Pharmacokinetic Enhancers (Boosters)—Escort for Drugs against Degrading Enzymes and Beyond



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
- inhibitors of hepatic drug-metabolizing enzymes such as CYP3A4
- inhibitors of drug-specific metabolizing enzymes
- inhibitors of bacterial β-lactamases as escort for β-lactam antibiotics
2. Antimicrobial Chemotherapy
2.1. Enhancers Targeting Bacterial Enzymes: β-Lactamase Inhibitors
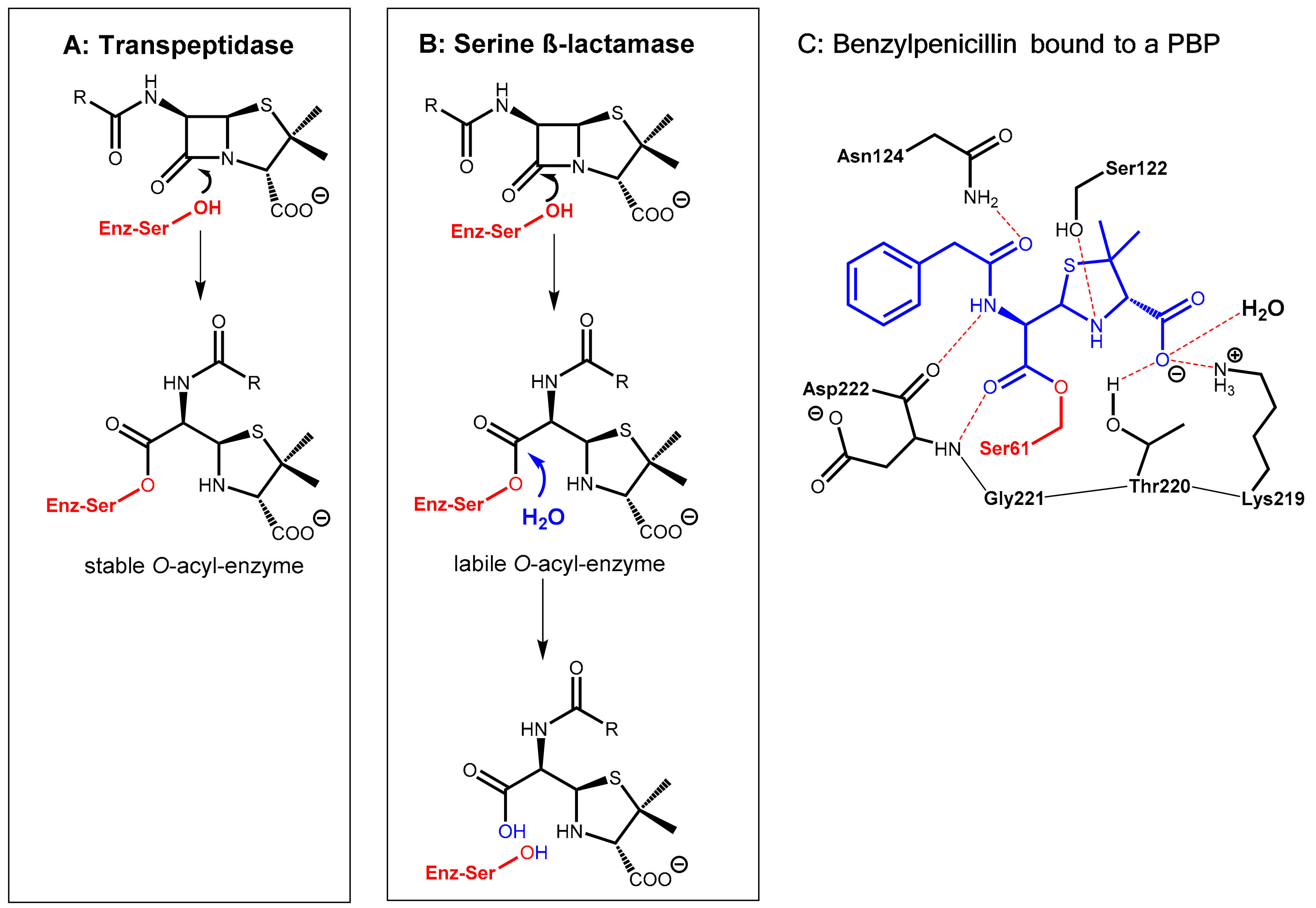
2.1.1. Background
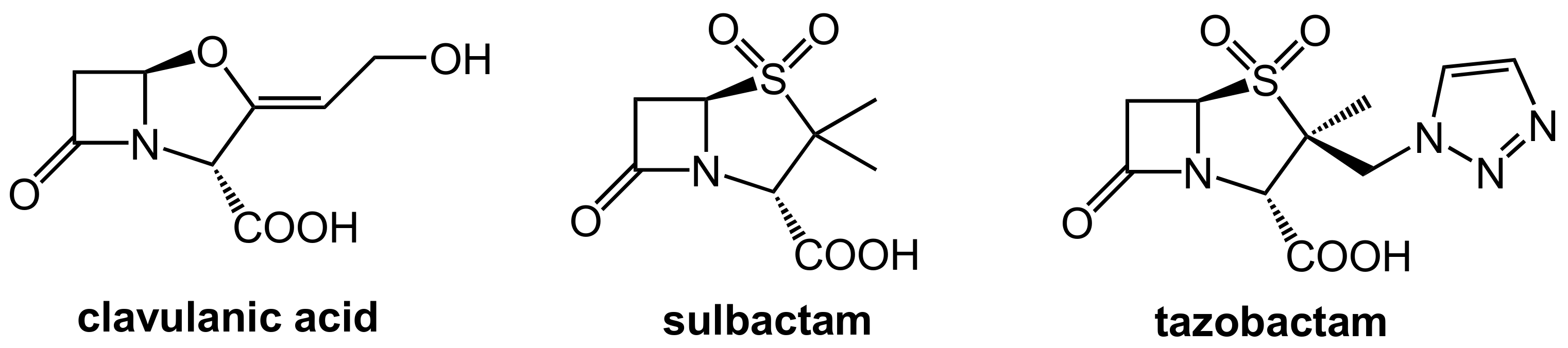
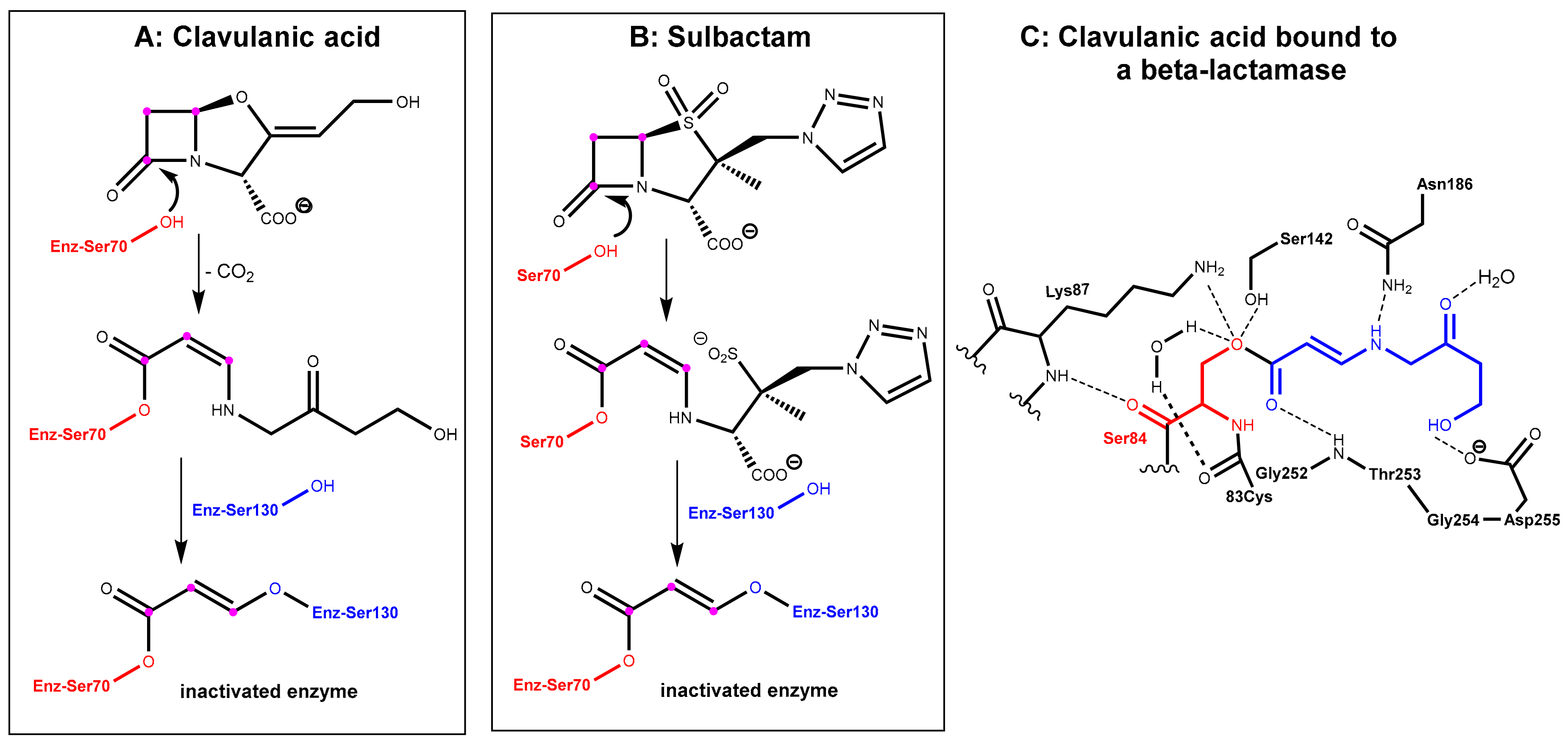
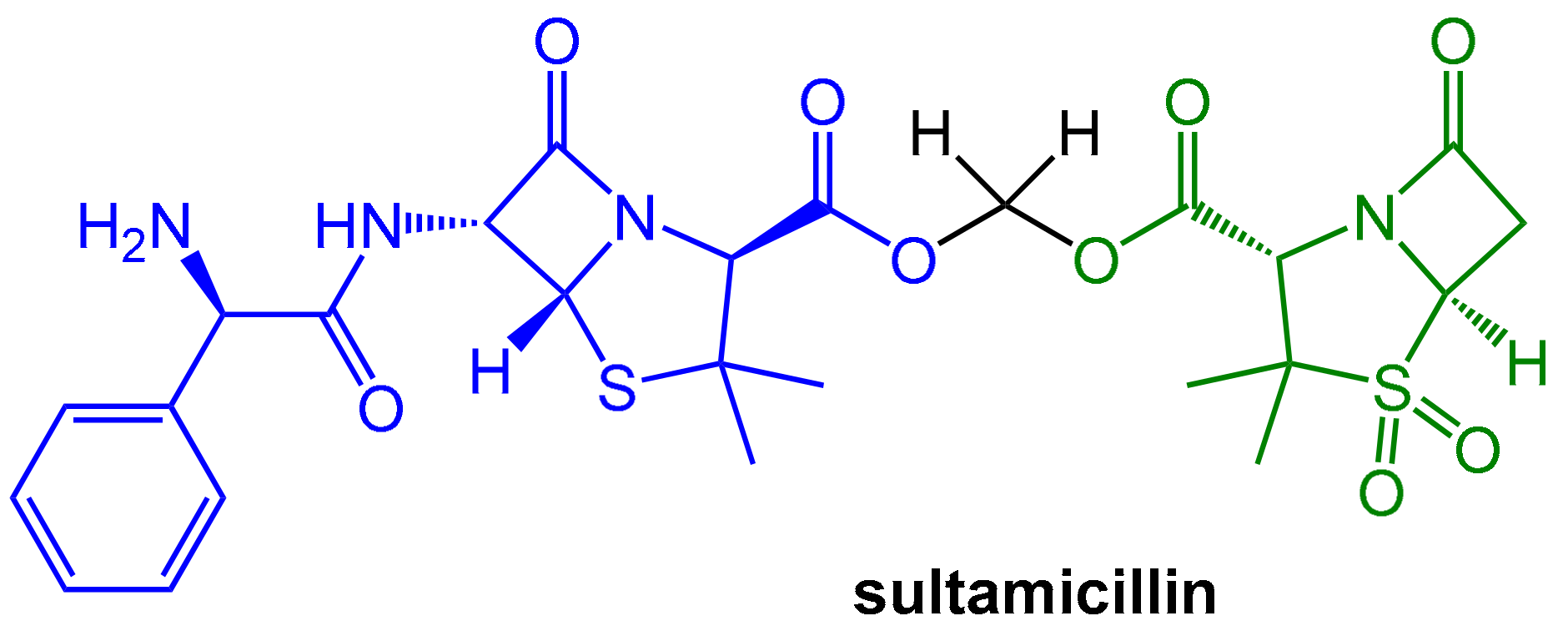
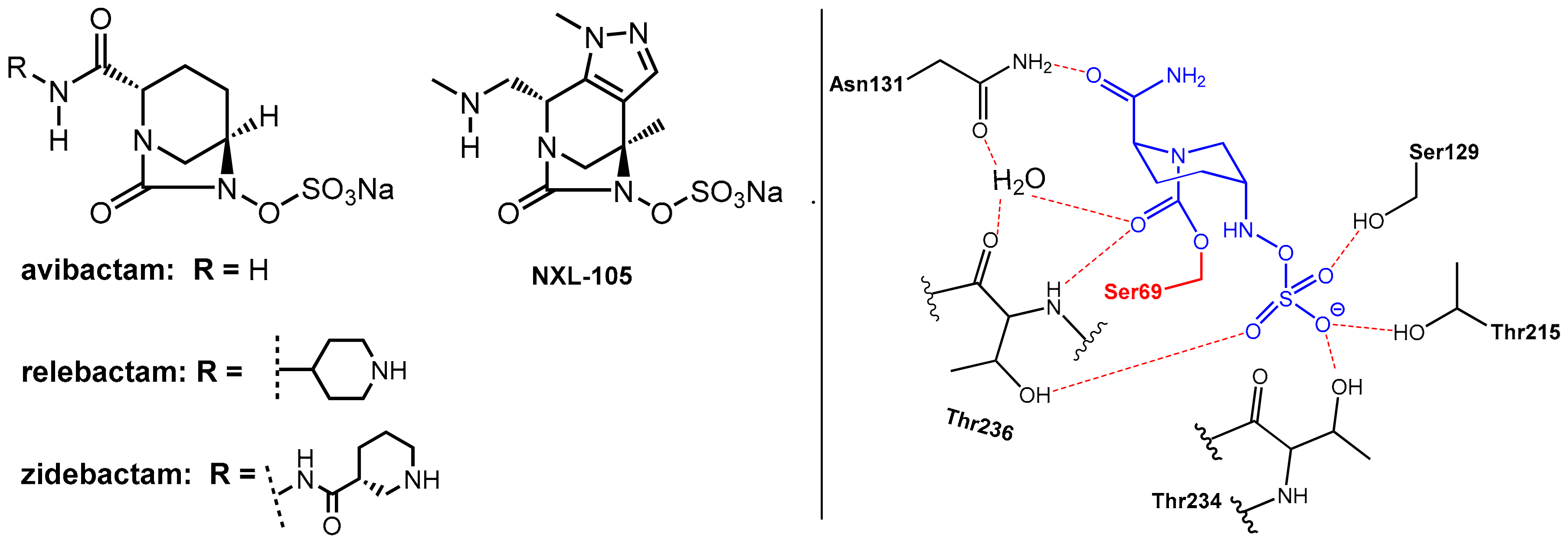
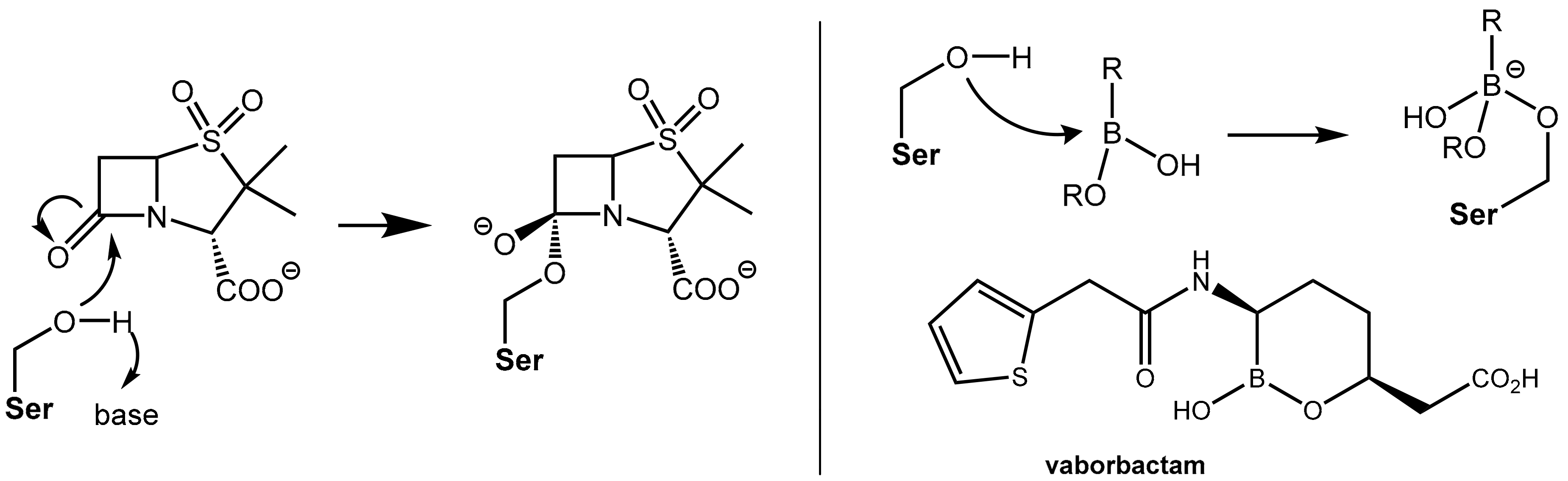
2.1.2. Irreversibly Binding β-Lactam-Type β-Lactamase Inhibitors
2.1.3. Reversibly Binding β-Lactamase Inhibitors
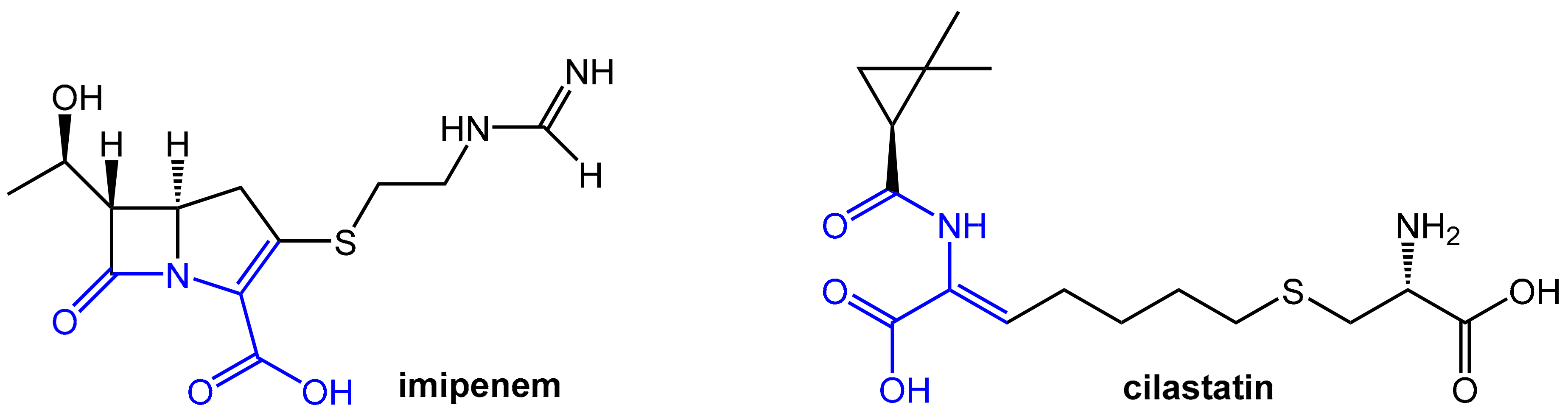
2.2. Enhancers Targeting Host Enzymes: Cilastatin
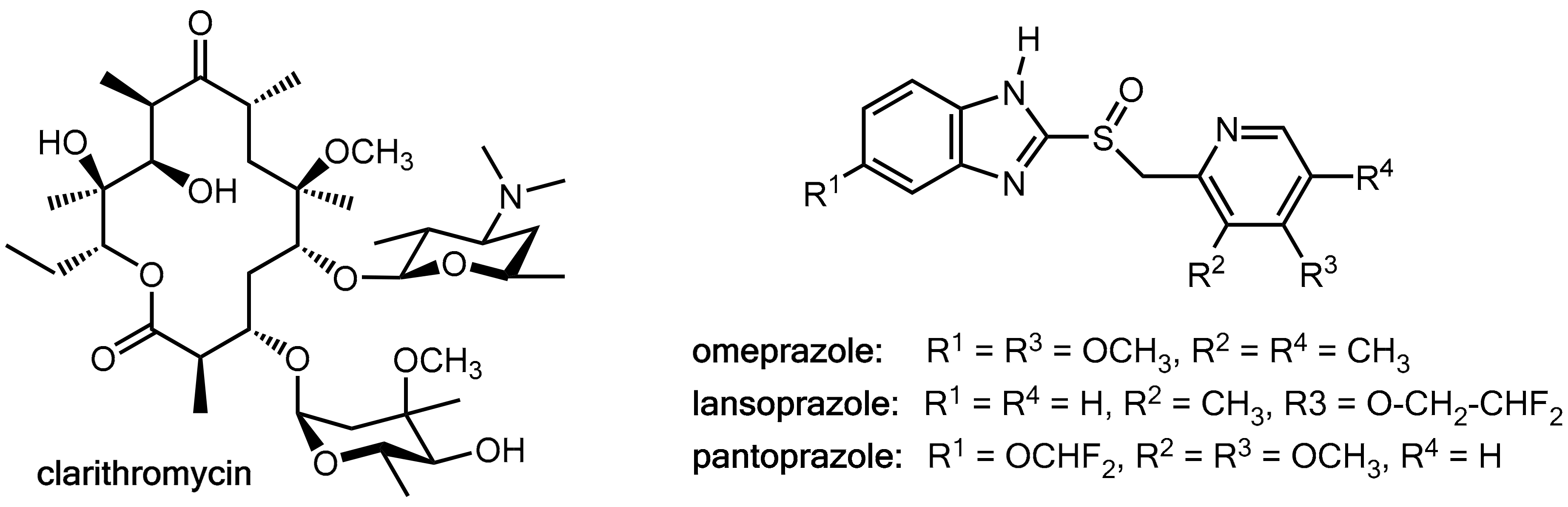
2.3. Fruitful Drug-Drug Interactions in Helicobacter pylori Eradication
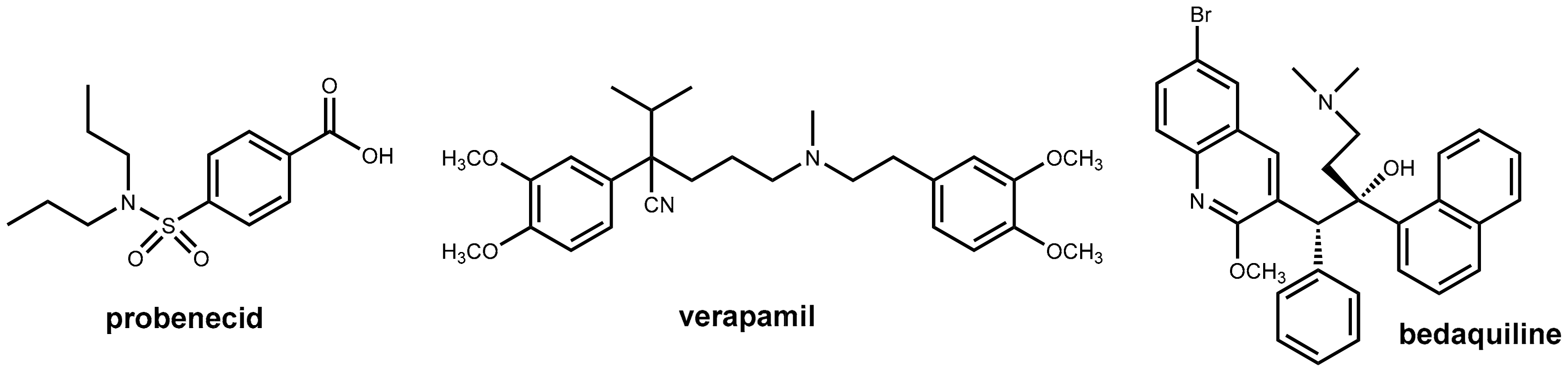
2.4. Enhancers for Antibiotics Beyond Enzyme Inhibition: Probenecid and Efflux Pump Inhibitors
2.4.1. Probenecid
2.4.2. Inhibitors of Bacterial Efflux Pumps (“Antibiotic Escort Molecules”, “Antibiotic Adjuvants”)
2.4.3. Pharmacokinetic Enhancement in Antitubercular Therapy (?): Bedaquiline Plus Verapamil
3. HIV Therapy: Inhibition of CYP3A4 for Boosting Antiretroviral Drugs
3.1. HIV Proteases
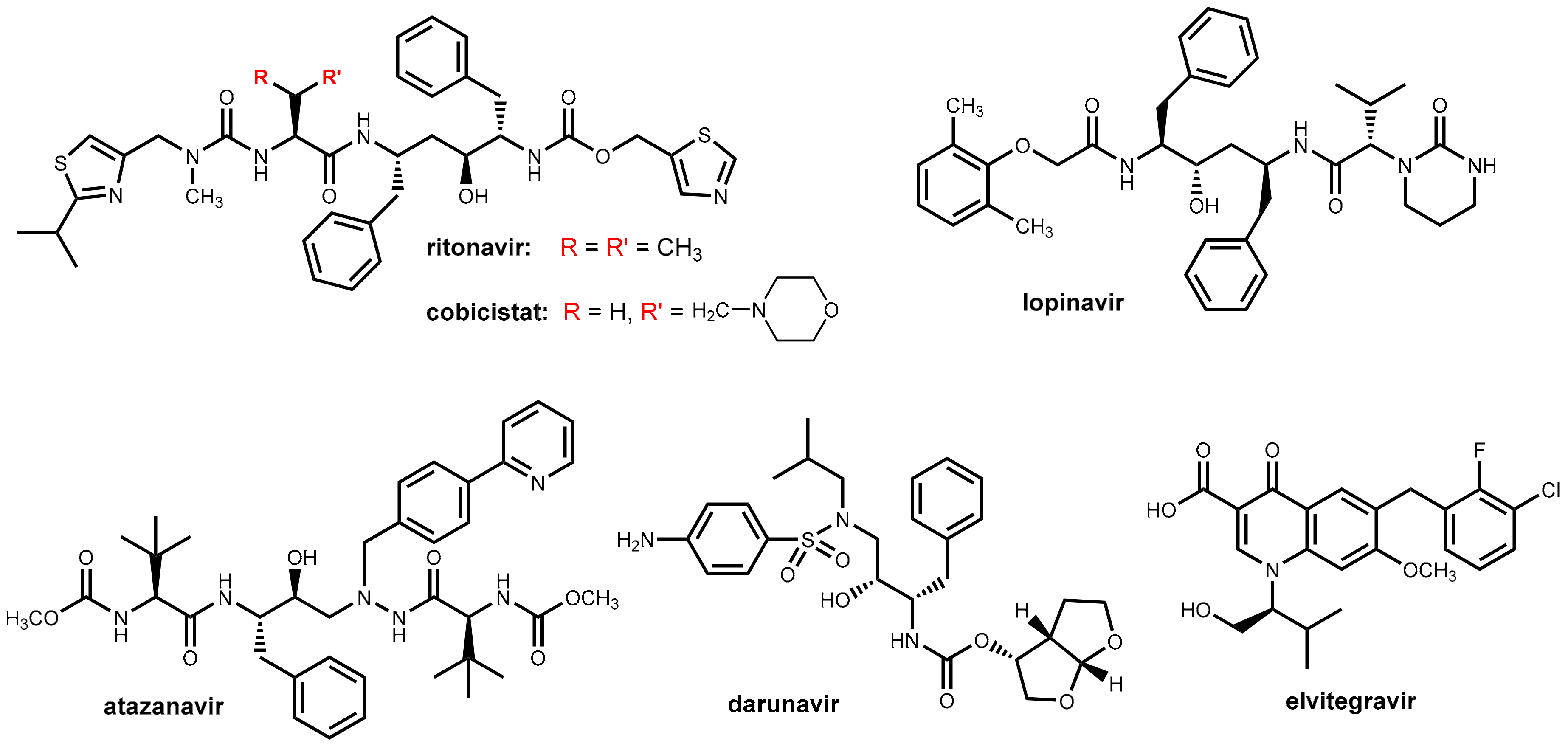
3.2. Ritonavir and Cobicistat
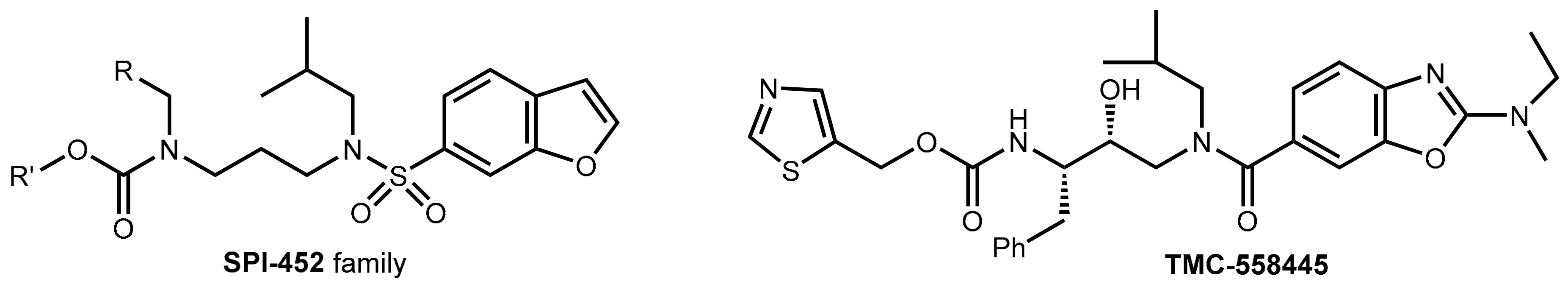
3.3. Development of New Boosters—A Long and Rocky Road
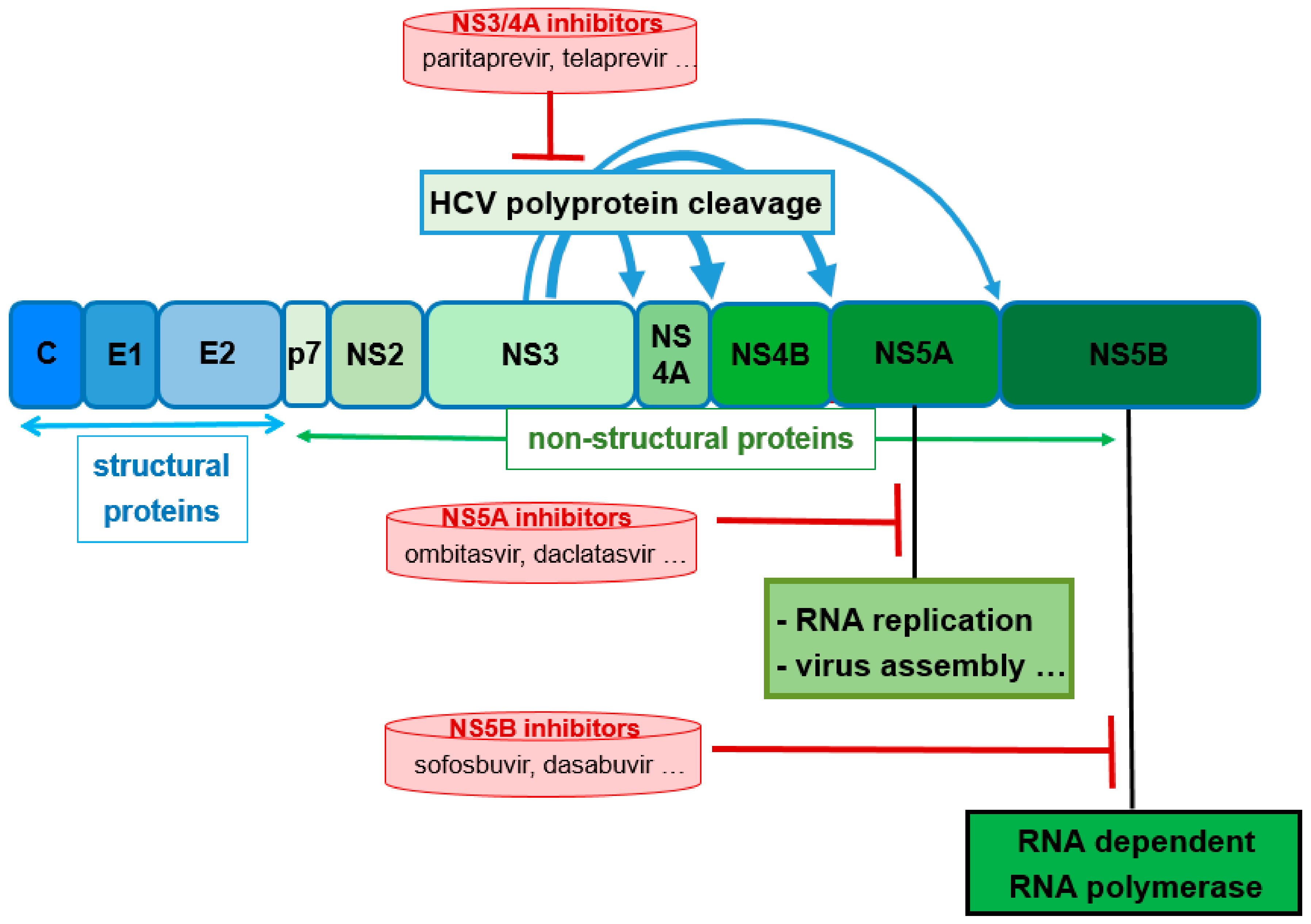
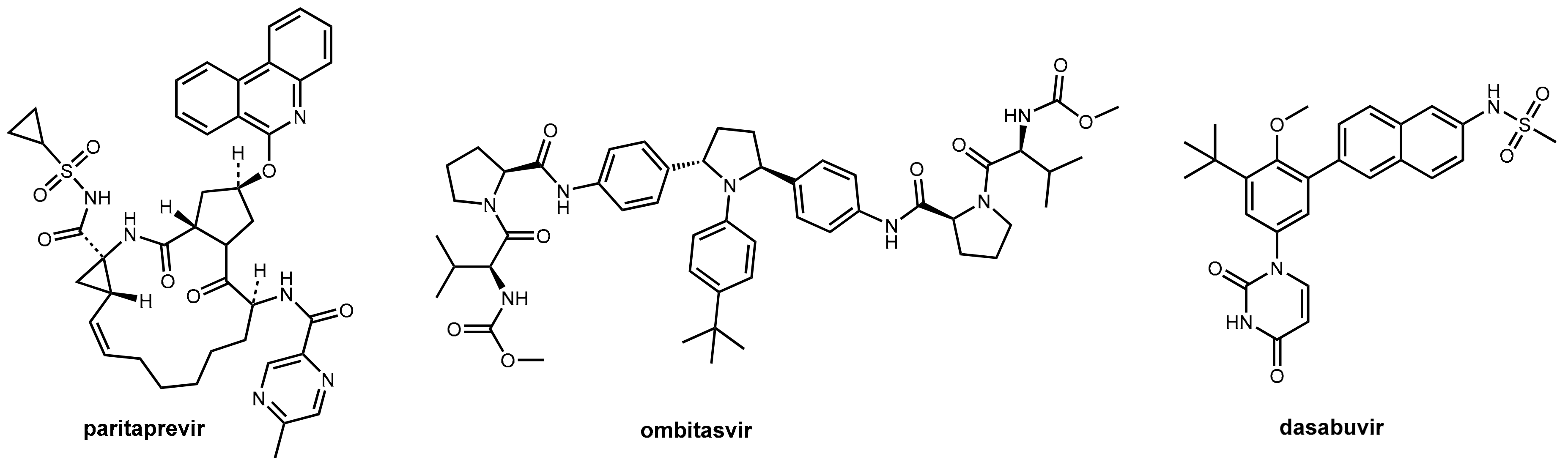
4. Ritonavir in Hepatitis C Therapy
5. Probenecid Reloaded for Antiviral Therapy
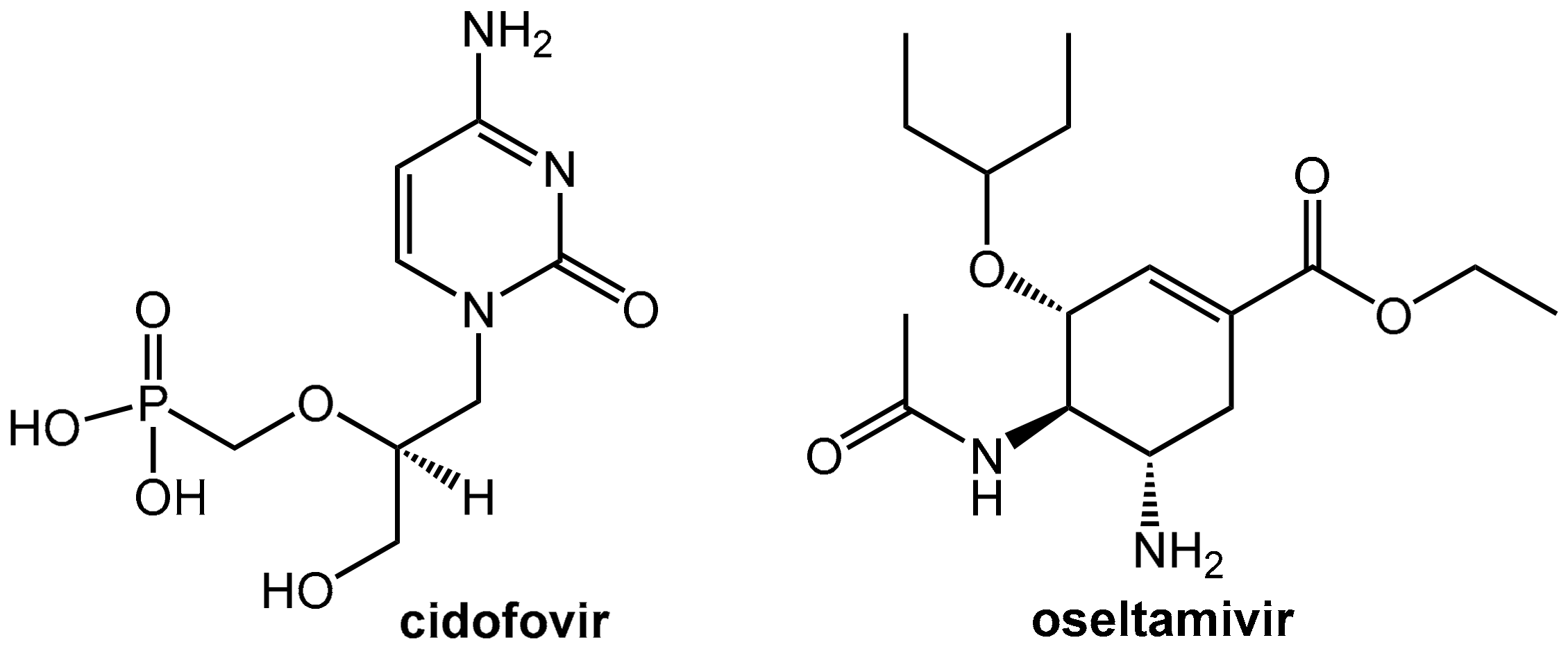
5.1. Reduction of the Nephrotoxicity of Cidofovir
5.2. Probenecid Plus Oseltamivir
6. Tumor Therapy
6.1. Pharmacokinetic Enhancers For Fluoropyrimidine Antimetabolites
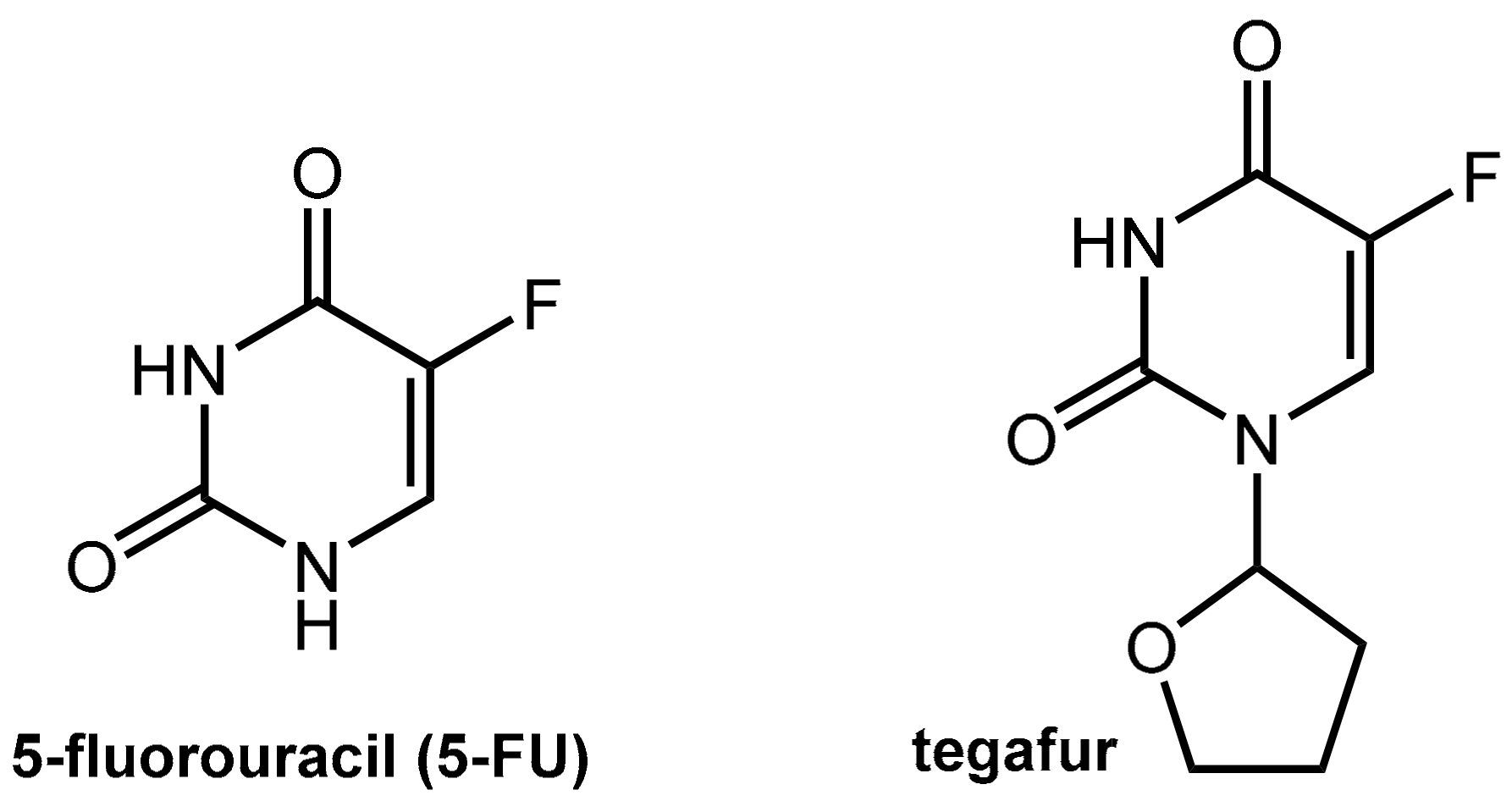
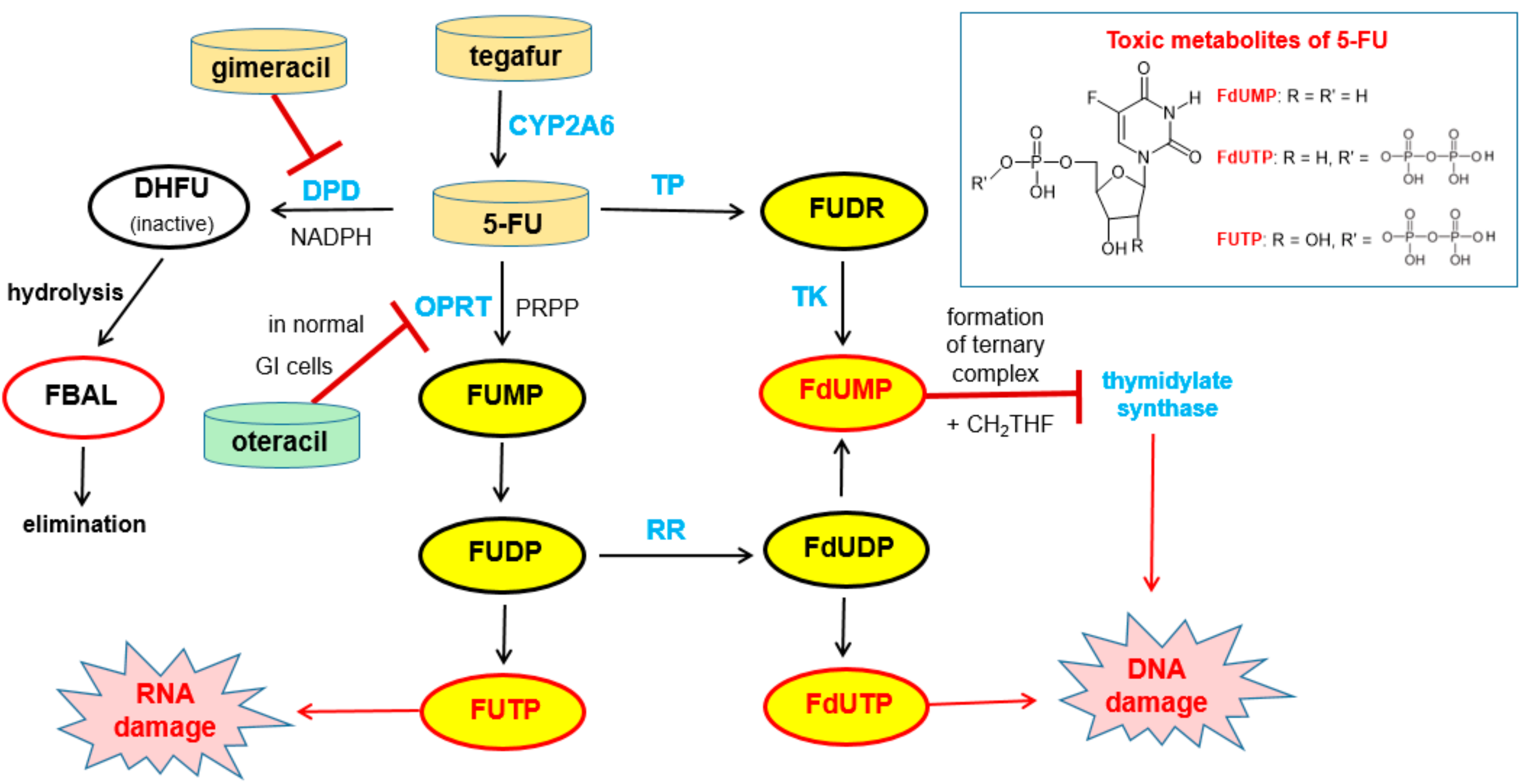
6.1.1. Metabolism of 5-Fluorouracil (5-FU) and Tegafur
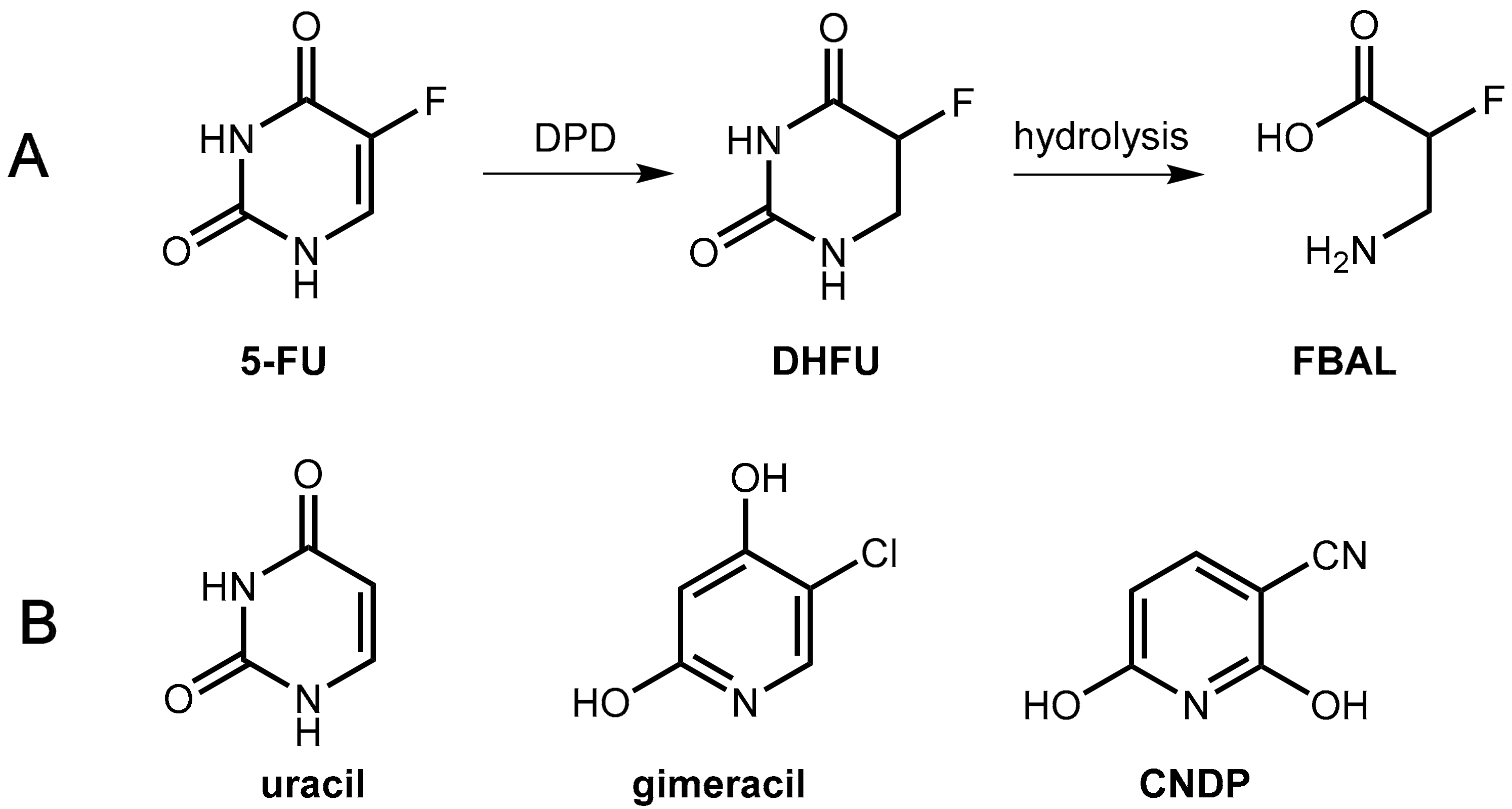
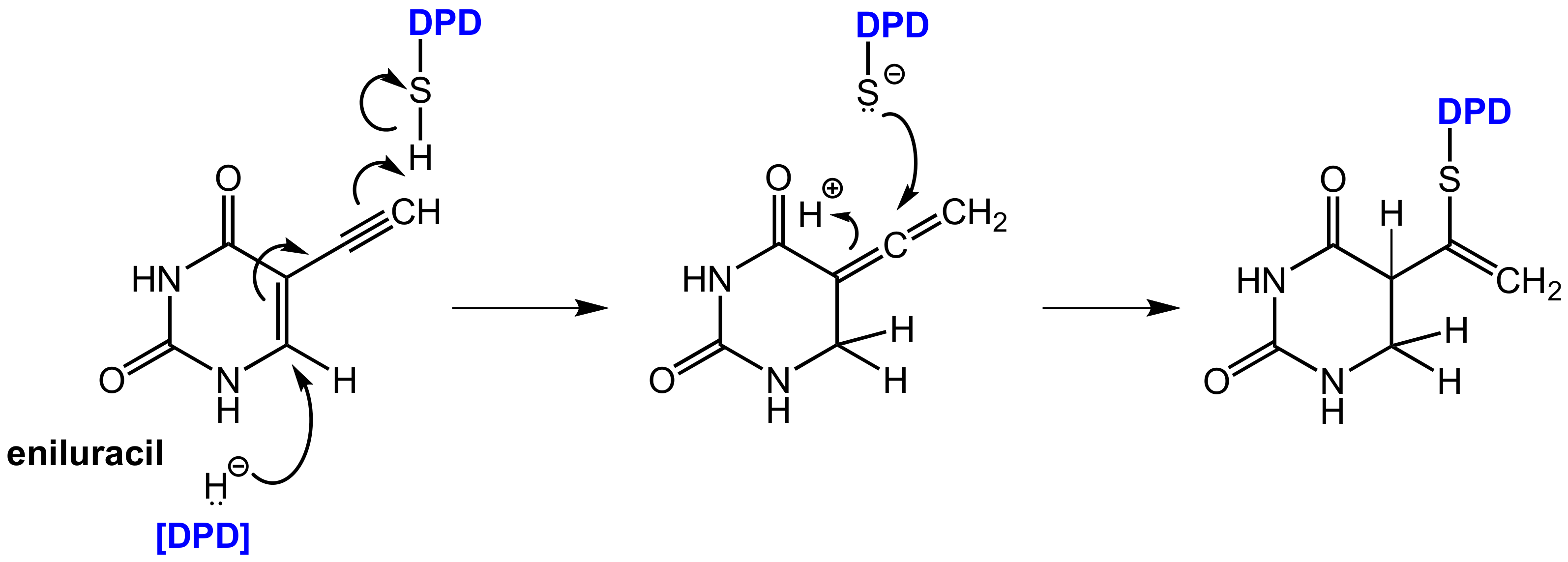
6.1.2. Inhibitors of Dihydropyrimidine Dehydrogenase (DPD)
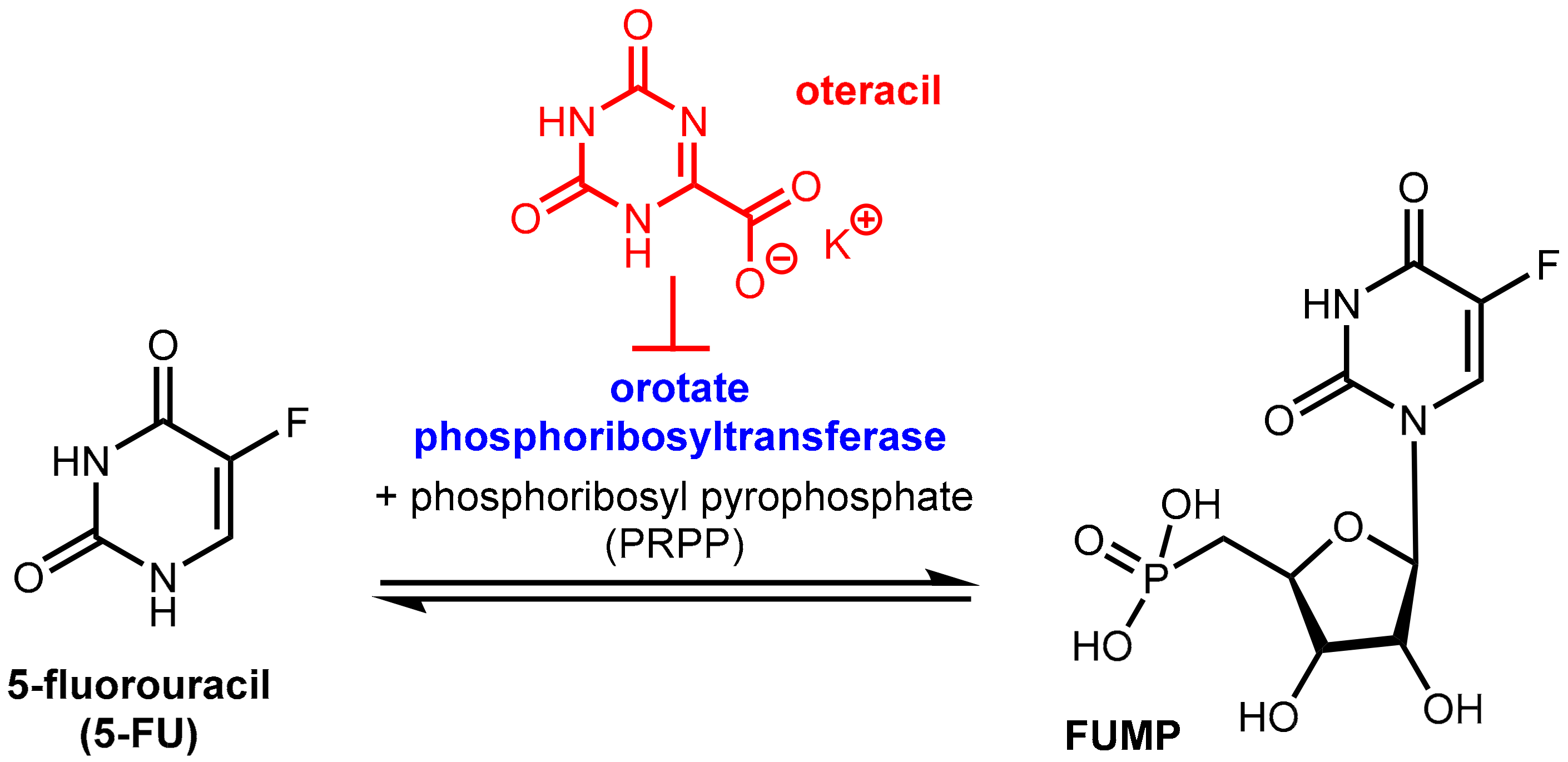
6.1.3. Inhibition of Orotate Phosphoribosyltransferase
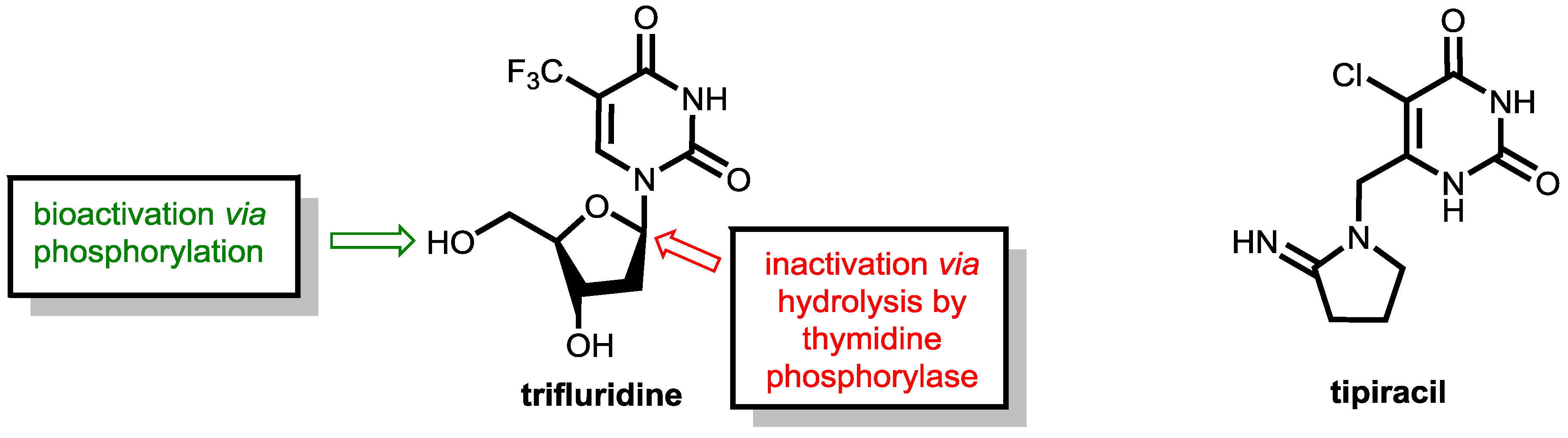
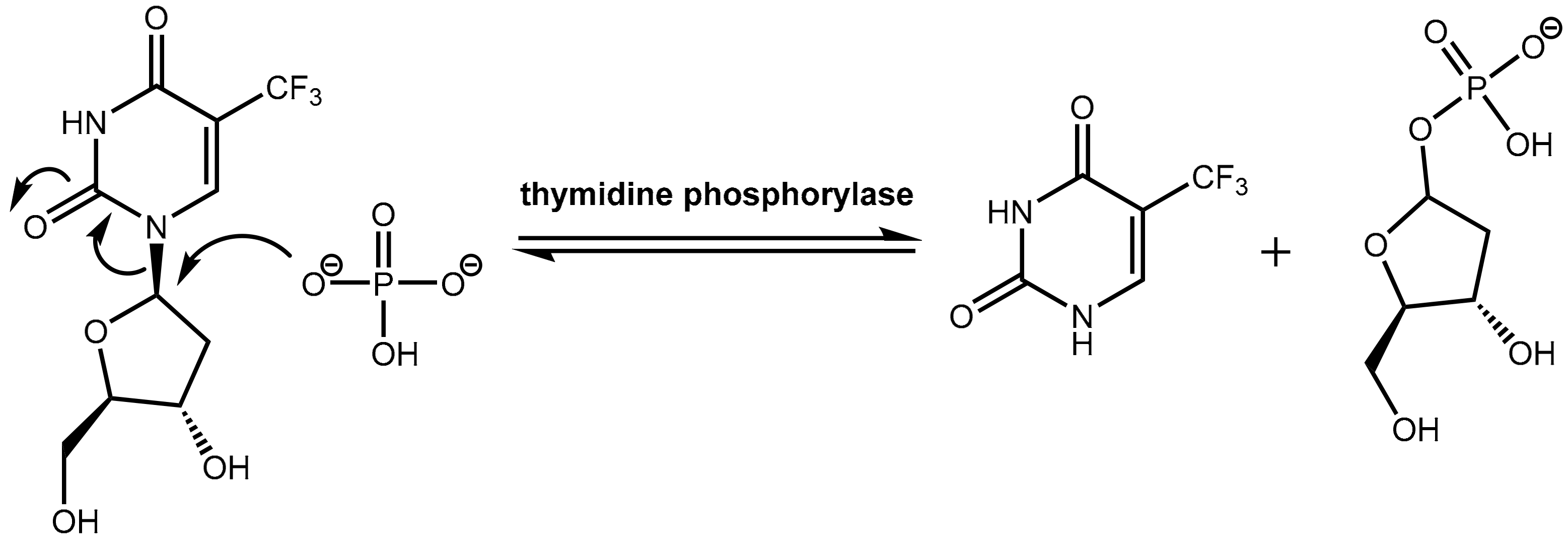
6.2. Inhibition of Thymidine Phosphorylase: Tipiracil As a Pharmacokinetic Enhancer for Trifluridine
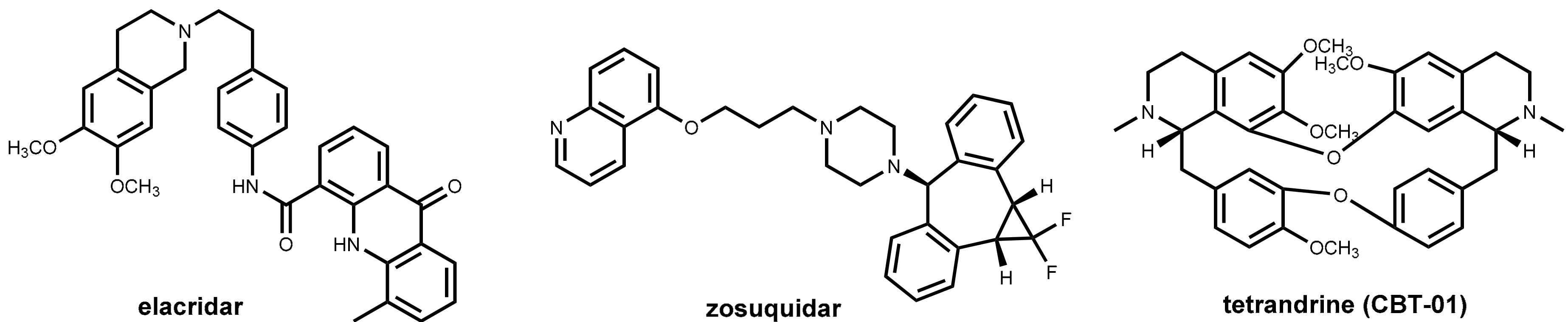
6.3. Efflux Pump Inhibitors In Cancer Therapy
7. Pharmacokinetic Enhancers for Levodopa
7.1. Parkinson’s Disease and Levodopa
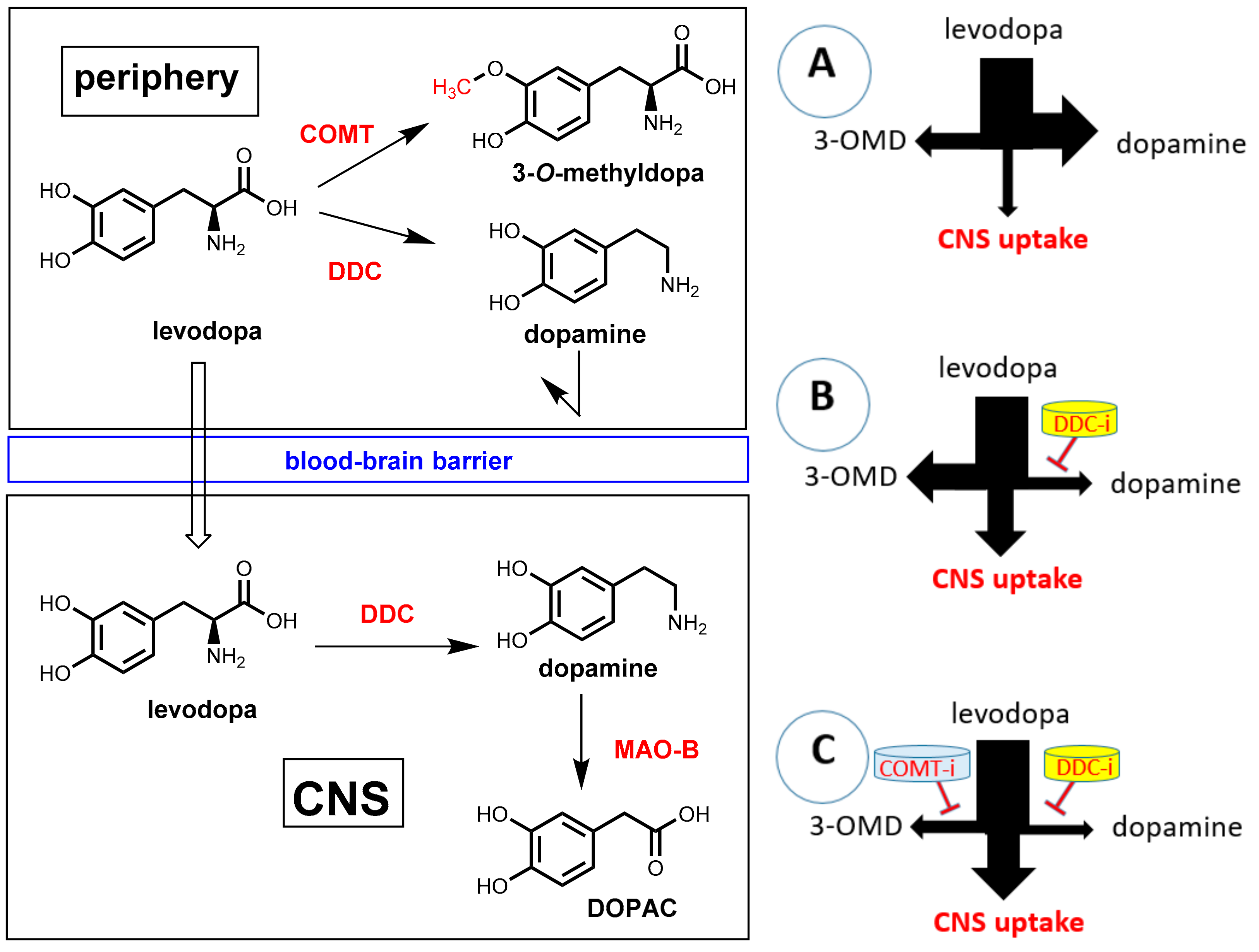
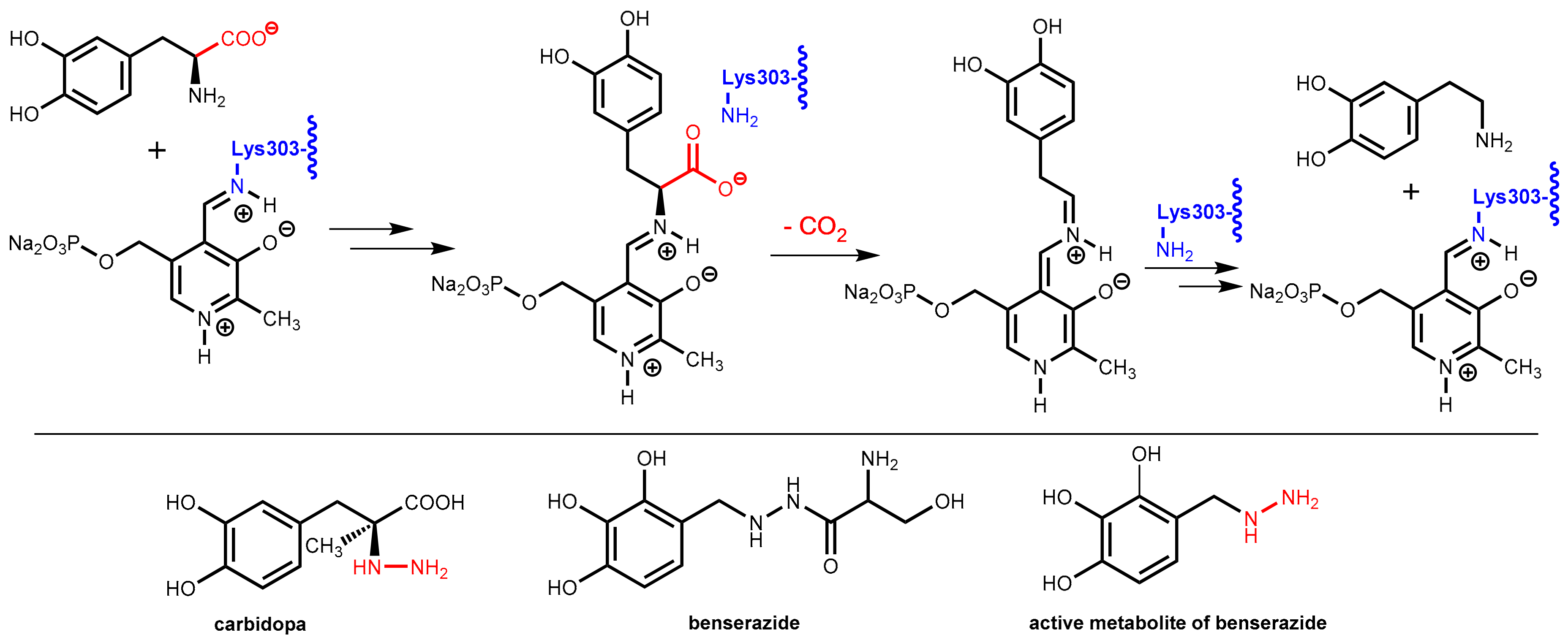
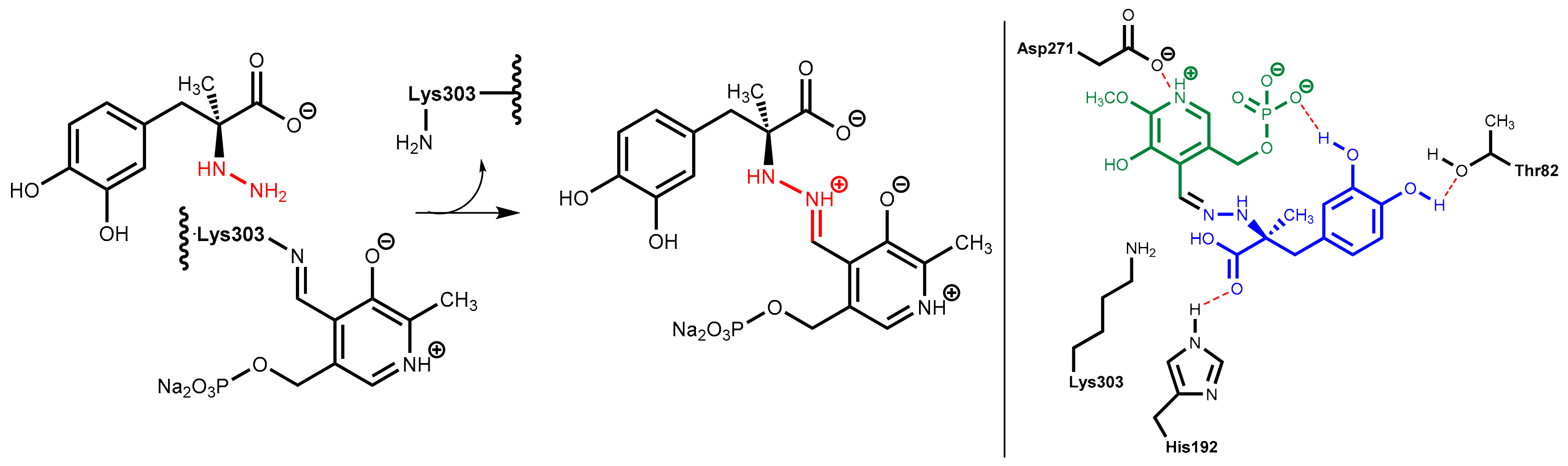
7.2. DOPA Decarboxylase Inhibitors: Carbidopa and Benserazide
7.3. COMT Inhibitors
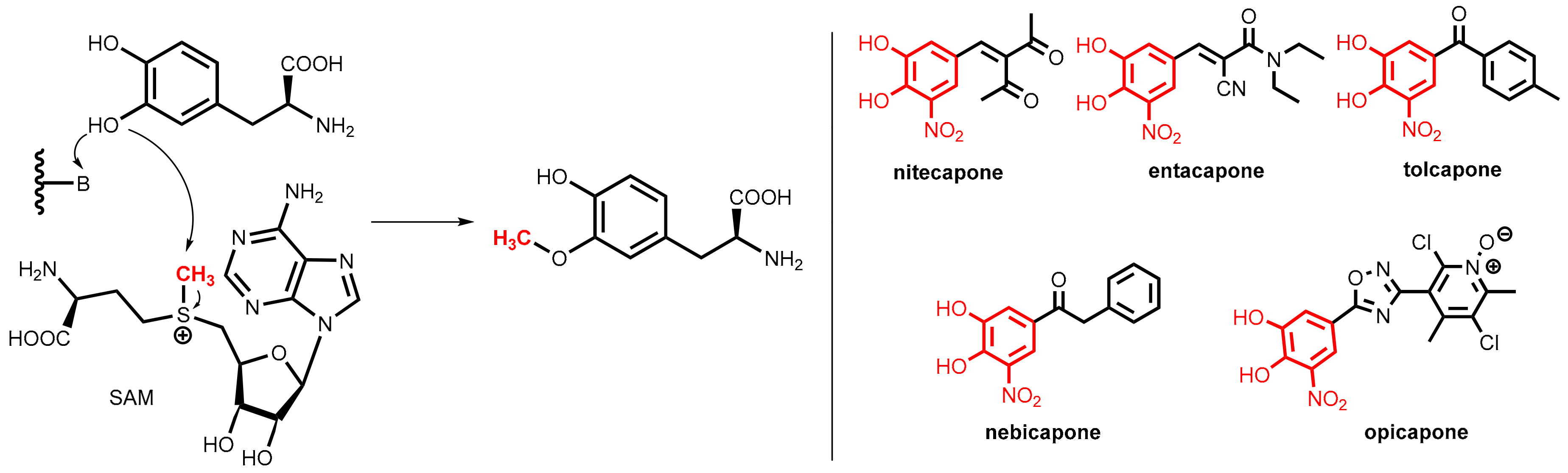
