Development of Topical/Transdermal Self-Emulsifying Drug Delivery Systems, Not as Simple as Expected



Abstract
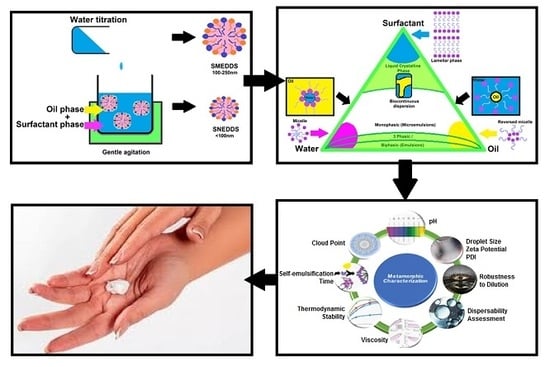
1. Introduction
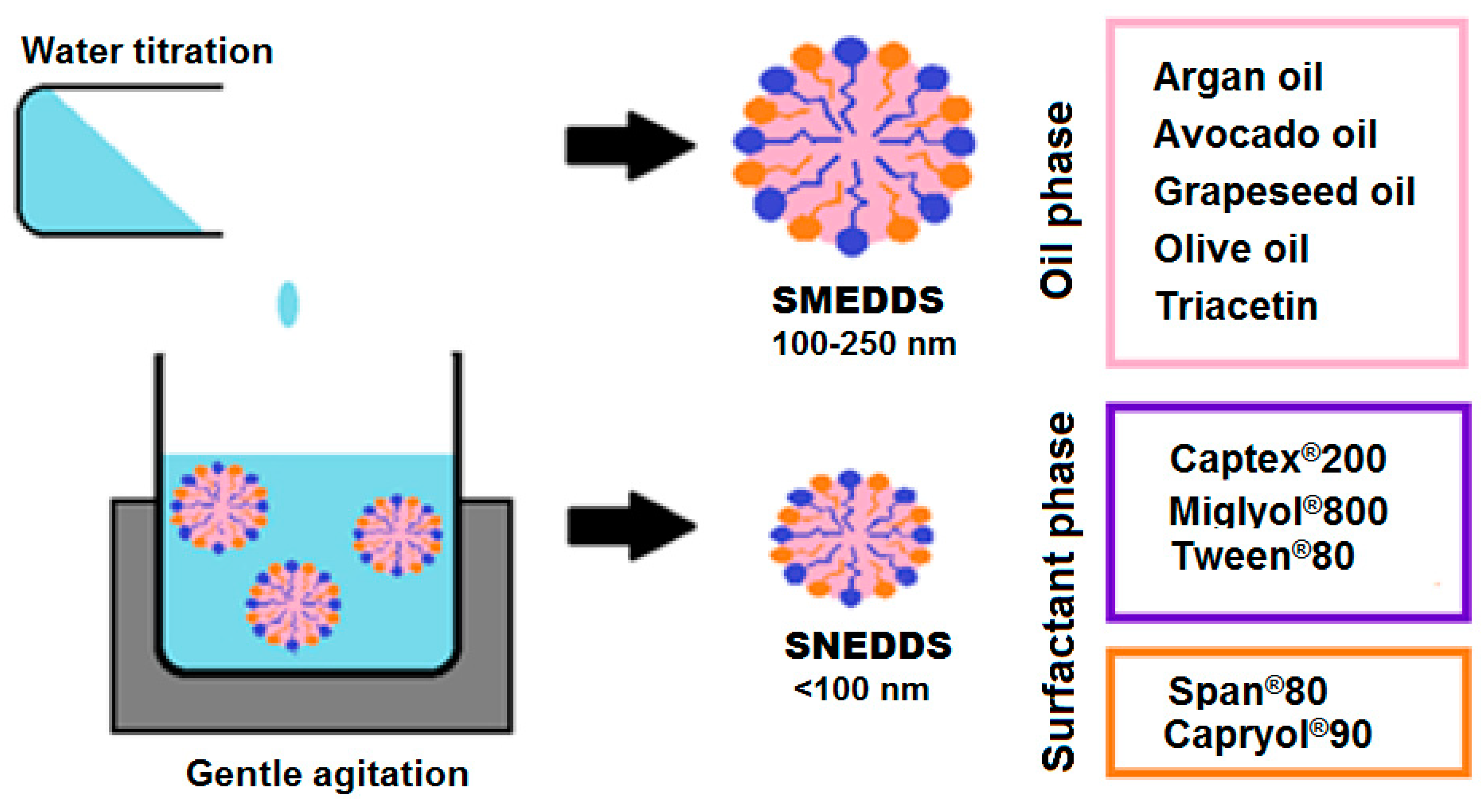
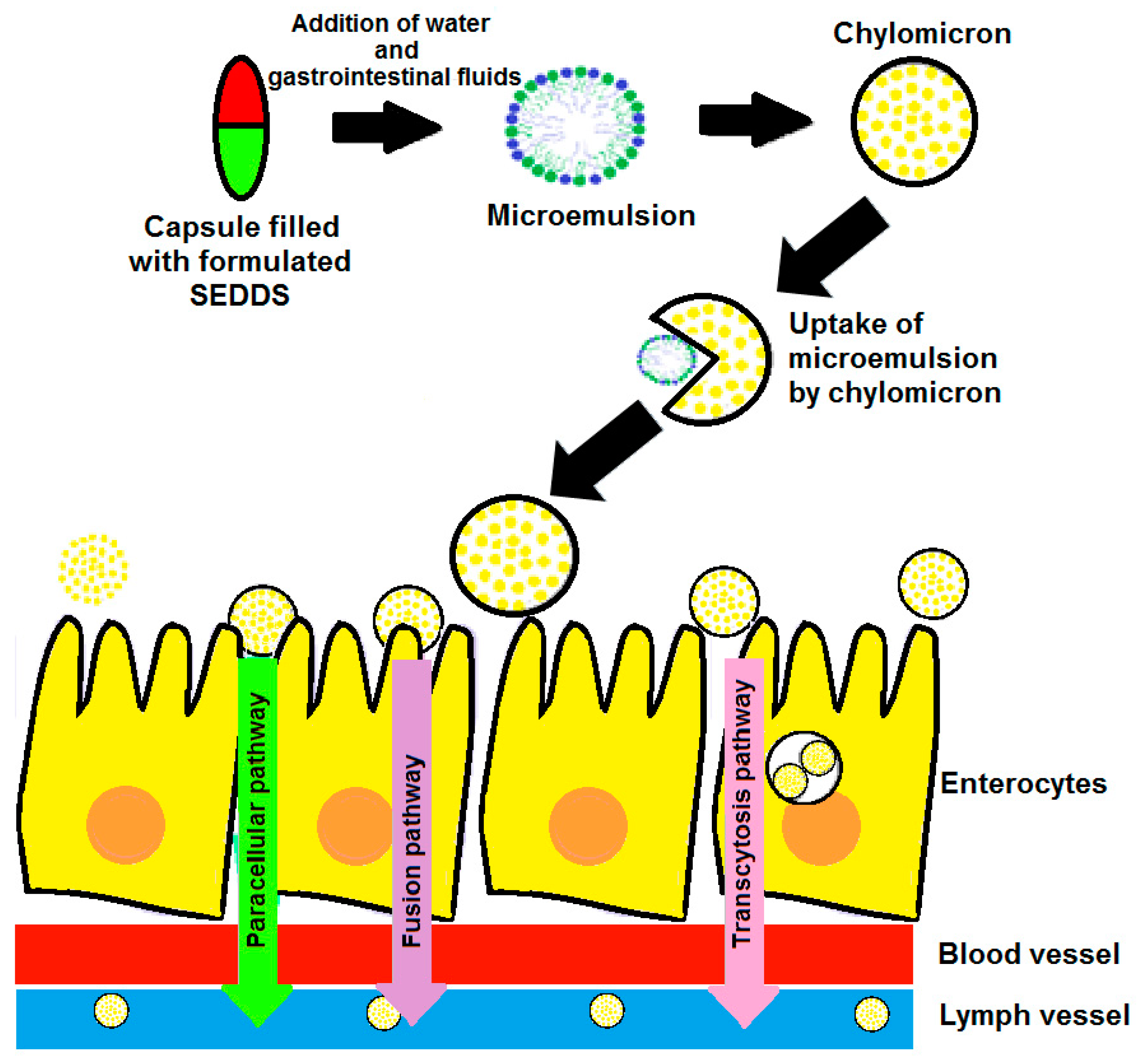
2. Mechanism of Spontaneous Emulsification
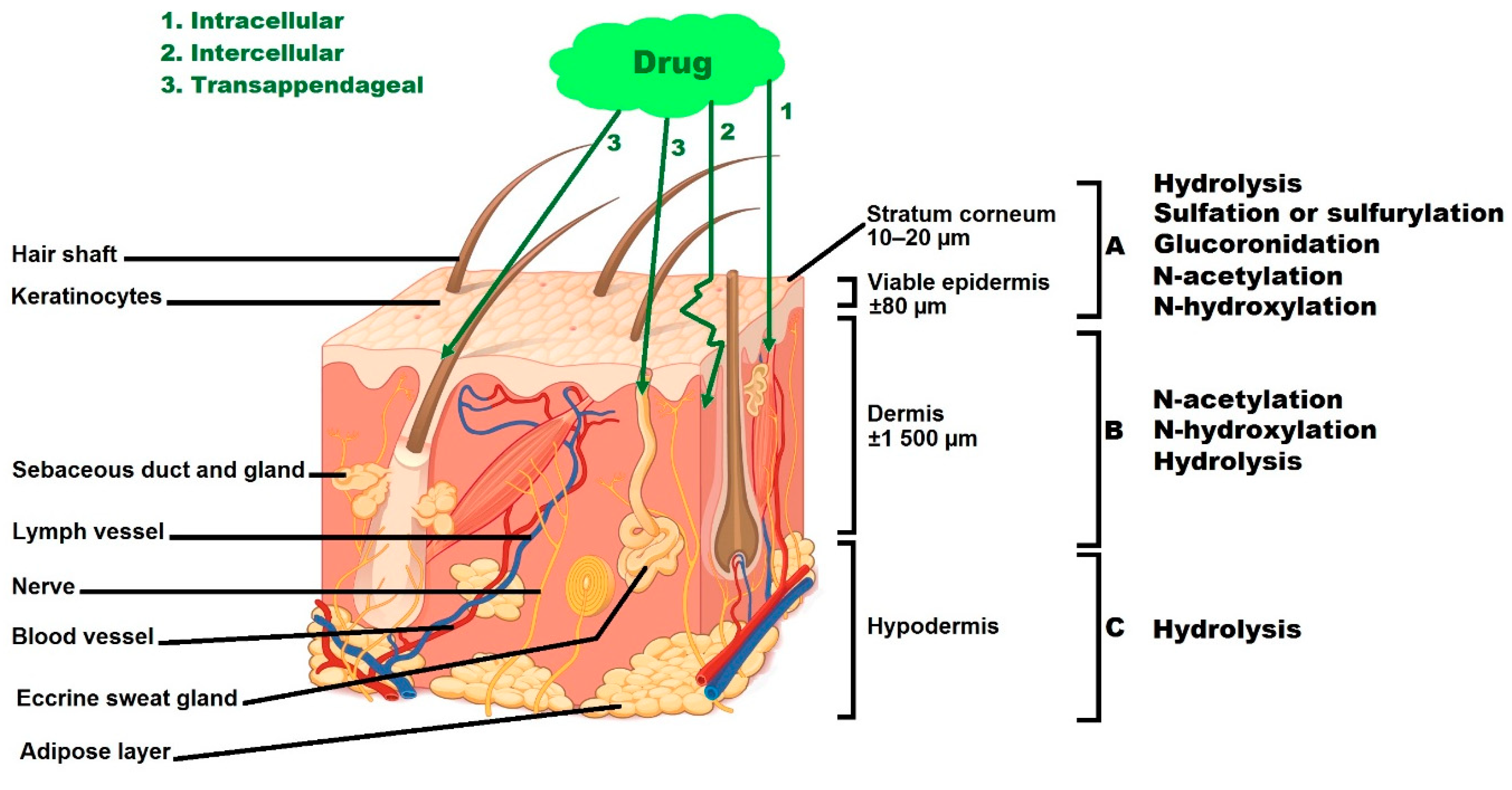
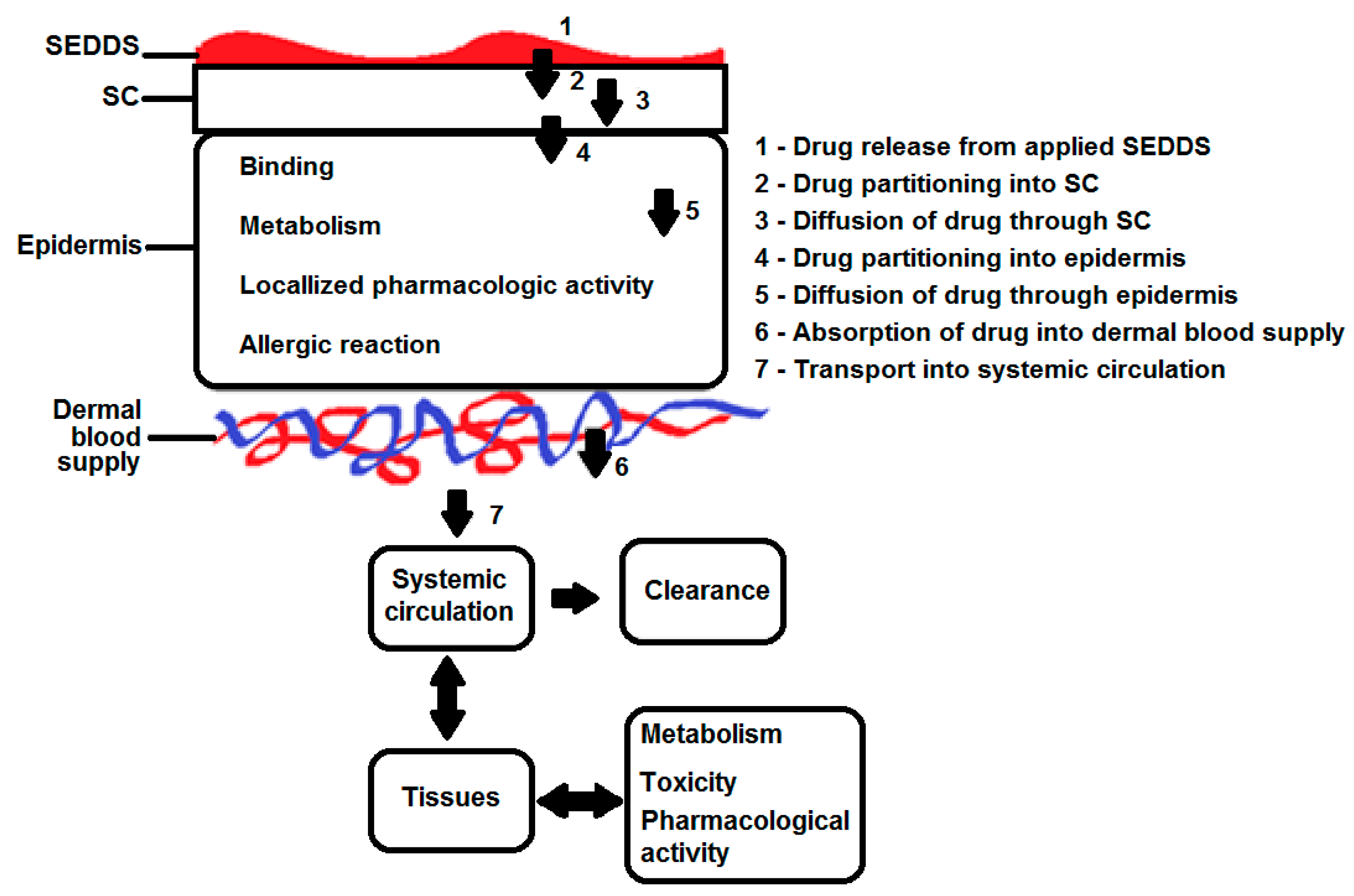
3. Skin, a Multi-Layered Organ
4. Excipients Fulfilling Different Roles Depending on the Route of Delivery
4.1. Active Compounds Incorporated in SEDDSs
4.2. The Oil Phase
4.3. Surfactants
4.4. Water
5. Compatibility of Topical/Transdermal SEDDS Excipients
6. Biocompatibility of Excipients Utilised to Establish Spontaneous Self-Emulsification
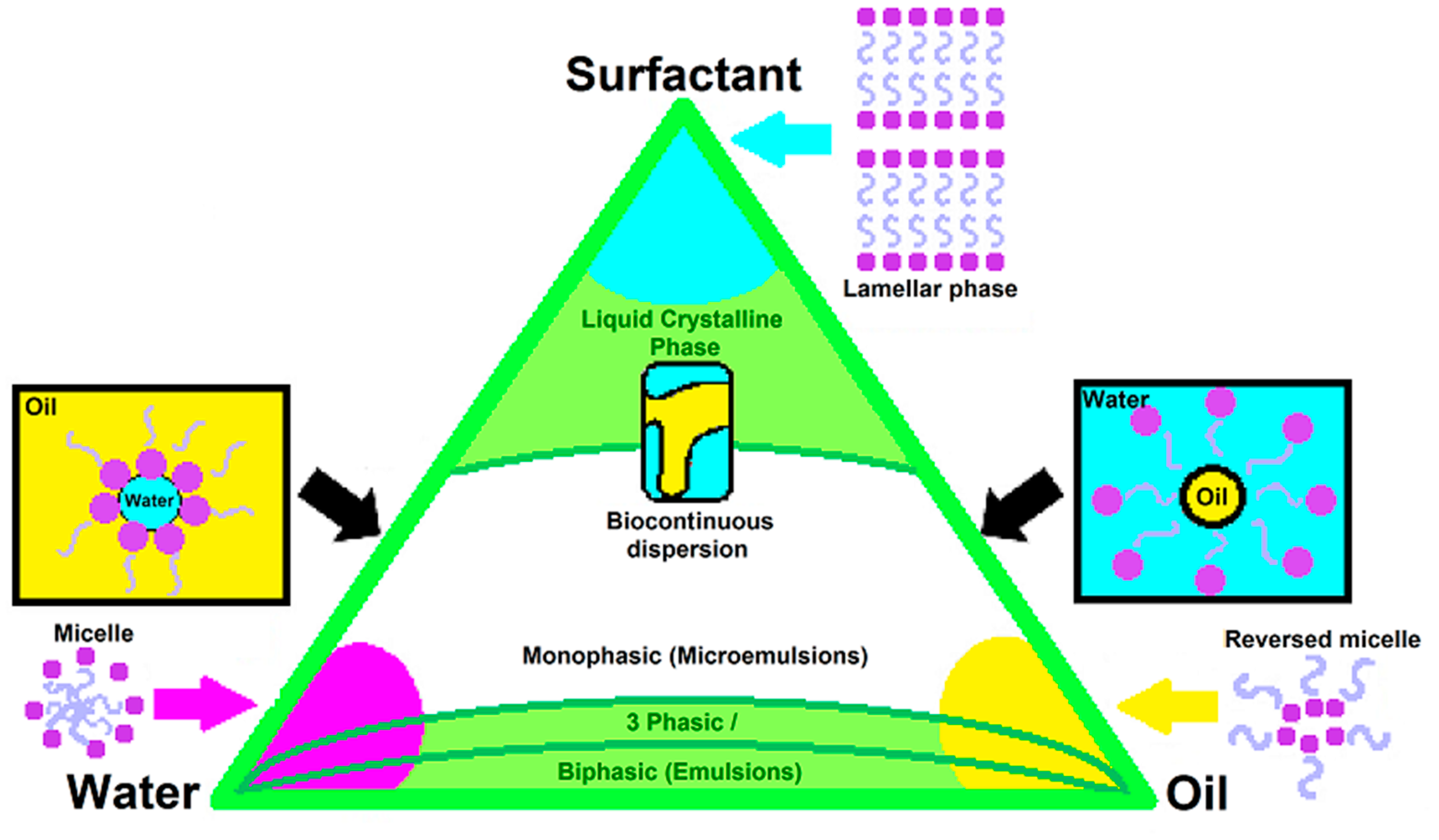
7. Pseudo-Ternary Phase Diagrams, Formerly Utilised Diagrams with Novel Potential
8. Metamorphic Characterisation of Topical/Transdermal SEDDSs Versus Orally Delivered SEDDSs
8.1. Evaluation of Droplet Sizes, Zeta Potential, and Polydispersity Index
8.2. Robustness to Dilution
8.3. Dispersibility Assessment
8.4. Self-Emulsification Time
8.5. Viscosity
8.6. Cloud Point Assessment
8.7. Thermodynamic Stability Studies
8.8. pH Measurement
9. Conclusions
Conflicts of Interest
References
- Pouton, C.W. Formulation of self-emulsifying drug delivery systems. Adv. Drug Deliv. Rev. 1997, 25, 47–58. [Google Scholar] [CrossRef]
- Mandić, J.; Pobirk, A.Z.; Vrečer, F.; Gašperlin, M. Overview of solidification techniques for self-emulsifying drug delivery systems from industrial perspective. Int. J. Pharm. 2017, 533, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Kazi, M.; Al-Swairi, M.; Ahmad, A.; Raish, M.; Alanazi, F.K.; Badran, M.M.; Ali Khan, A.; Alanazi, A.M.; Delwar Hussain, M. Evaluation of Self-Nanoemulsifying Drug Delivery Systems (SNEDDS) for Poorly Water-Soluble Talinolol: Preparation, in vitro and in vivo Assessment. Front. Pharmacol. 2019, 10, 459. [Google Scholar] [CrossRef] [PubMed]
- Kamal, M.M.; Nazzal, S. Development of a new class of sulforaphane-enabled self-emulsifying drug delivery systems (SFN-SEDDS) by high throughput screening: A case study with curcumin. Int. J. Pharm. 2018, 539, 147–156. [Google Scholar] [CrossRef]
- Ujhely, Z.; Vecsernyés, M.; Fehér, P.; Kósa, D.; Arany, P.; Nemes, D.; Sinka, D.; Vasvári, G.; Fenyvesi, F.; Váradi, J.; et al. Physico-chemical characterization of self-emulsifying drug delivery systems. Drug Discov. Today Technol. 2018, 27, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Dokania, S.; Joshi, A.K. Self-microemulsifying drug delivery system (SMEDDS)—Challenges and road ahead. Drug Deliv. 2015, 22, 675–690. [Google Scholar] [CrossRef]
- Kale, A.A.; Patravale, V.B. Design and evaluation of self-emulsifying drug delivery systems (SEDDS) of nimodipine. AAPS PharmSciTech 2008, 9, 191–196. [Google Scholar] [CrossRef]
- Fatouros, D.G.; Karpf, D.M.; Nielsen, F.S.; Mullertz, A. Clinical studies with oral lipid based formulations of poorly soluble compounds. Ther. Clin. Risk Manag. 2007, 3, 591–604. [Google Scholar]
- Pouton, C.W. Lipid formulations for oral administration of drugs: Non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur. J. Pharm. Sci. 2000, 11 (Suppl. 2), S93–S98. [Google Scholar] [CrossRef]
- Pouton, W.C.; Porter, J.H.C. Formulation of lipid-based delivery systems for oral administration: Materials, methods and strategies. Adv. Drug Deliv. Rev. 2008, 60, 625–637. [Google Scholar] [CrossRef]
- Chatterjee, B.; Almurisi, S.H.; Dukhan, A.A.M.; Mandal, U.K.; Sengupta, P. Controversies with self-emulsifying drug delivery system from pharmacokinetic point of view. Drug Deliv. 2016, 23, 3639–3652. [Google Scholar] [CrossRef] [PubMed]
- Syukri, Y.; Martien, R.; Lukitaningsih, E.; Nugroho, A.E. Novel self-nano emulsifying drug delivery system (SNEDDS) of andrographolide isolated from Andrographis paniculata Nees: Characterization, in-vitro and in-vivo assessment. J. Drug Deliv. Sci. Technol. 2018, 47, 514–520. [Google Scholar] [CrossRef]
- Xue, X.; Cao, M.; Qian, Y.; Chen, G. Preparation and optimization of rivaroxaban by self-nanoemulsifying drug delivery system (SNEDDS) for enhanced oral bioavailability and no food effect. AAPS PharmSciTech 2018, 19, 1847–1859. [Google Scholar] [CrossRef] [PubMed]
- Pouton, C.W. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef]
- Čerpnjak, K.; Zvonar, A.; Gašperlin, M.; Vrečer, F. Lipid-based systems as a promising approach for enhancing the bioavailability of poorly water-soluble drugs. Acta Pharm. 2013, 63, 427–445. [Google Scholar] [CrossRef]
