bugatti, S.; Bettari, L.; Cas, A.D.; Petrini, N.; Cas, L.D.; Metra, P.M. Nebivolol: Haemodynamic effects and clinical significance of combined β-blockade and nitric oxide release. Drugs 2010, 70, 41–56. [Google Scholar] [CrossRef]
- Zhou, J.; Yi, J.; Hu, N.N.; George, A.L.; Murray, K.T. Activation of protein kinase A modulates trafficking of the human cardiac sodium channel in Xenopus oocytes. Circ. Res. 2000, 87, 33–38. [Google Scholar] [CrossRef]
- Coppola, S.; Froio, S.; Chiumello, D. β-blockers in critically ill patients: From physiology to clinical evidence. Crit. Care 2015, 19, 83. [Google Scholar] [CrossRef]
- January, C.T.; Wann, L.S.; Alpert, J.S.; Calkins, H.; Cigarroa, J.E.; Cleveland, J.C.; Conti, J.B.; Ellinor, P.; Ezekowitz, M.D.; Field, M.E.; et al. AHA/ACC/HRS Guideline for the Management of Patients with Atrial Fibrillation. J. Am. Coll. Cardiol. 2014, 64, e1–e76. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Priori, S.G.; Spazzolini, C.; Moss, A.J.; Vincent, G.M.; Napolitano, C.; Denjoy, I.; Guicheney, P.; Breithardt, G.; Keating, M.T.; et al. Genotype-Phenotype Correlation in the Long-QT Syndrome. Circulation 2001, 103, 89–95. [Google Scholar] [CrossRef]
- Chatrath, R.; Bell, C.M.; Ackerman, M.J. β-blocker therapy failures in symptomatic probands with genotyped long-QT syndrome. Pediatr. Cardiol. 2004, 25, 459–465. [Google Scholar] [CrossRef]
- Hockalingam, P.; Crotti, L.; Girardengo, G.; Johnson, J.N.; Harris, K.M.; Van Der Heijden, J.F.; Hauer, R.N.; Beckmann, B.M.; Spazzolini, C.; Rordorf, R.; et al. Not all beta-blockers are equal in the management of long QT syndrome types 1 and 2: Higher recurrence of events under metoprolol. J. Am. Coll. Cardiol. 2012, 60, 2092–2099. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Moss, A.J.; Kaufman, E.S.; Shimizu, W.; Peterson, D.R.; Benhorin, J.; Lopes, C.; Towbin, J.A.; Spazzolini, C.; Crotti, L.; et al. Clinical Aspects of Type 3 Long-QT Syndrome: An International Multicenter Study. Circulation 2016, 134, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Zanchetti, A. Clinical pharmacodynamics of nebivolol: New evidence of nitric oxide-mediated vasodilating activity and peculiar haemodynamic properties in hypertensive patients. Blood Press 2004, 1, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, S.M.; Cay, S.; Cagirci, G.; Sen, N. Nebivolol therapy improves QTc and QTcd parameters in heart failure patients. Cardiovasc. J. Afr. 2012, 23, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Zipes, D.P.; Borggrefe, M.; Buxton, A.E.; Chaitman, B.; Fromer, M.; Gregoratos, G.; Klein, G.; Myerburg, R.J.; Quinones, M.A.; Roden, D.M.; et al. ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death-Executive Summary. J. Am. Coll. Cardiol. 2006, 48, e385–e484. [Google Scholar] [CrossRef]
- Steinberg, C.; Padfield, G.J.; Al-Sabeq, B.; Adler, A.; Yeung-Lai-Wah, J.A.; Kerr, C.R.; Deyell, M.W.; Andrade, J.G.; Bennett, M.T.; Yee, R.; et al. Experience with bisoprolol in long-QT1 and long-QT2 syndrome. J. Interv. Card. Electrophysiol. 2016, 47, 163–170. [Google Scholar] [CrossRef]
- Xie, W.; Xie, H.; Liu, F.; Li, W.; Dan, J.; Mei, Y.; Dan, L.; Xiao, X.; Li, J.; Chen, X. Propranolol induces apoptosis of human umbilical vein endothelial cells through downregulation of CD147. Br. J. Dermatol. 2013, 168, 739–748. [Google Scholar] [CrossRef]
- Natesan, V. Beta-adrenergic blocker treatment for COVID-19. Hypothesis 2020. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Agent | Mechanism of Action | Clinical Use |
|---|---|---|
| Non-selective β-adrenergic antagonists (first generation): | ||
| Propranolol | The same affinity for both the β1 and β2 receptors; membrane-stabilizing effect. | Hypertension, angina, supraventricular arrhythmia, ventricular arrhythmia, and myocardial infarction (MI). |