7.3.1. Catechol-O-Methyltransferase (COMT)
7.3.2. COMT Inhibitors
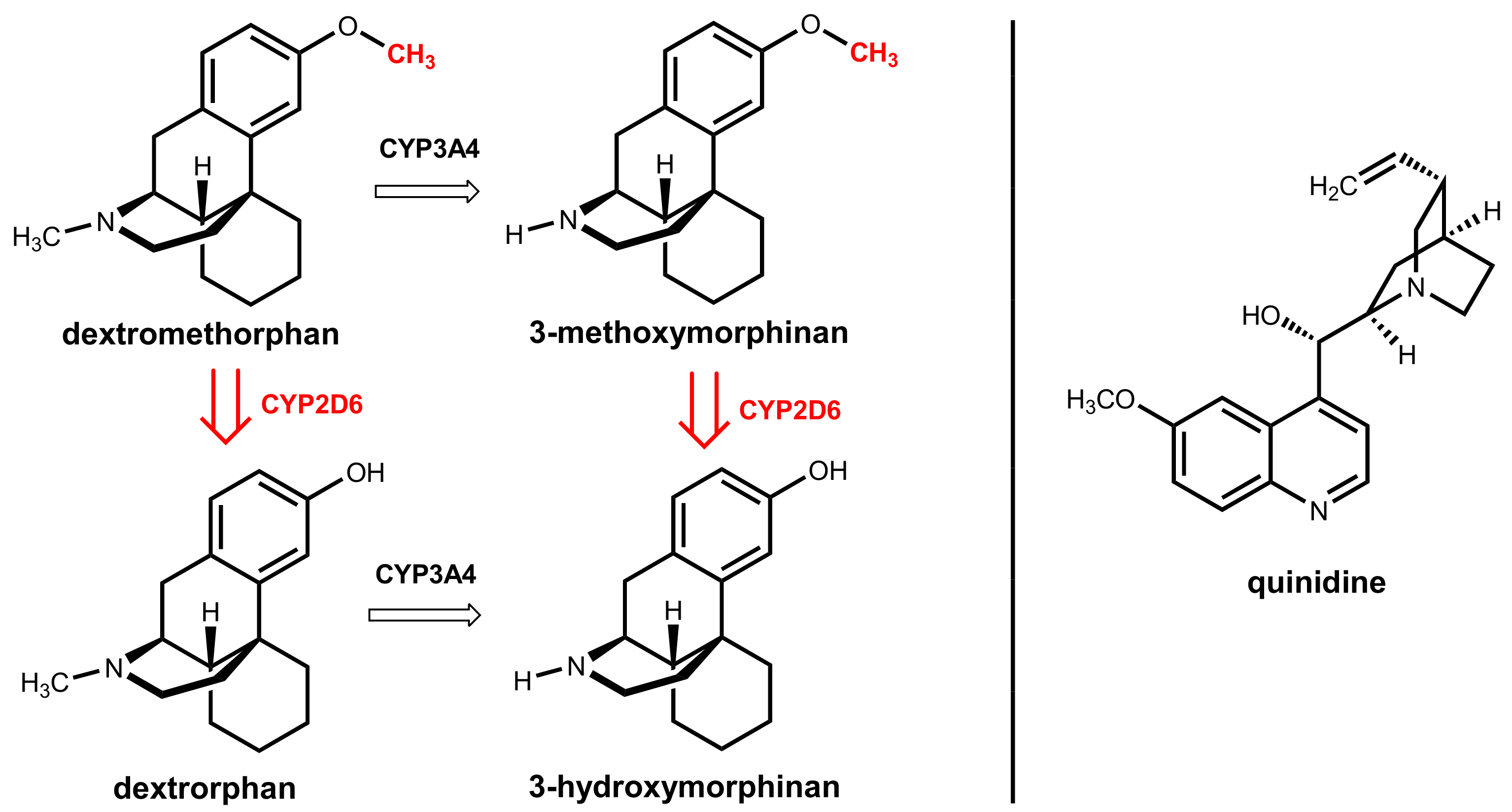
8. Dextromethorphan Plus Quinidine
9. Summary
- it allows for the administration of lower doses of the active agent while maintaining therapeutic levels at the relevant compartment
- it can eliminate variability in systemic exposure
- it can enable overcoming resistance problems in anti-infective and cancer therapy
- it reduces the extent of toxic side effects since lower daily doses of the primary therapeutic agent (drug) are administered
- it reduces the therapy costs since lower amounts of drugs are needed
- it can reduce the pill burden or dosing frequency and improves the medication adherence of patients
Author Contributions
Funding
Conflicts of Interest
References
- González-Bello, C. Antibiotic Adjuvants–A Strategy to Unlock Bacterial Resistance to Antibiotics. Bioorg. Med. Chem. Lett. 2017, 27, 4221–4228. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Abboud, M.I.; Markoulides, M.S.; Brem, J.; Schofield, C.J. The Road to Avibactam: The First Clinically Useful Non-β-Lactam Working Somewhat Like a β-Lactam. Future Med. Chem. 2016, 8, 1063–1084. [Google Scholar] [CrossRef] [PubMed]
- Padayatti, P.S.; Helfand, M.S.; Totir, M.A.; Carey, M.P.; Carey, P.R.; Bonomo, R.A.; van den Akker, F. High Resolution Crystal Structures of the Trans-Enamine Intermediates Formed by Sulbactam and Clavulanic Acid and E166a Shv-1 β-Lactamase. J. Biol. Chem. 2005, 280, 34900–34907. [Google Scholar] [CrossRef] [PubMed]
- Padayatti, P.S.; Helfand, M.S.; Totir, M.A.; Carey, M.P.; Hujer, A.M.; Carey, P.R.; Bonomo, R.A.; van den Akker, F. Tazobactam Forms a Stoichiometric Trans-Enamine Intermediate in the E166a Variant of Shv-1 β-Lactamase: 1.63 Å Crystal Structure. Biochemistry 2004, 43, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Lode, H.; Hampel, B.; Bruckner, G.; Koeppe, P. The Pharmacokinetics of Sultamicillin. APMIS Suppl. 1989, 5, 17–22. [Google Scholar]
- Shirley, M. Ceftazidime-Avibactam: A Review in the Treatment of Serious Gram-Negative Bacterial Infections. Drugs 2018, 78, 675–692. [Google Scholar] [CrossRef] [PubMed]
- Hecker, S.J.; Reddy, K.R.; Totrov, M.; Hirst, G.C.; Lomovskaya, O.; Griffith, D.C.; King, P.; Tsivkovski, R.; Sun, D.; Sabet, M.; et al. Discovery of a Cyclic Boronic Acid β-Lactamase Inhibitor (Rpx7009) with Utility vs Class A Serine Carbapenemases. J. Med. Chem. 2015, 58, 3682–3692. [Google Scholar] [CrossRef] [PubMed]
- Kahan, F.M.; Kropp, H.; Sundelof, J.G.; Birnbaum, J. Thienamycin: Development of Imipenem-Cilastatin. J. Antimicrob. Chemother. 1983, 12, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.J.; Forrester, L.J.; Zahler, W.L.; Burks, M. β-Lactamase Activity of Purified and Partially Characterized Human Renal Dipeptidase. J. Biol. Chem. 1984, 259, 14586–14590. [Google Scholar] [PubMed]