- Porter, C.J.H.; Pouton, C.W.; Cuine, J.F.; Charman, W.N. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv. Drug Deliv. Rev. 2008, 60, 673–691. [Google Scholar] [CrossRef]
- Rahman, M.A.; Hussain, A.; Hussain, M.S.; Mirza, M.A.; Iqbal, Z. Role of excipients in successful development of self-emulsifying/microemulsifying drug delivery system (SEDDS/SMEDDS). Drug Dev. Ind. Pharm. 2013, 39, 1–19. [Google Scholar] [CrossRef]
- Prajapat, M.D.; Patel, N.J.; Bariya, A.; Patel, S.S.; Butani, S.B. Formulation and evaluation of self-emulsifying drug delivery system for nimodipine, a BCS class II drug. J. Drug Deliv. Sci. Technol. 2017, 39, 59–68. [Google Scholar] [CrossRef]
- Leichner, C.; Baus, R.A.; Jelkmann, M.; Plautz, M.; Barthelmes, J.; Dünnhaupt, S.; Bernkop-Schnürch, A. In vitro evaluation of a self-emulsifying drug delivery system (SEDDS) for nasal administration of dimenhydrinate. Drug Deliv. Transl. Res. 2019, 14, 945–955. [Google Scholar] [CrossRef]
- Kauss, T.; Gaubert, A.; Tabaran, L.; Tonelli, G.; Phoeung, T.; Langlois, M.H.; White, N.; Cartwright, A.; Gomes, M.; Gaudin, K. Development of rectal self-emulsifying suspension of a moisture-labile water-soluble drug. Int. J. Pharm. 2018, 30, 283–291. [Google Scholar] [CrossRef]
- Rohrer, J.; Zupančič, O.; Hetényi, G.; Kurpiers, M.; Bernkop-Schnürch, A. Design and evaluation of SEDDS exhibiting high emulsifying properties. J. Drug Deliv. Sci. Technol. 2018, 44, 366–372. [Google Scholar] [CrossRef]
- Köllner, S.; Nardin, I.; Markt, R.; Griesser, J.; Prüfert, F.; Bernkop-Schnürch, A. Self-emulsifying drug delivery systems: Design of a novel vaginal delivery system for curcumin. Eur. J. Pharm. Biopharm. 2017, 115, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Pattewar, S.; Patil, D.; Sharma, S. Fabrication and characterization of self-microemulsifying mouth dissolving film for effective delivery of piroxicam. Indian J. Pharm. Sci. 2019, 81, 503–513. [Google Scholar] [CrossRef]
- ElKasabgy, N.A. Ocular supersaturated self-nanoemulsifying drug delivery systems (S-SNEDDS) to enhance econazole nitrate bioavailability. Int. J. Pharm. 2014, 460, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Yi, T.; Liu, Y. A new self-microemulsifying mouth dissolving film to improve the oral bioavailability of poorly water soluble drugs. Drug Dev. Ind. Pharm. 2013, 39, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Lupo, N.; Tkadlečková, V.N.; Jelkmann, M.; Laffleur, F.; Hetényi, G.; Kubová, K.; Bernkop-Schnürch, A. Self-emulsifying drug delivery systems: In vivo evaluation of their potential for oral vaccination. Acta Biomater. 2019, 94, 425–434. [Google Scholar] [CrossRef]
- Shaker, D.S.; Ishak, R.A.; Ghoneim, A.; Elhuoni, M.A. Nanoemulsion: A review on mechanisms for the transdermal delivery of hydrophobic and hydrophilic drugs. Sci. Pharm. 2019, 87, 17. [Google Scholar] [CrossRef]
- Bellefroid, C.; Lechanteur, A.; Evrard, B.; Piel, G. Lipid gene nanocarriers for the treatment of skin diseases: Current state-of-the-art. Eur. J. Pharm. Biopharm. 2019, 137, 95–111. [Google Scholar] [CrossRef]
- Nardin, I.; Köllner, S. Successful development of oral SEDDS: Screening of excipients from the industrial point of view. Adv. Drug Deliv. Rev. 2019, 142, 128–140. [Google Scholar] [CrossRef]
- Costagliola, C.; Semeraro, F.; Zeppa, L.; Bufalo, G.; Cardone, M.; Romano, M.; Ambrosone, L. Some physicochemical remarks on spontaneous emulsification of vitreal tamponades. BioMed Res. Int. 2014, 2014, 243056. [Google Scholar] [CrossRef]
- Silver, B.R.; Holub, K.; Mareček, V. Spontaneous emulsification at surfactantless liquid/liquid interfaces. J. Electroanal. Chem. 2017, 805, 91–97. [Google Scholar] [CrossRef]
- Solans, C.; Morales, D.; Homs, M. Spontaneous emulsification. Curr. Opin. Colloid Interface Sci. 2016, 22, 88–93. [Google Scholar] [CrossRef]
- Rani, S.; Rana, R.; Saraogi, G.K.; Kumar, V.; Gupta, U. Self-emulsifying oral lipid drug delivery systems: Advances and challenges. AAPS PharmSciTech 2019, 20, 129. [Google Scholar] [CrossRef] [PubMed]
- Gonҫalves, A.; Nikmaram, N.; Roohinejad, S.; Estevinho, B.N.; Rocha, F.; Greiner, R.; McClements, D.J. Production, properties and applications of solid self-emulsifying delivery systems (S-EDDS) in the food and pharmaceutical industries. Colloids Surf. 2018, 538, 108–126. [Google Scholar] [CrossRef]
- Feingold, K.R.; Schmuth, M.; Elias, P.M. The regulation of permeability barrier homeostasis. J. Investig. Dermatol. 2007, 127, 1574–1576. [Google Scholar] [CrossRef]
