
Development and Validation of Miglitol and Its Impurities by RP-HPLC and Characterization Using Mass Spectrometry Techniques

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Equipment
2.2.1. Mass Spectrometry
2.2.2. Chromatographic Conditions
2.2.3. Preparation of Solutions
2.2.4. Standard and Sample Preparation
Preparation of standard stock solution-1
Preparation of standard stock solution-2
Preparation of 0.15% (w/w) standard solution
Preparation of 0.05% (w/w) standard solution (Limit of quantitation (LOQ) solution)
Preparation of 0.02% w/w standard solution (Limit of detection (LOD) solution)
Preparation of miglitol sample solution
2.3. Method Development
2.3.1. Method Validation
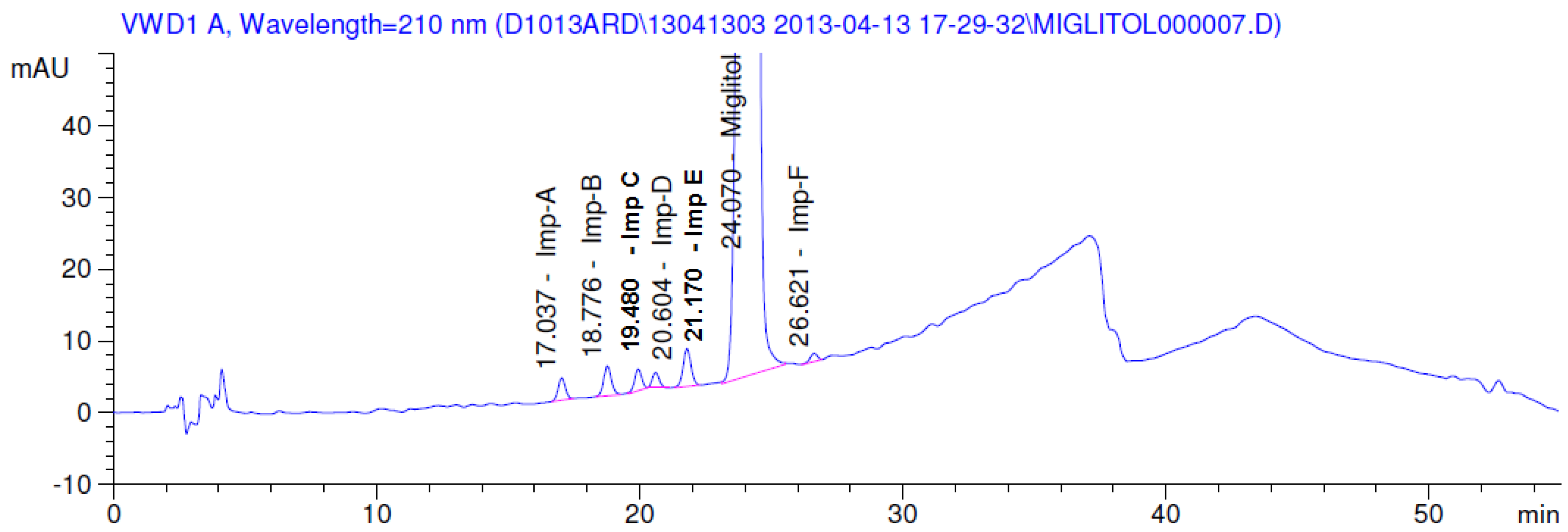
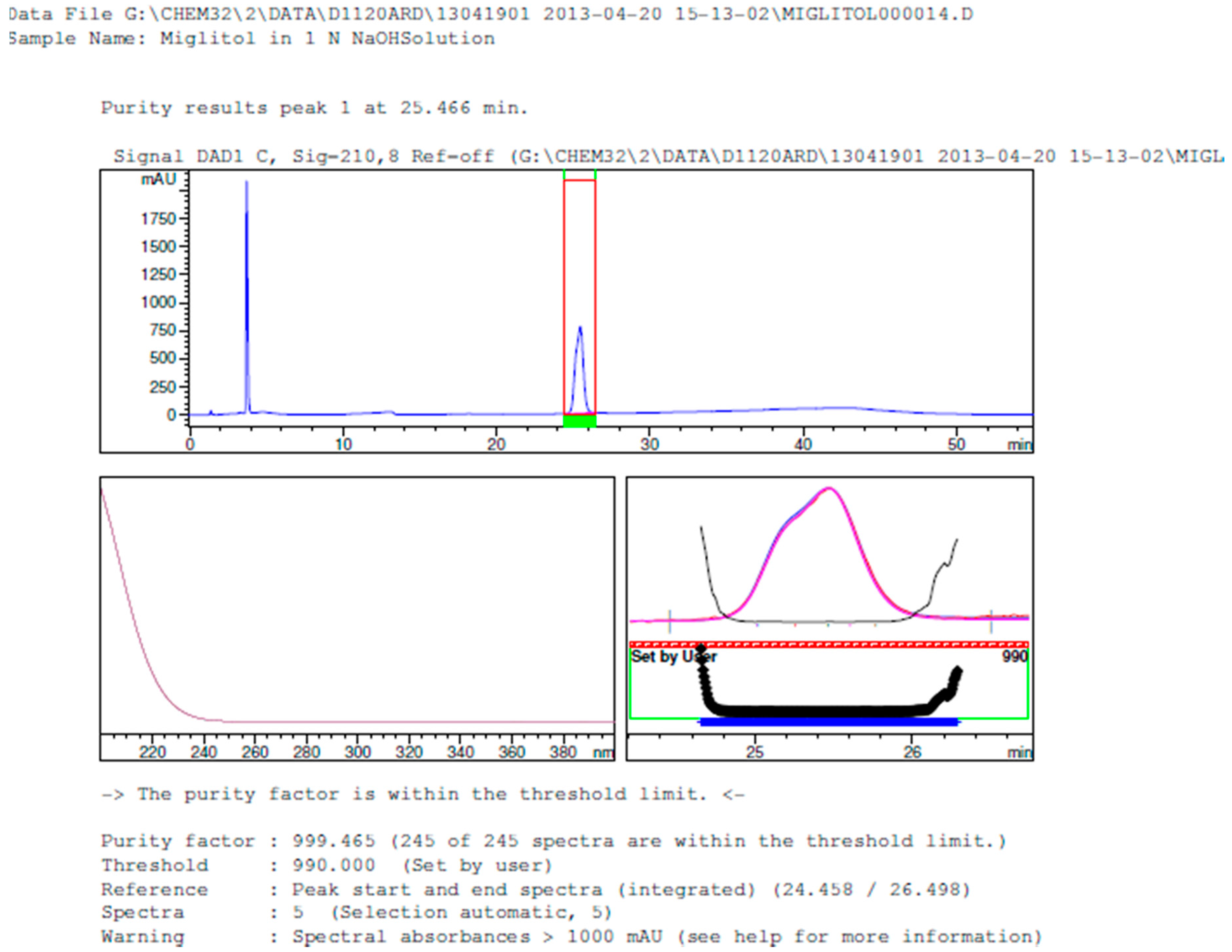