- Keynan, S.; Hooper, N.M.; Felici, A.; Amicosante, G.; Turner, A.J. The Renal Membrane Dipeptidase (Dehydropeptidase I) Inhibitor, Cilastatin, Inhibits the Bacterial Metallo-β-Lactamase Enzyme Cpha. Antimicrob. Agents Chemother. 1995, 39, 1629–1631. [Google Scholar] [CrossRef] [PubMed]
- Drusano, G.L. An Overview of the Pharmacology of Imipenem/Cilastatin. J. Antimicrob. Chemother. 1986, 18 (Suppl. E), 79–92. [Google Scholar] [CrossRef] [PubMed]
- Mainz, D.; Borner, K.; Koeppe, P.; Kotwas, J.; Lode, H. Pharmacokinetics of Lansoprazole, Amoxicillin and Clarithromycin after Simultaneous and Single Administration. J. Antimicrob. Chemother. 2002, 50, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Ushiama, H.; Echizen, H.; Nachi, S.; Ohnishi, A. Dose-Dependent Inhibition of Cyp3a Activity by Clarithromycin During Helicobacter Pylori Eradication Therapy Assessed by Changes in Plasma Lansoprazole Levels and Partial Cortisol Clearance to 6β-Hydroxycortisol. Clin. Pharmacol. Ther. 2002, 72, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Yasui-Furukori, N.; Uno, T.; Takahata, T.; Sugawara, K.; Munakata, A.; Tateishi, T. Effects of Clarithromycin on Lansoprazole Pharmacokinetics between Cyp2c19 Genotypes. Br. J. Clin. Pharmacol. 2005, 59, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Furuta, T.; Ohashi, K.; Kobayashi, K.; Iida, I.; Yoshida, H.; Shirai, N.; Takashima, M.; Kosuge, K.; Hanai, H.; Chiba, K.; et al. Effects of Clarithromycin on the Metabolism of Omeprazole in Relation to Cyp2c19 Genotype Status in Humans. Clin. Pharmacol. Ther. 1999, 66, 265–274. [Google Scholar] [CrossRef]
- Meyer, U.A. Metabolic Interactions of the Proton-Pump Inhibitors Lansoprazole, Omeprazole and Pantoprazole with Other Drugs. Eur. J. Gastroenterol. Hepatol. 1996, 8 (Suppl. 1), S21–S25. [Google Scholar] [CrossRef] [PubMed]
- Shirasaka, Y.; Sager, J.E.; Lutz, J.D.; Davis, C.; Isoherranen, N. Inhibition of Cyp2c19 and Cyp3a4 by Omeprazole Metabolites and Their Contribution to Drug-Drug Interactions. Drug Metab. Dispos. 2013, 41, 1414–1424. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, L.; Pazzucconi, F.; Ferrara, S.; Di Paolo, A.; Tacca, M.D.; Sirtori, C. Pharmacokinetic Interactions between Omeprazole/Pantoprazole and Clarithromycin in Health Volunteers. Pharmacol. Res. 2004, 49, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.H.; Woodward, R.; Peters, L.; Verwey, W.F.; Mattis, P.A. The Prolongation of Penicillin Retention in the Body by Means of Para-Aminohippuric Acid. Science 1944, 100, 107–108. [Google Scholar] [CrossRef] [PubMed]
- Robbins, N.; Koch, S.E.; Tranter, M.; Rubinstein, J. The History and Future of Probenecid. Cardiovasc. Toxicol. 2012, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, R.F.; Israili, Z.H.; Dayton, P.G. Clinical Pharmacokinetics of Probenecid. Clin. Pharmacokinet. 1981, 6, 135–151. [Google Scholar] [CrossRef] [PubMed]