- Li, J.; Xu, W.; Liang, Y.; Wang, H. The application of skin metabolomics in the context of transdermal drug delivery. Pharmacol. Rep. 2017, 69, 252–259. [Google Scholar] [CrossRef]
- Oesch, F.; Fabian, E.; Oesch-Bartlomowicz, B.; Werner, C.; Landsiedel, R. Drug-metabolizing enzymes in the skin of man, rat, and pig. Drug Metab. Rev. 2017, 39, 659–698. [Google Scholar] [CrossRef]
- Manevski, N.; Swart, P.; Balavenkatraman, K.K.; Bertschi, B.; Camenisch, G.; Kretz, O.; Schiller, H.; Walles, M.; Ling, B.; Wettstein, R.; et al. Phase II metabolism in human skin: Skin explants show full coverage for glucuronidation, sulfation, N-acetylation, catechol methylation, and glutathione conjugation. Drug Metab. Dispos. 2015, 43, 126–139. [Google Scholar] [CrossRef]
- Svensson, C.K. Biotransformation of drugs in human skin. Drug Metab. Dispos. 2007, 37, 247–253. [Google Scholar] [CrossRef]
- Mojumdar, E.H.; Pham, Q.D.; Topgaard, D.; Sparr, E. Skin hydration: Interplay between molecular dynamics, structure and water uptake in the stratum corneum. Sci. Rep. 2017, 7, 1–3. [Google Scholar] [CrossRef]
- Das, A.; Ahmed, A.B. Natural permeation enhancer for transdermal drug delivery system and permeation evaluation: A review. Asian J. Pharm. Clin. Res. 2017, 10, 5–9. [Google Scholar] [CrossRef]
- N’Da, D.D. Prodrug strategies for enhancing the percutaneous absorption of drugs. Molecules 2014, 19, 20780–20807. [Google Scholar] [CrossRef] [PubMed]
- Burger, C.; Gerber, M.; Du Preez, J.L.; Du Plessis, J. Optimised transdermal delivery of pravastatin. Int. J. Pharm. 2015, 496, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Jepps, O.G.; Dancik, Y.; Anissimov, Y.G.; Roberts, M.S. Modelling the human skin barrier—Towards a better understanding of dermal absorption. Adv. Drug Deliv. Rev. 2013, 65, 152–168. [Google Scholar] [CrossRef]
- King, M.J.; Michel, D.; Foldvari, M. Evidence for lymphatic transport of insulin by topically applied biphasic vesicles. J. Pharm. Pharmacol. 2003, 55, 1339–1344. [Google Scholar] [CrossRef]
- Gurram, A.K.; Deshpande, P.B.; Kar, S.S.; Nayak, U.Y.; Udupa, N.; Reddy, M.S. Role of components in the formation of self-microemulsifying drug delivery systems. Indian J. Pharm. Sci. 2015, 77, 249–257. [Google Scholar] [CrossRef]
- Tang, J.; Sun, J.; Cui, F.; Zhang, T.; Liu, X.; He, Z. Self-emulsifying drug delivery systems for improving oral absorption of ginkgo biloba extracts. Drug Deliv. 2008, 15, 477–484. [Google Scholar] [CrossRef]
- Schmid-Wendtner, M.H.; Korting, H.C. The pH of the skin surface and its impact on the barrier function. Skin Pharmacol. Physiol. 2006, 19, 296. [Google Scholar] [CrossRef]
- Nair, A.; Jacob, S.; Al-Dhubiab, B.; Attimarad, M.; Harsha, S. Basic considerations in the dermatokinetics of topical formulations. Braz. J. Pharm. Sci. 2013, 49, 423–434. [Google Scholar] [CrossRef]
- Holm, R. Bridging the gaps between academic research and industrial product developments of lipid-based formulations. Adv. Drug Deliv. Rev. 2019, 142, 118–127. [Google Scholar] [CrossRef]
- Naik, A.; Kalia, Y.N.; Guy, R.H. Transdermal drug delivery: Overcoming the skin’s barrier function. PSTT 2000, 3, 318–326. [Google Scholar] [CrossRef]
- Wen, H.; Jung, H.; Li, X. Drug delivery approaches in addressing clinical pharmacology-related issues: Opportunities and challenges. AAPS J. 2015, 17, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Parhi, R.; Swain, S. Transdermal evaporation drug delivery system: Concept to commercial products. Adv. Pharm. Bull. 2018, 8, 535. [Google Scholar] [CrossRef]
- Hauss, D.J.; Fogal, S.E.; Ficorilli, J.V.; Price, C.A.; Roy, T.; Jayaraj, A.A.; Keirns, J.J. Lipid-based delivery systems for improving the bioavailability and lymphatic transport of a poorly water-soluble LTB4 inhibitor. J. Pharm. Sci. 1998, 87, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Shizuki, M.; Miyoshi, K.; Sakai, T.; Hidaka, H.; Takamura, H.; Matoba, T. Relationship between the molecular structures and emulsification properties of edible oils. Biosci. Biotechnol. Biochem. 1994, 58, 1258–1261. [Google Scholar] [CrossRef]
- Lane, M.E. Skin penetration enhancers. Int. J. Pharm. 2013, 447, 12–21. [Google Scholar] [CrossRef]
- Van Zyl, L.; Du Preez, J.; Gerber, M.; Du Plessis, J.; Viljoen, J. Essential fatty acids as transdermal penetration enhancers. J. Pharm. Sci. 2016, 105, 188–193. [Google Scholar] [CrossRef]