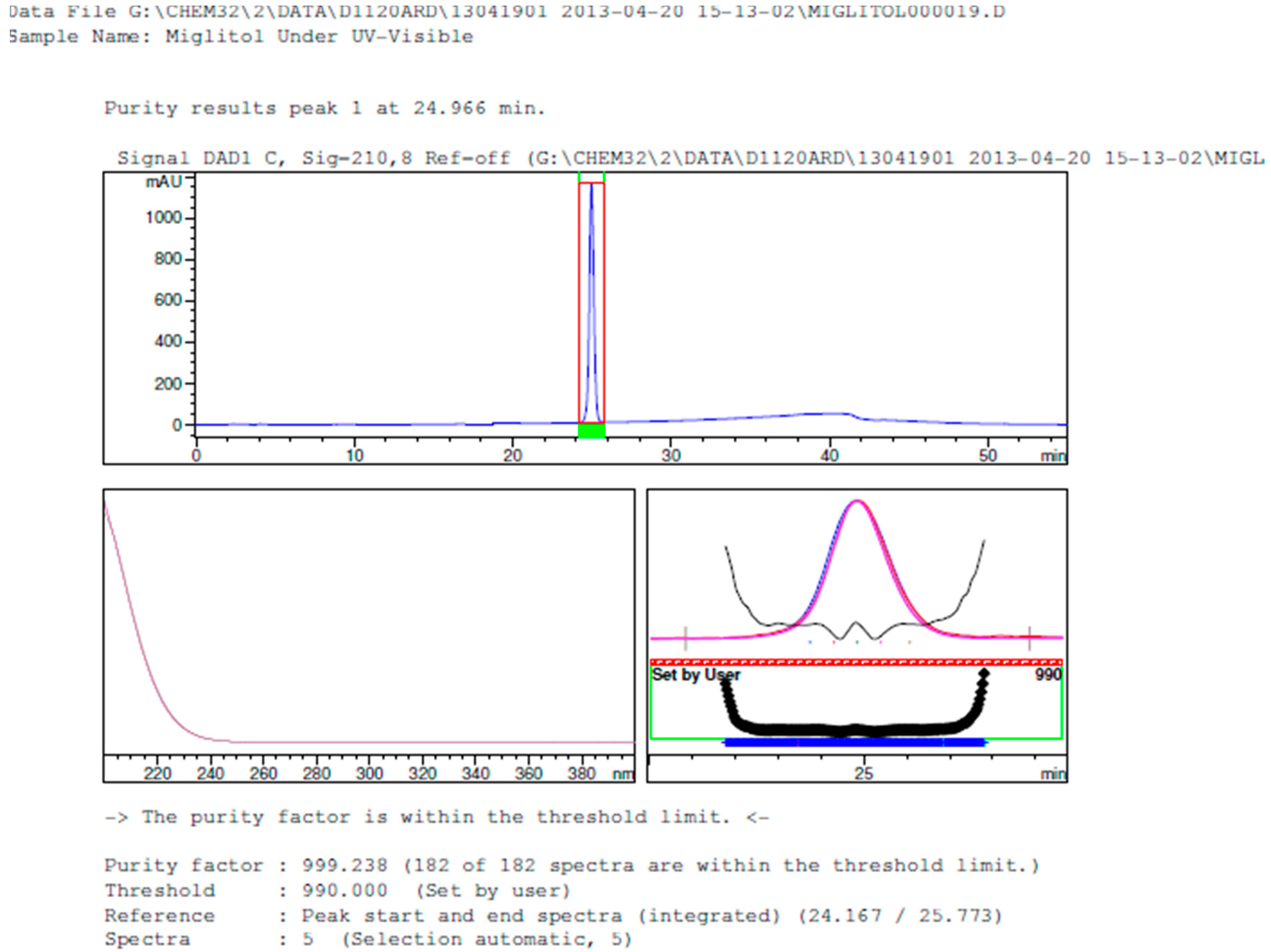
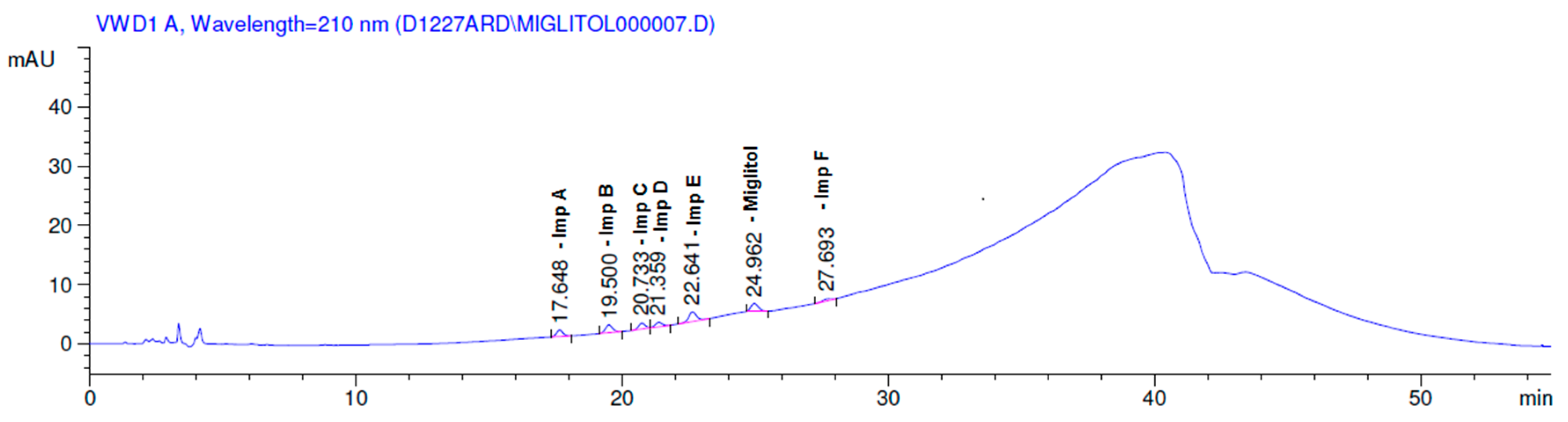
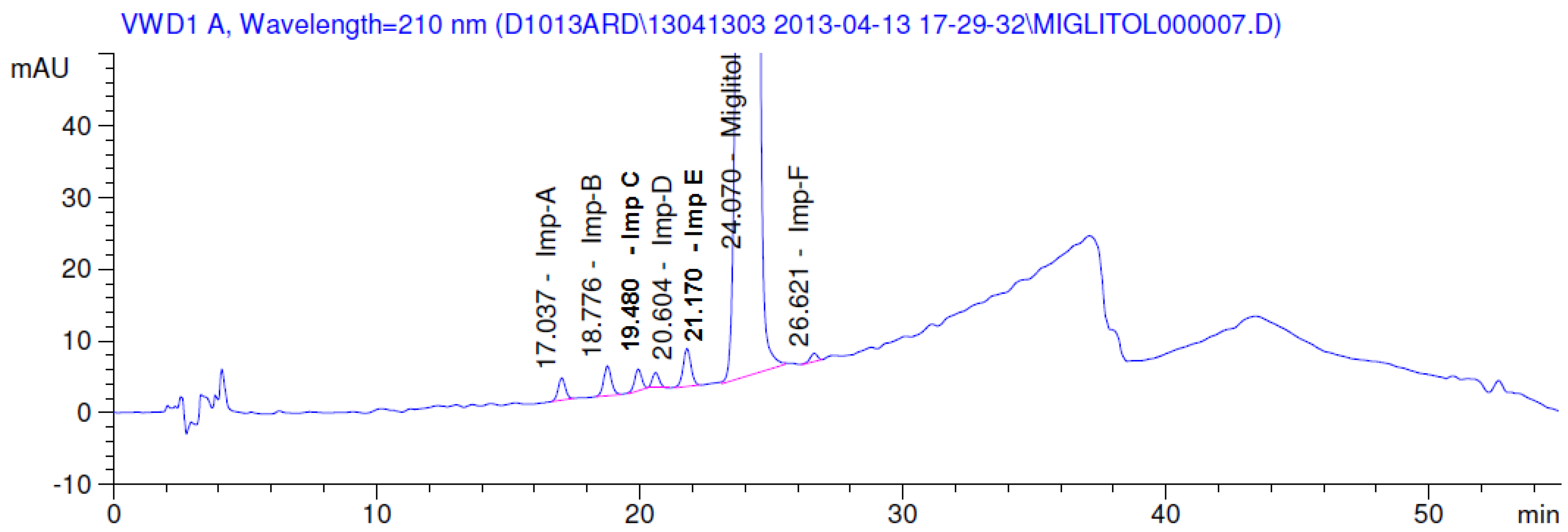
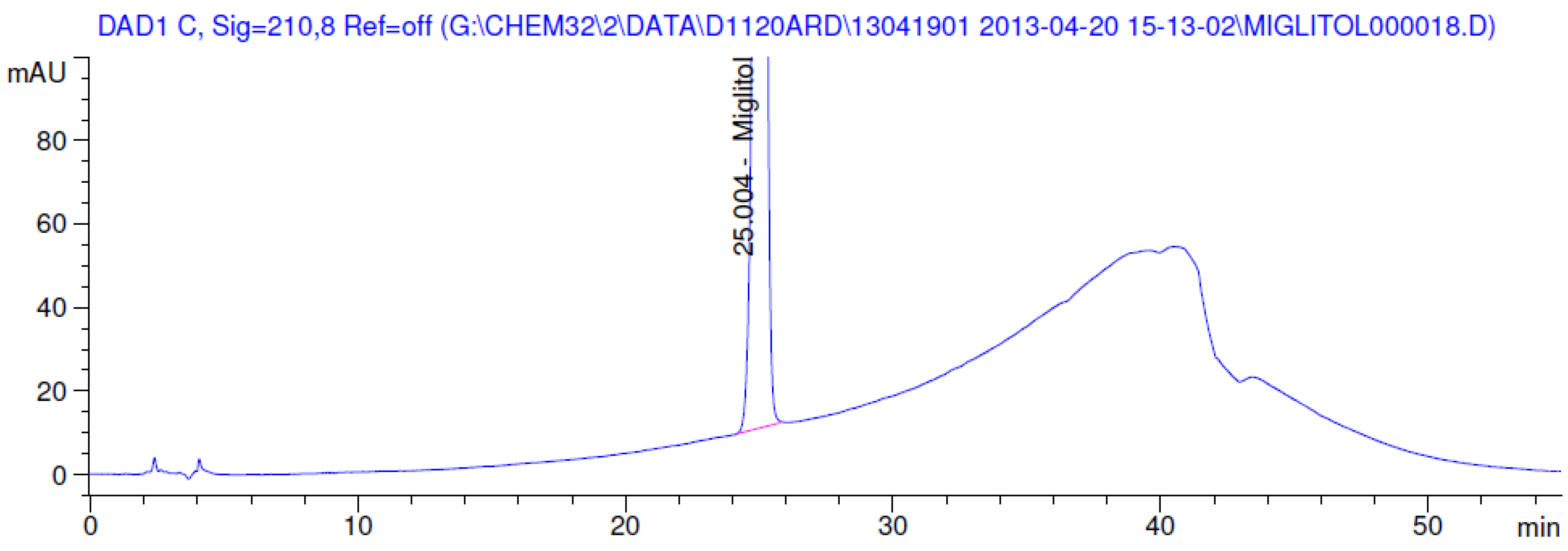
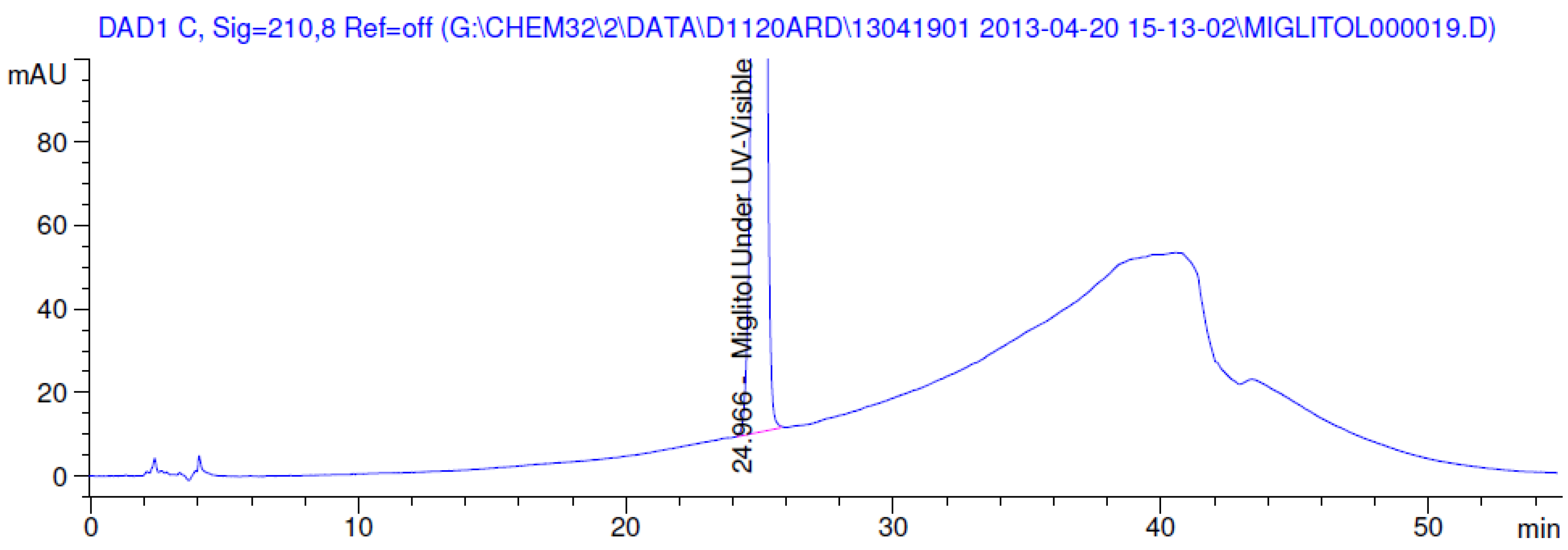