- Landersdorfer, C.B.; Kirkpatrick, C.M.; Kinzig, M.; Bulitta, J.B.; Holzgrabe, U.; Jaehde, U.; Reiter, A.; Naber, K.G.; Rodamer, M.; Sorgel, F. Competitive Inhibition of Renal Tubular Secretion of Ciprofloxacin and Metabolite by Probenecid. Br. J. Clin. Pharmacol. 2010, 69, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Lomovskaya, O.; Bostian, K.A. Practical Applications and Feasibility of Efflux Pump Inhibitors in the Clinic-A Vision for Applied Use. Biochem. Pharmacol. 2006, 71, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Spengler, G.; Kincses, A.; Gajdacs, M.; Amaral, L. New Roads Leading to Old Destinations: Efflux Pumps as Targets to Reverse Multidrug Resistance in Bacteria. Molecules 2017, 22, 468. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Venter, H.; Ma, S. Efflux Pump Inhibitors: A Novel Approach to Combat Efflux-Mediated Drug Resistance in Bacteria. Curr. Drug Targets 2016, 17, 702–719. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Cohen, K.A.; Winglee, K.; Maiga, M.; Diarra, B.; Bishai, W.R. Efflux Inhibition with Verapamil Potentiates Bedaquiline in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 574–576. [Google Scholar] [CrossRef] [PubMed]
- Pule, C.M.; Sampson, S.L.; Warren, R.M.; Black, P.A.; van Helden, P.D.; Victor, T.C.; Louw, G.E. Efflux Pump Inhibitors: Targeting Mycobacterial Efflux Systems to Enhance Tb Therapy. J. Antimicrob. Chemother. 2016, 71, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Tasneen, R.; Peloquin, C.A.; Almeida, D.V.; Li, S.Y.; Barnes-Boyle, K.; Lu, Y.; Nuermberger, E. Verapamil Increases the Bioavailability and Efficacy of Bedaquiline but Not Clofazimine in a Murine Model of Tuberculosis. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Larson, K.B.; Wang, K.; Delille, C.; Otofokun, I.; Acosta, E.P. Pharmacokinetic Enhancers in HIV Therapeutics. Clin. Pharmacokinet. 2014, 53, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Croxtall, J.D.; Perry, C.M. Lopinavir/Ritonavir: A Review of Its Use in the Management of HIV-1 Infection. Drugs 2010, 70, 1885–1915. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Chu, Y.; Wang, Y. HIV Protease Inhibitors: A Review of Molecular Selectivity and Toxicity. HIV AIDS (Auckl.) 2015, 7, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Von Richter, O.; Burk, O.; Fromm, M.F.; Thon, K.P.; Eichelbaum, M.; Kivisto, K.T. Cytochrome P450 3a4 and P-Glycoprotein Expression in Human Small Intestinal Enterocytes and Hepatocytes: A Comparative Analysis in Paired Tissue Specimens. Clin. Pharmacol. Ther. 2004, 75, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Renjifo, B.; van Wyk, J.; Salem, A.H.; Bow, D.; Ng, J.; Norton, M. Pharmacokinetic Enhancement in HIV Antiretroviral Therapy: A Comparison of Ritonavir and Cobicistat. AIDS Rev. 2015, 17, 37–46. [Google Scholar] [PubMed]
- Tseng, A.; Hughes, C.A.; Wu, J.; Seet, J.; Phillips, E.J. Cobicistat Versus Ritonavir: Similar Pharmacokinetic Enhancers but Some Important Differences. Ann. Pharmacother. 2017, 51, 1008–1022. [Google Scholar] [CrossRef] [PubMed]