- Viljoen, J.M.; Cowley, A.; Du Preez, J.; Gerber, M.; Du Plessis, J. Penetration enhancing effects of selected natural oils utilized in topical dosage forms. Drug Dev. Ind. Pharm. 2015, 41, 2045–2054. [Google Scholar] [CrossRef]
- Lundborg, M.; Wennberg, C.L.; Narangifard, A.; Lindahl, E.; Norlén, L. Predicting drug permeability through skin using molecular dynamics simulation. JCR 2018, 283, 269–279. [Google Scholar] [CrossRef]
- Knothe, G.; Dunn, R.O. A comprehensive evaluation of the melting points of fatty acids and esters determined by differential scanning calorimetry. J. Am. Oil Chem. Soc. 2009, 86, 843–856. [Google Scholar] [CrossRef]
- Chi, S.-C.; Park, E.S.; Kim, H. Effect of penetration enhancers on flurbiprofen permeation through rat skin. Int. J. Pharm. 1995, 126, 267–274. [Google Scholar] [CrossRef]
- Aungst, B.J. Structure/effect studies of fatty acids isomers as skin penetration enhancers and skin irritants. Pharm. Res. 1989, 3, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.K.; Zhong, L.; Santiago, J. Anti-inflammatory and skin barrier repair effects of topical application of some plant oils. Int. J. Mol. 2017, 19, 70. [Google Scholar] [CrossRef] [PubMed]
- ElMasry, S.R.; Hathout, R.M.; Abdel-Halim, M.; Mansour, S. In Vitro transdermal delivery of sesamol using oleic acid chemically-modified gelatin nanoparticles as a potential breast cancer medication. J. Drug Deliv. Sci. Technol. 2018, 48, 30–39. [Google Scholar] [CrossRef]
- Hadgraft, J.; Lane, M.E. Advanced topical formulations (ATF). Int. J. Pharm. 2016, 514, 52–57. [Google Scholar] [CrossRef]
- Longhi, R. Omega Fatty Acids in Brain and Neurological Health, 2nd ed.; Academic Press: London, UK, 2019; pp. 457–477. [Google Scholar]
- Cicero, N.; Albergamo, A.; Salvo, A.; Bua, G.D.; Bartolomeo, G.; Mangano, V.; Rotondo, A.; Di Stefano, V.; Di Bella, G.; Dugo, G. Chemical characterization of a variety of cold-pressed gourmet oils available on the Brazilian market. Food Res. Int. 2018, 109, 517–525. [Google Scholar] [CrossRef]
- Rueda, A.; Seiquer, I.; Olalla, M.; Giménez, R.; Lara, L.; Cabrera-Vique, C. Characterization of fatty acid profile of argan oil and other edible vegetable oils by gas chromatography and discriminant analysis. J. Chem. 2014, 843908. [Google Scholar] [CrossRef]
- Hu, W.; Fitzgerald, M.; Topp, B.; Alam, M.; O’Hare, T.J. A review of biological functions, health benefits, and possible de novo biosynthetic pathway of palmitoleic acid in macadamia nuts. J. Funct. Foods 2019, 62, 103520. [Google Scholar] [CrossRef]
- Vaughn, A.R.; Clark, A.K.; Sivamani, R.K.; Shi, V.Y. Natural oils for skin-barrier repair: Ancient compounds now backed by modern science. Am. J. Clin. Dermatol. 2018, 19, 103–117. [Google Scholar] [CrossRef]
- Gore, E.; Picard, C.; Savary, G. Spreading behavior of cosmetic emulsions: Impact of the oil phase. Biotribology 2018, 16, 17–24. [Google Scholar] [CrossRef]
- Vaz, S.; Silva, R.; Amaral, M.H.; Martins, E.; Lobo, J.S.; Silva, A.C. Evaluation of the biocompatibility and skin hydration potential of vitamin E-loaded lipid nanosystems formulations: In vitro and human in vivo studies. Colloids Surf. B 2019, 179, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Mojeiko, G.; de Brito, M.; Salata, G.C.; Lopes, L.B. Combination of microneedles and microemulsions to increase celecoxib topical delivery for potential application in chemoprevention of breast cancer. Int. J. Pharm. 2019, 560, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Kiselmann, C.; Dobler, D.; Schmidts, T.; Eicher, A.C.; Möbs, C.; Pfützner, W.; Runkel, F. Development of a skin-friendly microemulsion for dermal allergen-specific immunotherapy. Int. J. Pharm. 2018, 550, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Effendy, I.; Maibach, H.I. Surfactants and experimental irritant contact dermatitis. Contact Dermat. 1995, 33, 217–225. [Google Scholar] [CrossRef]
- Ou, B.; Huang, D.; Hampsch-Woodill, M.; Flanagan, J.A.; Deemer, E.K. Analysis of antioxidant activities of common vegetables employing oxygen radical absorbance capacity (ORAC) and ferric reducing antioxidant power (FRAP) assays: A comparative study. J. Agric. Food Chem. 2002, 5, 223–228. [Google Scholar] [CrossRef]
- Margetts, L.; Sawyer, R. Transdermal drug delivery: Principles and opioid therapy. BJA CEPD 2007, 7, 171–176. [Google Scholar] [CrossRef]
- Ibrahim, T.M.; Abdallah, M.H.; El-Megrab, N.A.; El-Nahas, H.M. Upgrading of dissolution and anti-hypertensive effect of Carvedilol via two combined approaches: Self-emulsification and liquisolid techniques. Drug Dev. Ind. Pharm. 2018, 44, 873–885. [Google Scholar] [CrossRef]