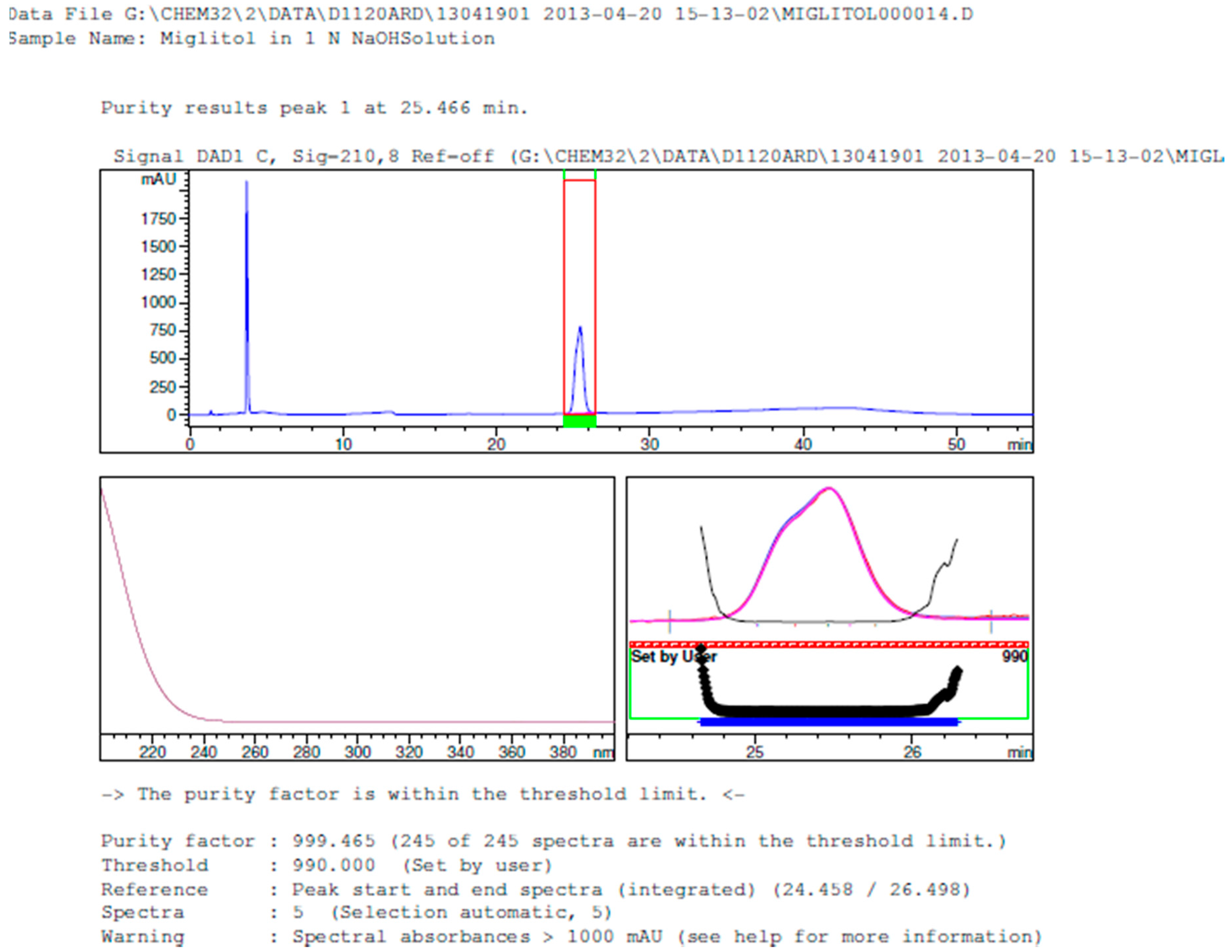
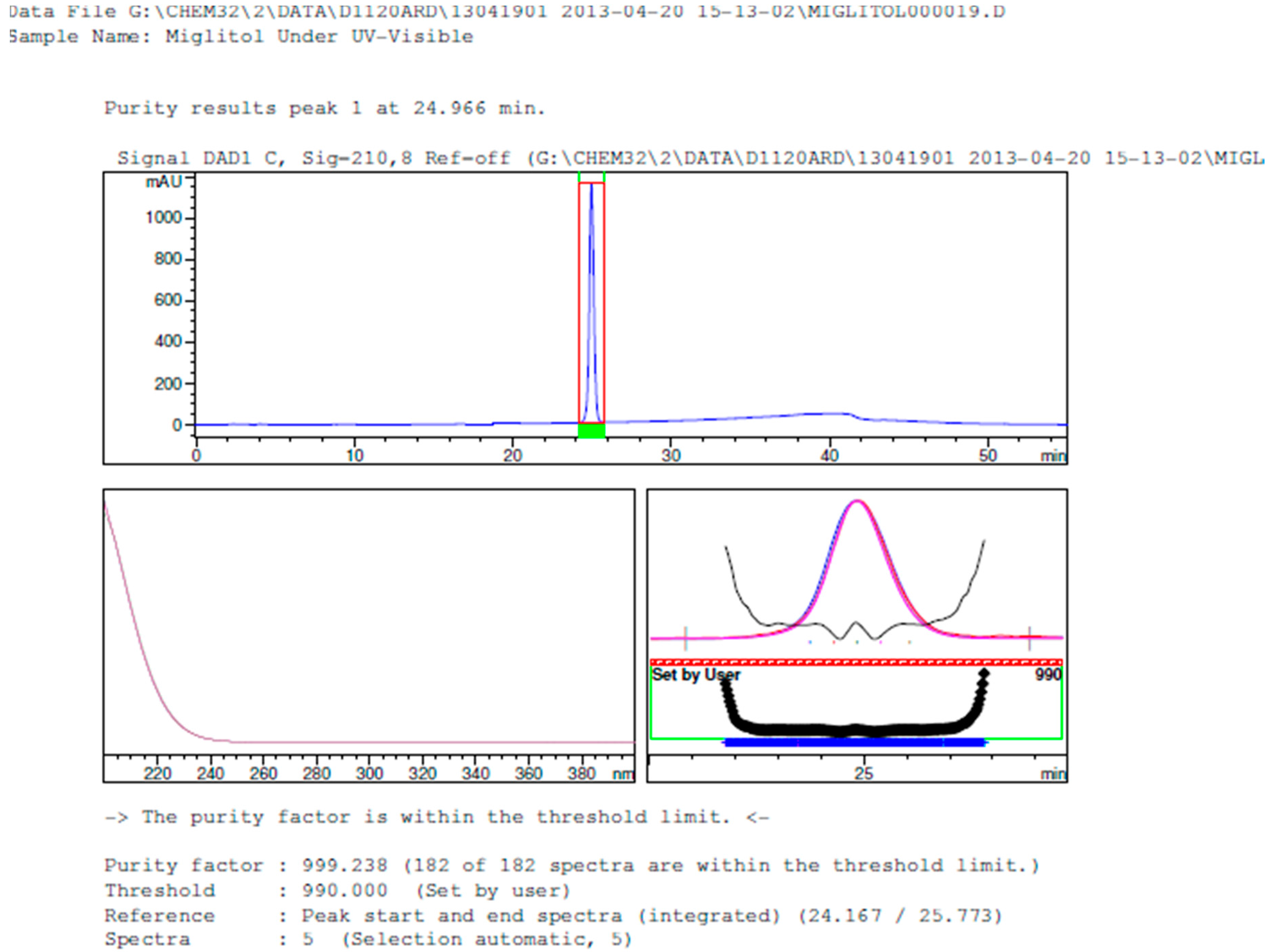
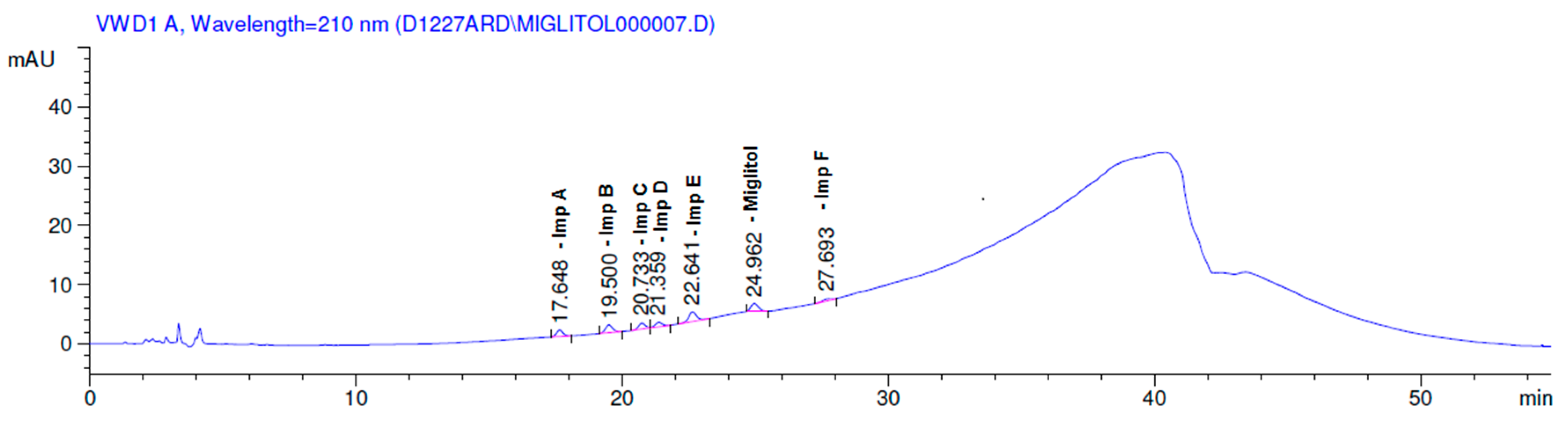
2.3.2. Specificity
2.3.3. Precision
2.3.4. Linearity
2.3.5. Accuracy