- Atta, M.G.; De Seigneux, S.; Lucas, G.M. Clinical Pharmacology in HIV Therapy. Clin. J. Am. Soc. Nephrol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hussaini, T. Paritaprevir/Ritonavir-Ombitasvir and Dasabuvir, the 3d Regimen for the Treatment of Chronic Hepatitis C Virus Infection: A Concise Review. Hepat. Med. 2016, 8, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Tran, T.; Chen, T.; Mikus, G.; Greenblatt, D.J. Inhibition of Human Cytochromes P450 in Vitro by Ritonavir and Cobicistat. J. Pharm. Pharmacol. 2017, 69, 1786–1793. [Google Scholar] [CrossRef] [PubMed]
- Lepist, E.-I.; Phan, T.K.; Roy, A.; Tong, L.; MacLennan, K.; Murray, B.; Ray, A.S. Cobicistat Boosts the Intestinal Absorption of Transport Substrates, Including HIV Protease Inhibitors and Gs-7340, in Vitro. Antimicrob. Agents Chemother. 2012, 56, 5409–5413. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Desai, M.C. Cobicistat and Ritonavir as Pharmacoenhancers for Antiviral Drugs. In Successful Strategies for the Discovery of Antiviral Drugs; Desai, M.C., Meanwell, N.A., Eds.; The Royal Society of Chemistry: London, UK, 2013; pp. 451–481. ISBN 978-1-84973-657-2. [Google Scholar]
- Harbeson, S.L.; Tung, R.D. Preparation of Azapeptide Derivatives as HIV Protease Inhibitors. U.S. Patent 815,880,5B2, 17 April 2012. [Google Scholar]
- Hezode, C.; Asselah, T.; Reddy, K.R.; Hassanein, T.; Berenguer, M.; Fleischer-Stepniewska, K.; Marcellin, P.; Hall, C.; Schnell, G.; Pilot-Matias, T.; et al. Ombitasvir Plus Paritaprevir Plus Ritonavir with or without Ribavirin in Treatment-Naive and Treatment-Experienced Patients with Genotype 4 Chronic Hepatitis C Virus Infection (Pearl-I): A Randomised, Open-Label Trial. Lancet 2015, 385, 2502–2509. [Google Scholar] [CrossRef]
- Keating, G. Ombitasvir/Paritaprevir/Ritonavir: A Review in Chronic HCV Genotype 4 Infection. Drugs 2016, 76, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Klibanov, O.M.; Gale, S.E.; Santevecchi, B. Ombitasvir/Paritaprevir/Ritonavir and Dasabuvir Tablets for Hepatitis C Virus Genotype 1 Infection. Ann. Pharmacother. 2015, 49, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Felmlee, D.J. Mechanisms of Hepatitis C Viral Resistance to Direct Acting Antivirals. Viruses 2015, 7, 6716–6729. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Marzioni, M.; Russo, P.; Aghemo, A.; Alberti, A.; Ascione, A.; Antinori, A.; Bruno, R.; Bruno, S.; Chirianni, A.; et al. Plus Ribavirin for Patients with Hepatitis C Virus Genotype 1 or 4 Infection with Cirrhosis (Abacus): A Prospective Observational Study. Lancet Gastroenterol. Hepatol. 2017, 2, 427–434. [Google Scholar] [CrossRef]