- Haque, T.; Talukder, M.M.U. Chemical enhancer: A simplistic way to modulate barrier function of the stratum corneum. Adv. Pharm. Bull. 2018, 8, 169–179. [Google Scholar] [CrossRef]
- Chintalapudi, R.; Murthy, T.E.G.K.; Lakshmi, K.R.; Manohar, G.G. Formulation, optimization, and evaluation of self-emulsifying drug delivery systems of nevirapine. Int. J. Pharm. Investig. 2015, 5, 205–213. [Google Scholar] [CrossRef]
- Viljoen, J.M.; Botes, D.; Steenekamp, J.H. Formulation and evaluation of selected transmucosal dosage forms containing a double fixed-dose of acyclovir and ketoconazole. Eur. J. Pharm. Sci. 2018, 111, 503–513. [Google Scholar] [CrossRef]
- Braissant, O.; Wirz, D.; Göpfert, B.; Daniels, A.U. Biomedical use of isothermal microcalorimeters. Sensors 2010, 10, 9369–9383. [Google Scholar] [CrossRef] [PubMed]
- Simončič, Z.; Zupančič, P.; Roškar, R.; Gartner, A.; Kogej, K.; Kmetec, V. Use of microcalorimetry in determination of stability of enalapril maleate and enalapril maleate tablet formulations. Int. J. Pharm. 2007, 342, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Ermita, D.A.C.; Valente, F.L.; Carlo-Reis, E.C.; Araújo, F.R.; Ribeiro, I.M.; Cintra, C.C.V.; Borges, A.P.B. Characterization and in vivo biocompatibility analysis of synthetic hydroxyapatite compounds associated with magnetite nanoparticles for a drug delivery system in osteomyelitis treatment. Res. Mater. 2020, 5, 100063. [Google Scholar] [CrossRef]
- Naahidi, S.; Jafari, M.; Logan, M.; Wang, Y.; Yuan, Y.; Bae, H.; Dixon, B.; Chen, P. Biocompatibility of hydrogel-based scaffolds for tissue engineering applications. Biotechnol. Adv. 2017, 35, 530–544. [Google Scholar] [CrossRef]
- Naahidi, S.; Jafari, M.; Edalat, F.; Raymond, K.; Khademhosseini, A.; Chen, P. Biocompatibility of engineered nanoparticles for drug delivery. J. Control. Release 2013, 166, 182–194. [Google Scholar] [CrossRef]
- Kohane, D.S.; Langer, R. Biocompatibility and drug delivery systems. Chem. Sci. 2010, 1, 441–446. [Google Scholar] [CrossRef]
- Atarés, L.; Chiralt, A. Essential oils as additives in biodegradable films and coatings for active food packaging. Trends Food Sci. Technol. 2016, 48, 51–62. [Google Scholar] [CrossRef]
- McWilliam, V.; Peters, R.; Tang, M.L.; Dharmage, S.; Ponsonby, A.L.; Gurrin, L.; Perrett, K.; Koplin, J.; Allen, K.J.; Dwyer, T.; et al. Patterns of tree nut sensitization and allergy in the first 6 years of life in a population-based cohort. J. Allergy Clin. Immunol. 2019, 143, 644–650. [Google Scholar] [CrossRef]
- Ménard, N.; Tsapis, N.; Poirier, C.; Arnauld, T.; Moine, L.; Lefoulon, F.; Péan, J.; Fattal, E. Drug solubilization and in vitro toxicity evaluation of lipoamino acid surfactants. Int. J. Pharm. 2012, 423, 312–320. [Google Scholar] [CrossRef]
- Dimitrijevic, D.; Shaw, A.J.; Florence, A.T. Effects of some non-ionic surfactants on transepithelial permeability in Caco-2 cells. J. Pharm. Pharmacol. 2000, 52, 157–162. [Google Scholar] [CrossRef]
- Sigward, E.; Mignet, N.; Rat, P.; Dutot, M.; Muhamed, S.; Guigner, J.; Scherman, D.; Brossard, D.; Crauste-Manciet, S. Formulation and cytotoxicity evaluation of new self-emulsifying multiple W/O/W nanoemulsions. Int. J. Nanomed. 2013, 8, 611–625. [Google Scholar] [CrossRef][Green Version]
- Nemes, D.; Ujhelyi, Z.; Arany, P.; Pető, A.; Feher, P.; Varadi, J.; Fenyvesi, F.; Vecsernyes, M.; Bacskay, I. Biocompatibility investigation of different pharmaceutical excipients used in liquid dosage forms. Pharmazie 2018, 73, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Festing, S.; Wilkinson, R. The ethics of animal research. Talking Point on the use of animals in scientific research. EMBO Rep. 2007, 8, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Ashmawy, A.M.; Ayoub, I.M.; Eldahshan, O.A. Chemical composition, cytotoxicity and molecular profiling of Cordia africana Lam. on human breast cancer cell line. Nat. Prod. Res. 2020. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Tamura, M.; Tahara, Y.; Kamiya, N.; Goto, M. Ionic liquid-in-oil microemulsion as a potential carrier of sparingly soluble drug: Characterization and cytotoxicity evaluation. Int. J. Pharm. 2010, 400, 243–250. [Google Scholar] [CrossRef]
- Jalil, A.; Asim, M.H.; Akkus, Z.B.; Schoenthaler, M.; Matuszczak, B.; Bernkop-Schnürch, A. Self-emulsifying drug delivery systems comprising chlorhexidine and alkyl-EDTA: A novel approach for augmented antimicrobial activity. J. Mol. Liq. 2019, 295, 111649. [Google Scholar] [CrossRef]