2.3.6. Solution Stability and Mobile Phase Stability
2.3.7. Robustness
3. Results and Discussion
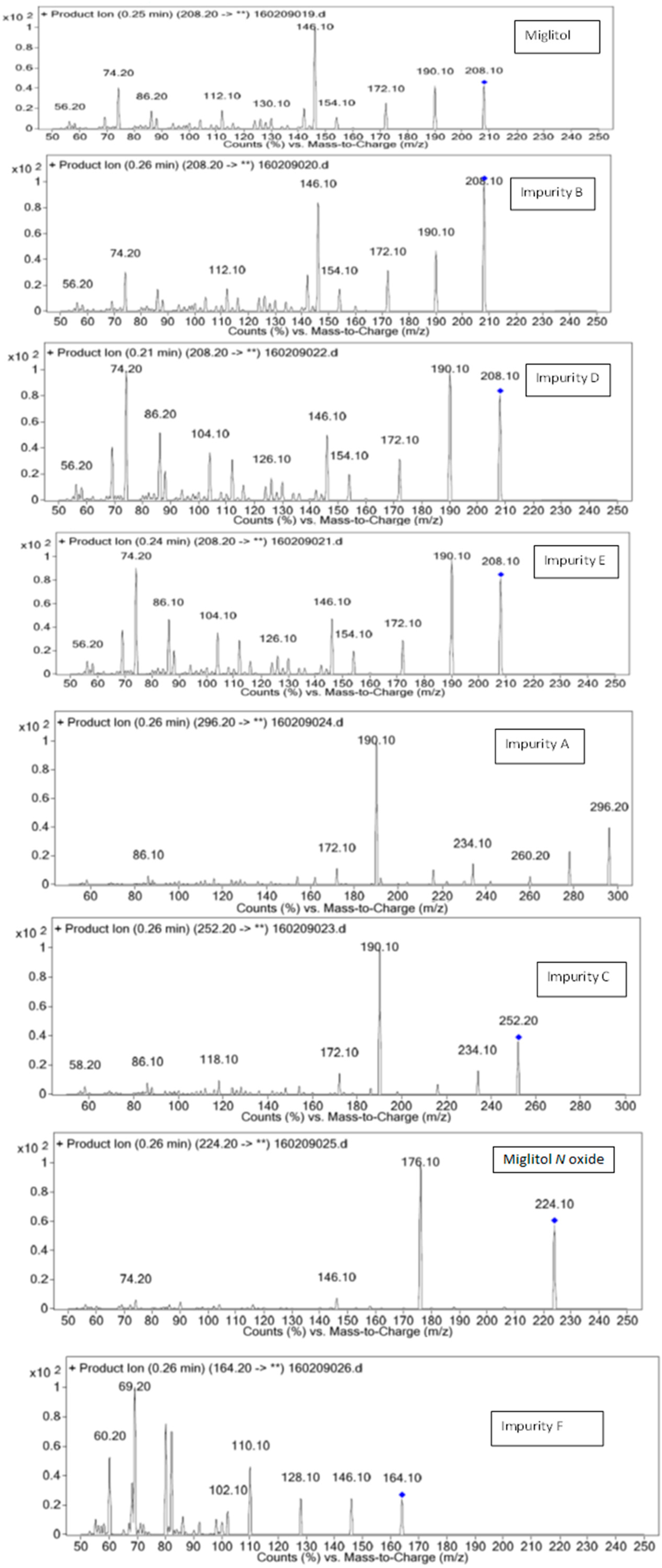
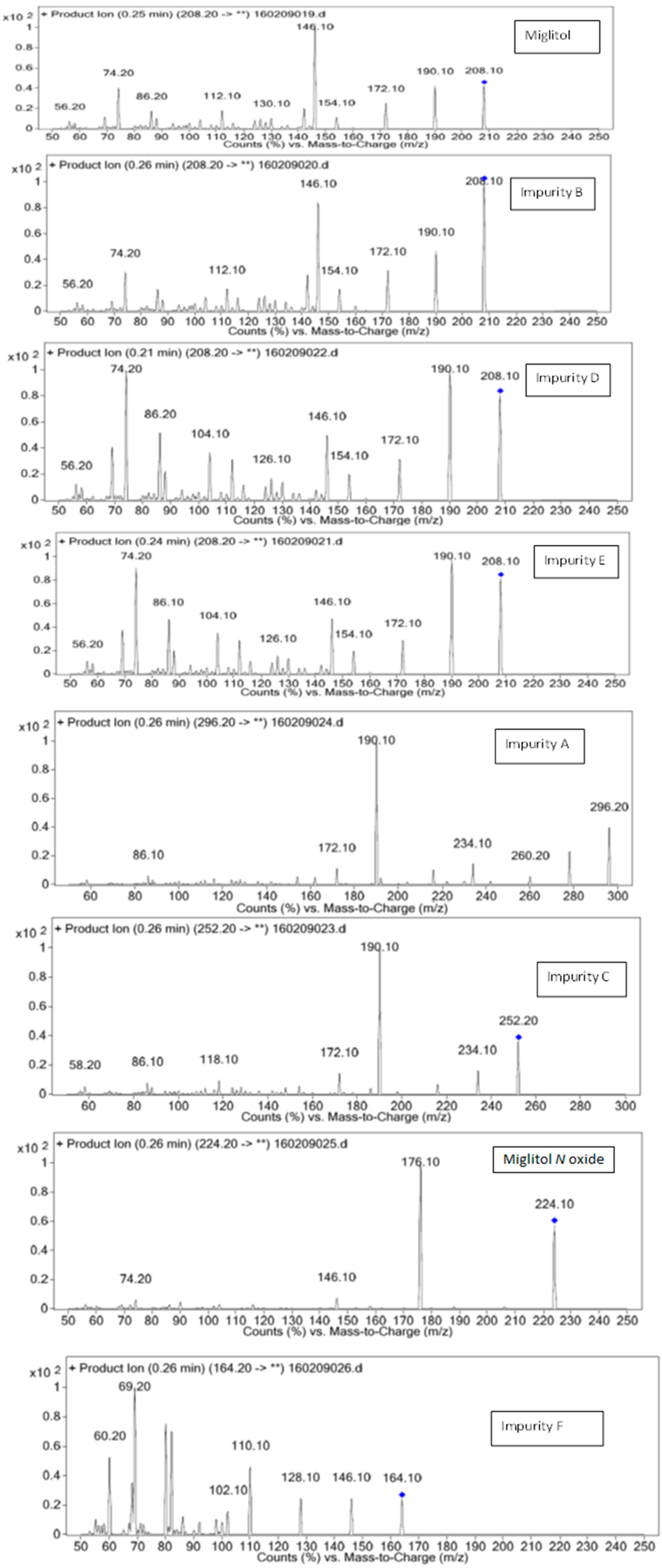