- Ouwerkerk-Mahadevan, S.; Snoeys, J.; Peeters, M.; Beumont-Mauviel, M.; Simion, A. Drug-Drug Interactions with the NS3/4a Protease Inhibitor Simeprevir. Clin. Pharmacokinet. 2016, 55, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Cundy, K.C.; Petty, B.G.; Flaherty, J.; Fisher, P.E.; Polis, M.A.; Wachsman, M.; Lietman, P.S.; Lalezari, J.P.; Hitchcock, M.J.; Jaffe, H.S. Clinical Pharmacokinetics of Cidofovir in Human Immunodeficiency Virus-Infected Patients. Antimicrob. Agents Chemother. 1995, 39, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Unadkat, J.D.; Kirby, B.J.; Endres, C.J.; Zolnerciks, J.K. The Impact and in Vitro to in Vivo Prediction of Transporter-Based Drug–Drug Interactions in Humans. In Enzyme- and Transporter-Based Drug-Drug Interactions; Pang, K.S., Rodrigues, A.D., Eds.; Springer: New York, NY, USA, 2010; pp. 517–553. [Google Scholar] [CrossRef]
- Uwai, Y.; Ida, H.; Tsuji, Y.; Katsura, T.; Inui, K.-I. Renal Transport of Adefovir, Cidofovir, and Tenofovir by Slc22a Family Members (Hoat1, Hoat3, and Hoct2). Pharm. Res. 2007, 24, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.L.; Rodriguez, C.A.; Mucci, M.; Ingrosso, A.; Duncan, B.A.; Nickens, D.J. Pharmacokinetics and Renal Effects of Cidofovir with a Reduced Dose of Probenecid in HIV-Infected Patients with Cytomegalovirus Retinitis. J. Clin. Pharmacol. 2003, 43, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Butler, D. Wartime Tactic Doubles Power of Scarce Bird-Flu Drug. Nature 2005, 438, 6. [Google Scholar] [CrossRef] [PubMed]
- Hill, G.; Cihlar, T.; Oo, C.; Ho, E.S.; Prior, K.; Wiltshire, H.; Barrett, J.; Liu, B.; Ward, P. The Anti-Influenza Drug Oseltamivir Exhibits Low Potential to Induce Pharmacokinetic Drug Interactions Via Renal Secretion—Correlation of in Vivo and in Vitro Studies. Drug Metab. Disposition 2002, 30, 13–19. [Google Scholar] [CrossRef]
- Schilsky, R.L.; Kindler, H.L. Eniluracil: An Irreversible Inhibitor of Dihydropyrimidine Dehydrogenase. Expert Opin. Investig. Drugs 2000, 9, 1635–1649. [Google Scholar] [CrossRef] [PubMed]
- Paff, M.T.; Baccanari, D.P.; Davis, S.T.; Cao, S.; Tansik, R.L.; Rustum, Y.M.; Spector, T. Preclinical Development of Eniluracil: Enhancing the Therapeutic Index and Dosing Convenience of 5-Fluorouracil. Invest. New Drugs 2000, 18, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in Medicinal Chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of Action and Clinical Strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Sanford, M. S-1 (Teysuno(R)): A Review of Its Use in Advanced Gastric Cancer in Non-Asian Populations. Drugs 2013, 73, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Nadal, J.C.; Van Groeningen, C.J.; Pinedo, H.M.; Peters, G.J. In Vivo Potentiation of 5-Fluorouracil by Leucovorin in Murine Colon Carcinoma. Biomed. Pharmacother. 1988, 42, 387–393. [Google Scholar] [PubMed]
- Diasio, R.B. Oral DPD-Inhibitory Fluoropyrimidine Drugs. Oncology (Williston Park) 2000, 14, 19–23. [Google Scholar] [PubMed]
- Peters, G.J.; Noordhuis, P.; Van Kuilenburg, A.B.; Schornagel, J.H.; Gall, H.; Turner, S.L.; Swart, M.S.; Voorn, D.; Van Gennip, A.H.; Wanders, J.; et al. Pharmacokinetics of S-1, an Oral Formulation of Ftorafur, Oxonic Acid and 5-Chloro-2,4-Dihydroxypyridine (Molar Ratio 1:0.4:1) in Patients with Solid Tumors. Cancer Chemother. Pharmacol. 2003, 52, 1–12. [Google Scholar] [CrossRef] [PubMed]