- Ke, Z.; Hou, X.; Jia, X.-B. Design and optimisation of self-nanoemulsifying drug delivery systems for improved bioavailability of cyclovirobuxine D. Drug Des. Dev. Ther. 2016, 10, 2049–2060. [Google Scholar] [CrossRef]
- Dash, P.N.; Habibuddin, M.; Humaira, T.; Ramesh, D. Design, optimisation and elevation of glipizide solid self-nanoemulsifying drug delivery for enhanced solubility and dissolution. SPJ 2015, 23, 528–540. [Google Scholar] [CrossRef]
- Hegde, R.R.; Verma, A.; Ghosh, A. Microemulsion: New insights into the ocular drug delivery. ISRN Pharm. 2013. [Google Scholar] [CrossRef]
- Lépori, C.M.O.; Correa, N.M.; Silber, J.J.; Chávez, F.V.; Falcone, R.D. Interfacial properties modulated by the water confinement in reverse micelles created by the ionic liquid-like surfactant bmim-AOT. Soft Matter 2019, 15, 947–955. [Google Scholar] [CrossRef]
- Silva, O.F.; Fernández, M.A.; Silber, J.J.; De Rossi, R.H.; Correa, N.M. Inhibited phenol ionisation in reverse micelles: Confinement effect at the nanometre scale. ChemPhysChem 2012, 3, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Barauch, B.; Roden, J.M.; Sedgnick, M.; Correa, N.M.; Levinger, N.E.; Crans, D.S. When is water not water? Exploring water confined in large reverse micelles using a highly charged inorganic molecular probe. J. Am. Chem. Soc. 2006, 128, 12758–12765. [Google Scholar] [CrossRef]
- Kogan, A.; Garti, N. Microemulsions as transdermal drug delivery vehicles. Adv. Colloid Interface Sci. 2006, 16, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Messih, H.A.; Ishak, R.A.; Geneidi, A.S.; Mansour, S. Tailoring novel soft nano-vesicles ‘Flexosomes’ for enhanced transdermal drug delivery: Optimization, characterization and comprehensive ex vivo–in vivo evaluation. Int. J. Pharm. 2019, 560, 101–115. [Google Scholar] [CrossRef] [PubMed]
- El Zaafarany, G.M.; Awad, G.A.; Holayel, S.M.; Mortada, N.D. Role of edge activators and surface charge in developing ultradeformable vesicles with enhanced skin delivery. Int. J. Pharm. 2010, 397, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Clegg, P.S.; Li, T.; Rumble, K.A.; Tavacoli, J.W. Bijels formed by direct mixing. Soft Matter 2017, 13, 4824–4829. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.G.; White, K.A.J.; Delgado-Charro, M.B. A mechanistic approach to modelling the formation of a drug reservoir in the skin. Math. Biosci. 2016, 281, 36–45. [Google Scholar] [CrossRef][Green Version]
- Ita, K.B. Transdermal drug delivery: Progress and challenges. J. Drug Deliv. Sci. Technol. 2014, 24, 245–250. [Google Scholar] [CrossRef]
- Czajkowska-Kośnik, A.; Szekalska, M.; Winnicka, K. Nanostructured lipid carriers: A potential use for skin drug delivery systems. Pharmacol. Rep. 2019, 71, 156–166. [Google Scholar] [CrossRef]
- Hardiningtyas, S.D.; Nagao, S.; Yamamoto, E.; Shirakigawa, N.; Wakabayashi, R.; Goto, M.; Ijima, H.; Kamiya, N. A non-sized gel in-in-oil suspension for transcutaneous protein delivery. Int. J. Pharm. 2019, 567, 118495. [Google Scholar] [CrossRef]
- Kaur, R.; Ajitha, M. Formulation of transdermal nanoemulsion gel drug delivery system of lovastatin and its in vivo characterization in glucocorticoid induced osteoporosis rat model. J. Drug Deliv. Sci. Technol. 2019, 52, 968–978. [Google Scholar] [CrossRef]
- Zhou, X.; Hao, Y.; Yuan, L.; Pradhan, S.; Shrestha, K.; Pradhan, O.; Liu, H.; Li, W. Nano-formulations for transdermal drug delivery: A review. CCL 2018, 29, 1713–1724. [Google Scholar] [CrossRef]
- Ali, H.H.; Hussein, A.A. Oral solid self-nanoemulsifying drug delivery systems of candesartan citexetil: Formulation, characterization and in vitro drug release studies. AAPS Open 2017, 3, 6. [Google Scholar] [CrossRef]
- Gupta, V.; Trivedi, P. Lipid Nanocarriers for Drug Targeting, 1st ed.; William Andrew Applied Science Publishers: Kidlington, UK, 2018; pp. 563–627. [Google Scholar]
- Ding, Z.; Jiang, Y.; Liu, X. Nanotechnology-Based Targeted Drug Delivery System for Brain Tumors, 1st ed.; Academic Press: London, UK, 2018; pp. 327–358. [Google Scholar]
- Sharma, S.; Shukla, P.; Misra, A.; Mishra, P.R. Colloid and Interface Science in Pharmaceutical Research and Development, 1st ed.; Elsevier: Kidlington, UK, 2014; pp. 149–172. [Google Scholar]
- Gumustas, M.; Sengel-Turk, C.T.; Gumustas, A.; Ozkan, S.A.; Uslu, B. Multifunctional Systems for Combined Delivery, Biosensing and Diagnostics, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 67–108. [Google Scholar]