| Nadolol | The same affinity for both the β1 and β2 receptors; no sympathomimetic or membrane-stabilizing actions. | Hypertension, angina, and long QT syndrome (LQTS). |
| Timolol | The same affinity for both the β1 and β2 receptors; no sympathomimetic or membrane-stabilizing actions. | Hypertension, congestive heart failure (HF), acute and MI. |
| Cardio-selective β1-adrenergic antagonists (second generation): | ||
| Metoprolol | No sympathomimetic or membrane-stabilizing actions. | Essential hypertension, angina, tachycardia, HF, vasovagal syncope, and secondary preventions after MI. |
| Atenolol | No sympathomimetic or membrane-stabilizing actions. | Hypertension, angina, coronary heart disease, arrhythmias, and minimization of the risk of complications after MI. |
| Esmolol | Little sympathomimetic activity and no membrane-stabilizing action. | Used when short duration is needed or in critically ill patients where rapid removal may be necessary. |
| Acebutolol | Some sympathomimetic and membrane-stabilizing actions. | Hypertension, atrial and ventricular arrhythmias, and acute MI in high-risk patients |
| Bisoprolol | No sympathomimetic or membrane-stabilizing actions; higher degree of β1 selectivity than metoprolol or atenolol. | HF, hypertension, MI, and arrhythmias. |
| β-adrenergic antagonists with additional cardiovascular effects (third generation—also have vasodilatory actions): | ||
| Labetalol | Inhibitor of the α1 and β (β1 and β2) receptors; partial agonist activity at β2 and also blocks neuronal uptake of norephinephrine (cocaine-like). | Chronic hypertension or hypertensive emergencies. |
| Carvedilol | Inhibits α1, β1, and β2 akin to labetalol, but also has antioxidant and anti-inflammatory properties; has membrane-stabilizing action, but no sympathomimetic activity. | Generating vasodilation and anti-inflammatory effects, may help in the treatment of HF; approved for treatment in hypertension, congestive HF, and LV-dysfunction after MI. |
| Celiprolol | Antagonist of α2 and β1 antagonist, and partial agonist of β2; also an α2 antagonist and promotes nitric oxide (NO) production. | Minimization of heart rate and blood pressure; used to medicate hypertension and angina. |
| Nebivolol | β1 antagonist with endothelial vasodilatory effect via stimulation of β3 receptors. | Antioxidant action and neutral or effective in carbohydrate and lipid metabolism; approved for treating hypertension. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heriansyah, T.; Nur Chomsy, I.; Febrianda, L.; Farahiya Hadi, T.; Andri Wihastuti, T. The Potential Benefit of Beta-Blockers for the Management of COVID-19 Protocol Therapy-Induced QT Prolongation: A Literature Review. Sci. Pharm. 2020, 88, 55. https://doi.org/10.3390/scipharm88040055
Heriansyah T, Nur Chomsy I, Febrianda L, Farahiya Hadi T, Andri Wihastuti T. The Potential Benefit of Beta-Blockers for the Management of COVID-19 Protocol Therapy-Induced QT Prolongation: A Literature Review. Scientia Pharmaceutica. 2020; 88(4):55. https://doi.org/10.3390/scipharm88040055
Chicago/Turabian StyleHeriansyah, Teuku, Indah Nur Chomsy, Lyra Febrianda, Tjut Farahiya Hadi, and Titin Andri Wihastuti. 2020. "The Potential Benefit of Beta-Blockers for the Management of COVID-19 Protocol Therapy-Induced QT Prolongation: A Literature Review" Scientia Pharmaceutica 88, no. 4: 55. https://doi.org/10.3390/scipharm88040055
APA StyleHeriansyah, T., Nur Chomsy, I., Febrianda, L., Farahiya Hadi, T., & Andri Wihastuti, T. (2020). The Potential Benefit of Beta-Blockers for the Management of COVID-19 Protocol Therapy-Induced QT Prolongation: A Literature Review. Scientia Pharmaceutica, 88(4), 55. https://doi.org/10.3390/scipharm88040055