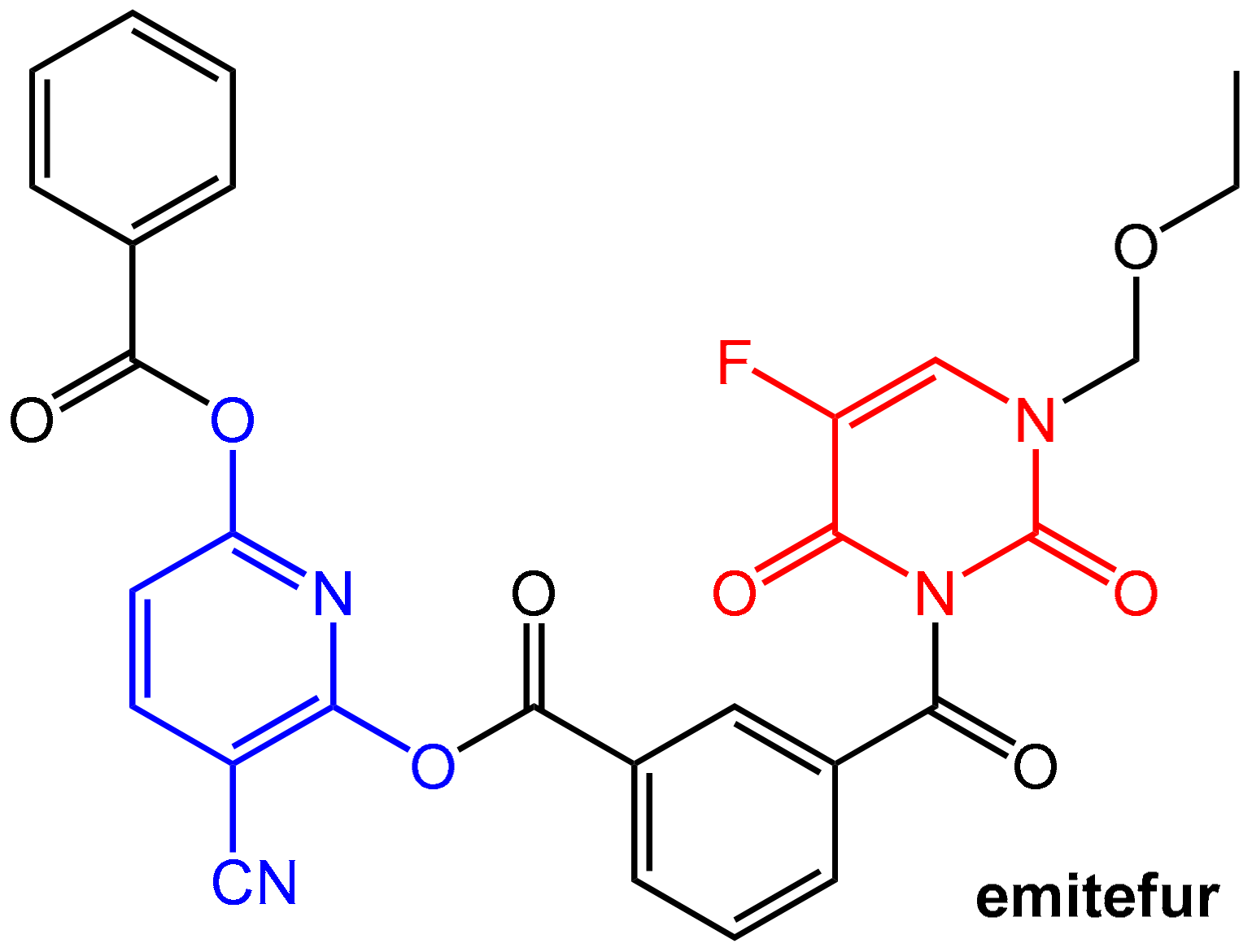
- Sugimachi, K.; Maehara, Y. A Phase Ii Trial of a New 5-Fluorouracil Derivative, Bof-A2 (Emitefur), for Patients with Advanced Gastric Cancer. Surg. Today 2000, 30, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.J.; Chestnut, W.G.; Merrill, B.M.; Spector, T. Mechanism-Based Inactivation of Dihydropyrimidine Dehydrogenase by 5-Ethynyluracil. J. Biol. Chem. 1992, 267, 5236–5242. [Google Scholar] [PubMed]
- Shirasaka, T.; Shimamoto, Y.; Fukushima, M. Inhibition by Oxonic Acid of Gastrointestinal Toxicity of 5-Fluorouracil without Loss of Its Antitumor Activity in Rats. Cancer Res. 1993, 53, 4004–4009. [Google Scholar] [PubMed]
- Kish, T.; Uppal, P. Trifluridine/Tipiracil (Lonsurf) for the Treatment of Metastatic Colorectal Cancer. P&T 2016, 41, 314–325. [Google Scholar]
- Puthiamadathil, J.M.; Weinberg, B.A. Emerging Combination Therapies for Metastatic Colorectal Cancer-Impact of Trifluridine/Tipiracil. Cancer Manag. Res. 2017, 9, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, M.; Suzuki, N.; Emura, T.; Yano, S.; Kazuno, H.; Tada, Y.; Yamada, Y.; Asao, T. Structure and Activity of Specific Inhibitors of Thymidine Phosphorylase to Potentiate the Function of Antitumor 2′-Deoxyribonucleosides. Biochem. Pharmacol. 2000, 59, 1227–1236. [Google Scholar] [CrossRef]
- Joshi, P.; Vishwakarma, R.A.; Bharate, S.B. Natural Alkaloids as P-Gp Inhibitors for Multidrug Resistance Reversal in Cancer. Eur. J. Med. Chem. 2017, 138, 273–292. [Google Scholar] [CrossRef] [PubMed]
- Dewanjee, S.; Dua, T.K.; Bhattacharjee, N.; Das, A.; Gangopadhyay, M.; Khanra, R.; Joardar, S.; Riaz, M.; Feo, V.; Zia-Ul-Haq, M. Natural Products as Alternative Choices for P-Glycoprotein (P-Gp) Inhibition. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Qing, Z.X.; Huang, J.L.; Yang, X.Y.; Liu, J.H.; Cao, H.L.; Xiang, F.; Cheng, P.; Zeng, J.G. Anticancer and Reversing Multidrug Resistance Activities of Natural Isoquinoline Alkaloids and Their Structure-Activity Relationship. Curr. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Robey, R.W.; Chen, C.C.; Draper, D.; Luchenko, V.; Barnett, D.; Oldham, R.K.; Caluag, Z.; Frye, A.R.; Steinberg, S.M.; et al. A Pharmacodynamic Study of the P-Glycoprotein Antagonist Cbt-1® in Combination with Paclitaxel in Solid Tumors. Oncologist 2012, 17, e512–e523. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, H.; Nishikawa, N.; Nagai, M.; Tsujii, T.; Yabe, H.; Kubo, M.; Ieiri, I.; Nomoto, M. Pharmacokinetics of Levodopa/Benserazide Versus Levodopa/Carbidopa in Healthy Subjects and Patients with Parkinson’s Disease. Neurol. Clin. Neurosci. 2015, 3, 68–73. [Google Scholar] [CrossRef]