- Carter, P.; Narasimhan, B.; Wang, Q. Biocompatible nanoparticles and vesicular systems in transdermal drug delivery for various skin diseases. Int. J. Pharm. 2019, 555, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Umerska, A.; Cassisa, V.; Matougui, N.; Joly-Guillou, M.L.; Eveillard, M.; Saulnier, P. Antibacterial action of lipid nanocapsules containing fatty acids or monoglycerides as co-surfactants. Eur. J. Pharm. Biopharm. 2016, 108, 100–110. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M. Impact of particle size and polydispersity index on the clinical applications of lipidic nanocarrier systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Czajkowska-Kośnik, A.; Szekalska, M.; Amelian, A.; Szymańska, E.; Winnicka, K. Development and evaluation of liquid and solid self-emulsifying drug delivery systems for atorvastatin. Molecules 2015, 20, 21010–21022. [Google Scholar] [CrossRef]
- Sheshala, R.; Anuar, N.K.; Samah, N.H.A.; Wong, T.W. In vitro drug dissolution/permeation testing of nanocarriers for skin application: A comprehensive review. AAPS PharmSciTech 2019, 20, 164. [Google Scholar] [CrossRef]
- Nasr, A.; Gardouh, A.; Ghorab, M. Novel solid self-nanoemulsifying drug delivery system (S-SNEDDS) for oral delivery of olmesartan medoxomil: Design, formulation, pharmacokinetic and bioavailability evaluation. Pharmaceutics 2016, 8, 20. [Google Scholar] [CrossRef]
- Kathe, K.; Kathpalia, H. Film forming systems for topical and transdermal drug delivery. Asian J. Pharm. Sci. 2017, 12, 487–497. [Google Scholar] [CrossRef]
- Chaudhari, K.S.; Akamanchi, K.G. Novel bicephalous heterolipid based self-microemulsifying drug delivery system for solubility and bioavailability enhancement of efavirenz. Int. J. Pharm. 2019, 560, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Perazzo, A.; Preziosi, V.; Guido, S. Phase inversion emulsification: Current understanding and applications. Adv. Colloid Interface Sci. 2015, 222, 581–599. [Google Scholar] [CrossRef] [PubMed]
- Eid, A.M.; El-Enshasy, H.A.; Aziz, R.; Elmarzugi, N.A. The preparation and evaluation of self-nanoemulsifying systems containing Swietenia oil and an examination of its anti-inflammatory effects. Int. J. Nanomed. 2014, 9, 4685. [Google Scholar] [CrossRef][Green Version]
- Nowak, E.; Kovalchuk, N.M.; Che, Z.; Simmons, M.J. Effect of surfactant concentration and viscosity of outer phase during the coalescence of a surfactant-laden drop with a surfactant-free drop. Colloids Surf. A Physicochem. Eng. Asp. 2016, 505, 124–131. [Google Scholar] [CrossRef]
- Akilu, S.; Baheta, A.T.; Kadirgama, K.; Padmanabhan, E.; Sharma, K.V. Viscosity, electrical and thermal conductivities of ethylene and propylene glycol-based β-SiC nanofluids. J. Mol. Liq. 2019, 284, 780–792. [Google Scholar] [CrossRef]
- Mendonsa, N.S.; Pradhan, A.; Sharma, P.; Prado, R.M.; Murthy, S.N.; Kundu, S.; Repka, M.A. A quality by design approach to develop topical creams via hot-melt extrusion technology. Eur. J. Pharm. Sci. 2019, 136, 104948. [Google Scholar] [CrossRef]
- Picchi, D.; Poesio, P.; Ullmann, A.; Brauner, N. Characteristics of stratified flows of Newtonian/non-Newtonian shear-thinning fluids. Int. J. Multiph. Flow 2017, 97, 109–133. [Google Scholar] [CrossRef]
- Kondratiev, A.; Ilyushechkin, A. Flow behaviour of crystallising coal ash slags: Shear viscosity, non-newtonian flow and temperature of critical viscosity. Fuel 2018, 224, 783–800. [Google Scholar] [CrossRef]
- Shanthilal, J.; Bhattacharya, S. Characterisation of time-independent and time-dependent rheological behaviour simultaneously by multiple loop experimentation. J. Food Sci. Technol. 2016, 53, 4106–4109. [Google Scholar] [CrossRef]
- Nguyen, H.X.; Puri, A.; Banga, A.K. Methods to simulate rubbing of topical formulation for in vitro skin permeation studies. Int. J. Pharm. 2017, 519, 22–33. [Google Scholar] [CrossRef]
- Mewis, J.; Wagner, N.J. Thixotropy. Colloid Interface Sci. 2009, 147, 214–227. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, E. Strategies to control and inhibit the flocculation of protein-stabilized oil-in-water emulsions. Food Hydrocoll. 2019, 96, 209–223. [Google Scholar] [CrossRef]
- Rognon, P.G.; Einav, I.; Gay, C. Flowing resistance and dilatancy of dense suspensions: Lubrication and repulsion. J. Fluid Mech. 2011, 689, 75–96. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, M.; Xuan, S.; Jiang, W.; Cao, S.; Gong, X. Shear dependent electrical property of conductive shear thickening fluid. Mater. Des. 2017, 121, 92–100. [Google Scholar] [CrossRef]