- Daidone, F.; Montioli, R.; Paiardini, A.; Cellini, B.; Macchiarulo, A.; Giardina, G.; Bossa, F.; Borri Voltattorni, C. Identification by Virtual Screening and in Vitro Testing of Human Dopa Decarboxylase Inhibitors. PLoS ONE 2012, 7, e31610. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M.; Lyseng-Williamson, K.A. Tolcapone: A Review of Its Use in the Management of Parkinson’s Disease. CNS Drugs 2005, 19, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Burkhard, P.; Dominici, P.; Borri-Voltattorni, C.; Jansonius, J.N.; Malashkevich, V.N. Structural Insight into Parkinson’s Disease Treatment from Drug-Inhibited Dopa Decarboxylase. Nat. Struct. Biol. 2001, 8, 963–967. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.E.; Soares-da-Silva, P. Medicinal Chemistry of Catechol O-Methyltransferase (Comt) Inhibitors and Their Therapeutic Utility. J. Med. Chem. 2014, 57, 8692–8717. [Google Scholar] [CrossRef] [PubMed]
- Schrag, A. Entacapone in the Treatment of Parkinson’s Disease. Lancet Neurol. 2005, 4, 366–370. [Google Scholar] [CrossRef]
- Bonifacio, M.J.; Torrao, L.; Loureiro, A.I.; Palma, P.N.; Wright, L.C.; Soares-da-Silva, P. Pharmacological Profile of Opicapone, a Third-Generation Nitrocatechol Catechol-O-Methyl Transferase Inhibitor, in the Rat. Br. J. Pharmacol. 2015, 172, 1739–1752. [Google Scholar] [CrossRef] [PubMed]
- Annus, A.; Vecsei, L. Spotlight on Opicapone as an Adjunct to Levodopa in Parkinson’s Disease: Design, Development and Potential Place in Therapy. Drug Des. Devel. Ther. 2017, 11, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Gordin, A.; Kaakkola, S.; Teravainen, H. Clinical Advantages of COMT Inhibition with Entacapone-A Review. J. Neural. Transm. (Vienna) 2004, 111, 1343–1363. [Google Scholar] [CrossRef] [PubMed]
- Garnock-Jones, K.P. Dextromethorphan/Quinidine: In Pseudobulbar Affect. CNS Drugs 2011, 25, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A. Dextromethorphan/Quinidine: A Novel Dextromethorphan Product for the Treatment of Emotional Lability. Expert Opin. Pharmacother. 2006, 7, 2581–2598. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Britto, M.R.; Valderhaug, K.L.; Wedlund, P.J.; Smith, R.A. Dextromethorphan: Enhancing Its Systemic Availability by Way of Low-Dose Quinidine-Mediated Inhibition of Cytochrome P4502D6. Clin. Pharmacol. Ther. 1992, 51, 647–655. [Google Scholar] [CrossRef] [PubMed]




























© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krauß, J.; Bracher, F. Pharmacokinetic Enhancers (Boosters)—Escort for Drugs against Degrading Enzymes and Beyond. Sci. Pharm. 2018, 86, 43. https://doi.org/10.3390/scipharm86040043
Krauß J, Bracher F. Pharmacokinetic Enhancers (Boosters)—Escort for Drugs against Degrading Enzymes and Beyond. Scientia Pharmaceutica. 2018; 86(4):43. https://doi.org/10.3390/scipharm86040043
Chicago/Turabian StyleKrauß, Jürgen, and Franz Bracher. 2018. "Pharmacokinetic Enhancers (Boosters)—Escort for Drugs against Degrading Enzymes and Beyond" Scientia Pharmaceutica 86, no. 4: 43. https://doi.org/10.3390/scipharm86040043
APA StyleKrauß, J., & Bracher, F. (2018). Pharmacokinetic Enhancers (Boosters)—Escort for Drugs against Degrading Enzymes and Beyond. Scientia Pharmaceutica, 86(4), 43. https://doi.org/10.3390/scipharm86040043