- Ma, P.; Zeng, Q.; Tai, K.; He, X.; Yao, Y.; Hong, X.; Yuan, F. Development of stable curcumin nanoemulsions: Effects of emulsifier type and surfactant-to-oil ratios. J. Food Sci. Technol. 2018, 55, 3485–3497. [Google Scholar] [CrossRef] [PubMed]
- Zupančič, O.; Rohrer, J.; Thanh Lam, H.; Grießinger, J.A.; Bernkop-Schnürch, A. Development and in vitro characterization of self-emulsifying drug delivery system (SEDDS) for oral opioid peptide delivery. Drug Dev. Ind. Pharm. 2017, 43, 1694–1702. [Google Scholar] [CrossRef] [PubMed]
- AboulFotouh, K.; Allam, A.A.; El-Badry, M.; El-Sayed, A.M. Role of self-emulsifying drug delivery systems in optimizing the oral delivery of hydrophilic macromolecules and reducing interindividual variability. Colloids Surf. B 2018, 167, 82–92. [Google Scholar] [CrossRef]
- Heylings, J.R.; Davies, D.J.; Burton, R. Dermal absorption of testosterone in human and pig skin in vitro. Toxicol. Vitro 2018, 48, 71–77. [Google Scholar] [CrossRef]
- Liu, H.; Han, G.; Zhang, H.; Liu, Q.; Kong, B. Improving the physical and oxidative stability of emulsions based on the interfacial electrostatic effects between porcine bone protein hydrolysates and porcine bone protein hydrolysate-rutin conjugates. Food Hydrocoll. 2019, 94, 418–427. [Google Scholar] [CrossRef]
- Salimi, E.; Le-Vinh, B.; Zahir-Jouzdani, F.; Matuszczak, B.; Ghaee, A.; Bernkop-Schnürch, A. Self-emulsifying drug delivery systems changing their zeta-potential via a flip-flop mechanism. Int. J. Pharm. 2018, 550, 200–206. [Google Scholar] [CrossRef]
- Leonaviciute, G.; Adamovic, N.T.; Lam, H.T.; Rohrer, J.; Partenhauser, A. Bernkop-Schnürch, A. Self-emulsifying drug delivery systems (SEDDS): Proof-of-concept how to make them mucoadhesive. Eur. J. Pharm. Biopharm. 2017, 112, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Valicherla, G.R.; Dave, K.M.; Syed, A.A.; Riyazuddin, M.; Gupta, A.P.; Singh, A.; Mitra, K.; Datta, D.; Gayen, J.R. Formulation optimization of Docetaxel loaded self-emulsifying drug delivery system to enhance bioavailability and anti-tumor activity. Sci. Rep. 2016, 6, 26895. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.; Conejos-Sanchez, I.; Griffin, B.T.; O’Driscoll, C.M.; Alonso, M.J. Lipid-based nanocarriers for oral peptide delivery. Adv. Drug. Deliv. Rev. 2016, 106, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.; Dudhe, R.; Sharma, P.K. Nanoemulsion: An advanced mode of drug delivery system. 3 Biotech 2015, 5, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, R.R.; Osmani, R.A.; Ghodake, P.P.; Shaikh, S.M.; Chavan, S.R. Nanoemulsion: A review on novel profusion in advanced drug delivery. IJPBR 2014, 2, 122–127. [Google Scholar] [CrossRef]
- Khadke, S.; Roces, C.B.; Cameron, A.; Devitt, A.; Perrie, Y. Formulation and manufacturing of lymphatic targeting liposomes using microfluidics. JCR 2019, 307, 211–220. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Grading | Description of Visual Observation |
|---|---|
| Grade A | Swift emulsification, presents a clear/bluish appearance (60 s). |
| Grade B | Rapid emulsion, with bluish appearance (60 s). |
| Grade C | Emulsion exhibits fine, milky appearance (120 s). |
| Grade D | Dull, greyish-white appearance with an additional oily layer at emulsion surface together with slow emulsification (>120 s). |
| Grade E | Poor or minimal emulsification noted with large oil droplets noticed on the surface. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Staden, D.; du Plessis, J.; Viljoen, J. Development of Topical/Transdermal Self-Emulsifying Drug Delivery Systems, Not as Simple as Expected. Sci. Pharm. 2020, 88, 17. https://doi.org/10.3390/scipharm88020017
van Staden D, du Plessis J, Viljoen J. Development of Topical/Transdermal Self-Emulsifying Drug Delivery Systems, Not as Simple as Expected. Scientia Pharmaceutica. 2020; 88(2):17. https://doi.org/10.3390/scipharm88020017
Chicago/Turabian Stylevan Staden, Daniélle, Jeanetta du Plessis, and Joe Viljoen. 2020. "Development of Topical/Transdermal Self-Emulsifying Drug Delivery Systems, Not as Simple as Expected" Scientia Pharmaceutica 88, no. 2: 17. https://doi.org/10.3390/scipharm88020017
APA Stylevan Staden, D., du Plessis, J., & Viljoen, J. (2020). Development of Topical/Transdermal Self-Emulsifying Drug Delivery Systems, Not as Simple as Expected. Scientia Pharmaceutica, 88(2), 17. https://doi.org/10.3390/scipharm88020017