3.1. Mass Spectrometry Interpretation of Miglitol, 1-Dzeoxynojirimycin and Other Impurities
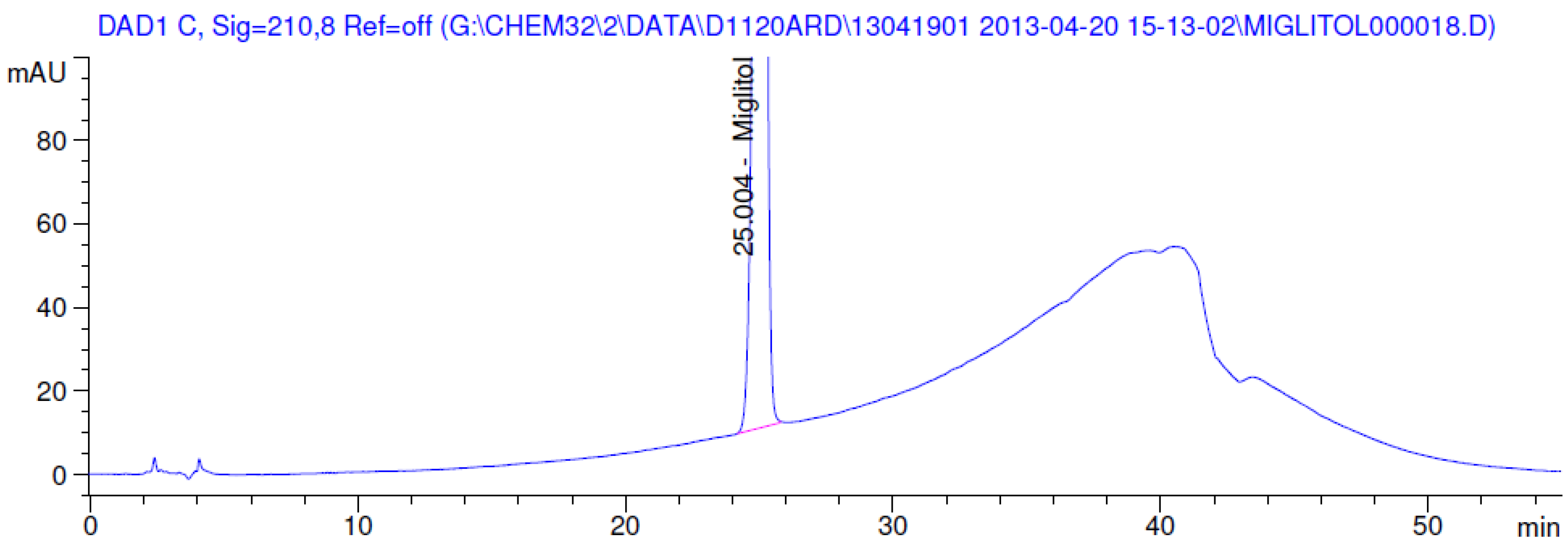
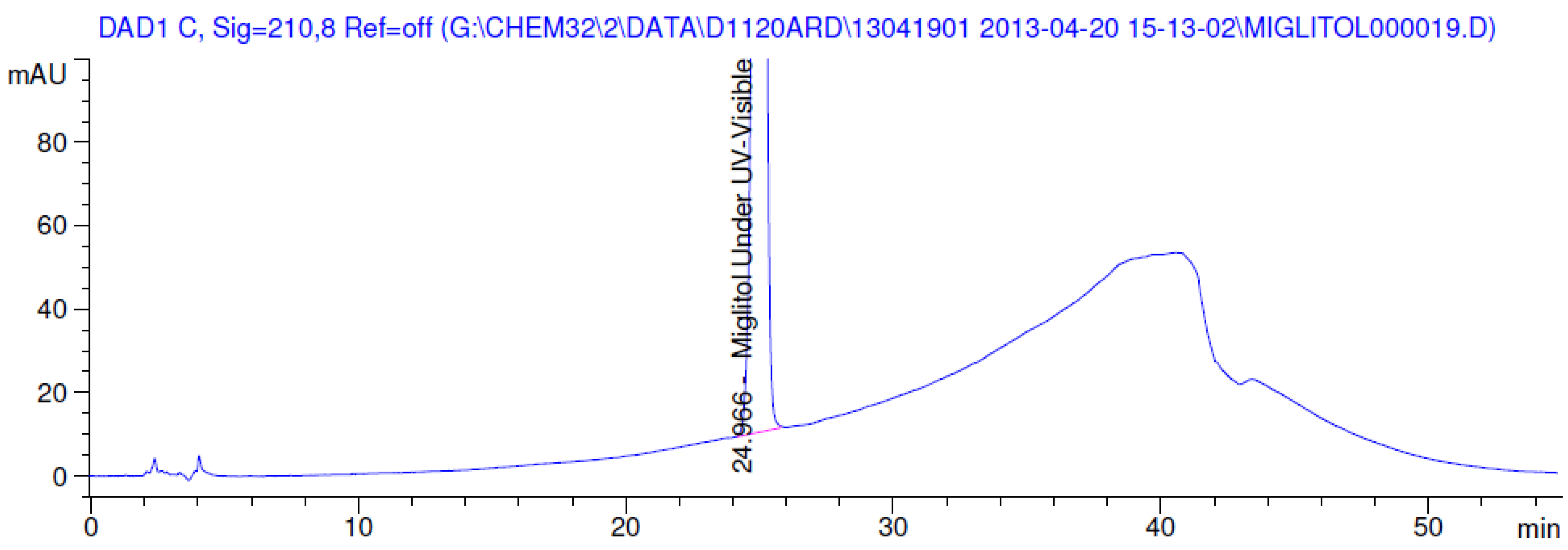
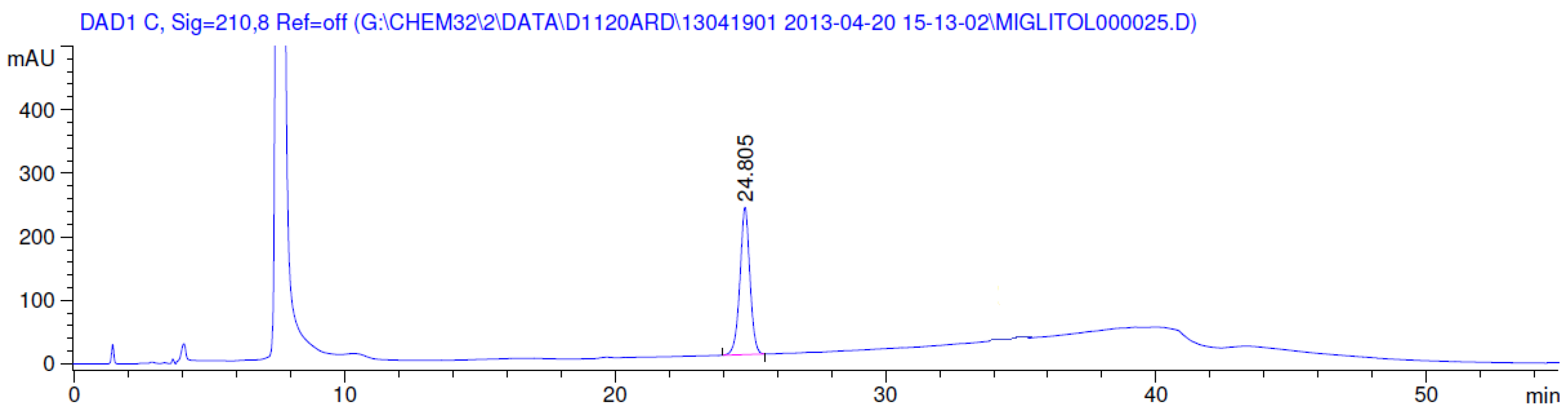
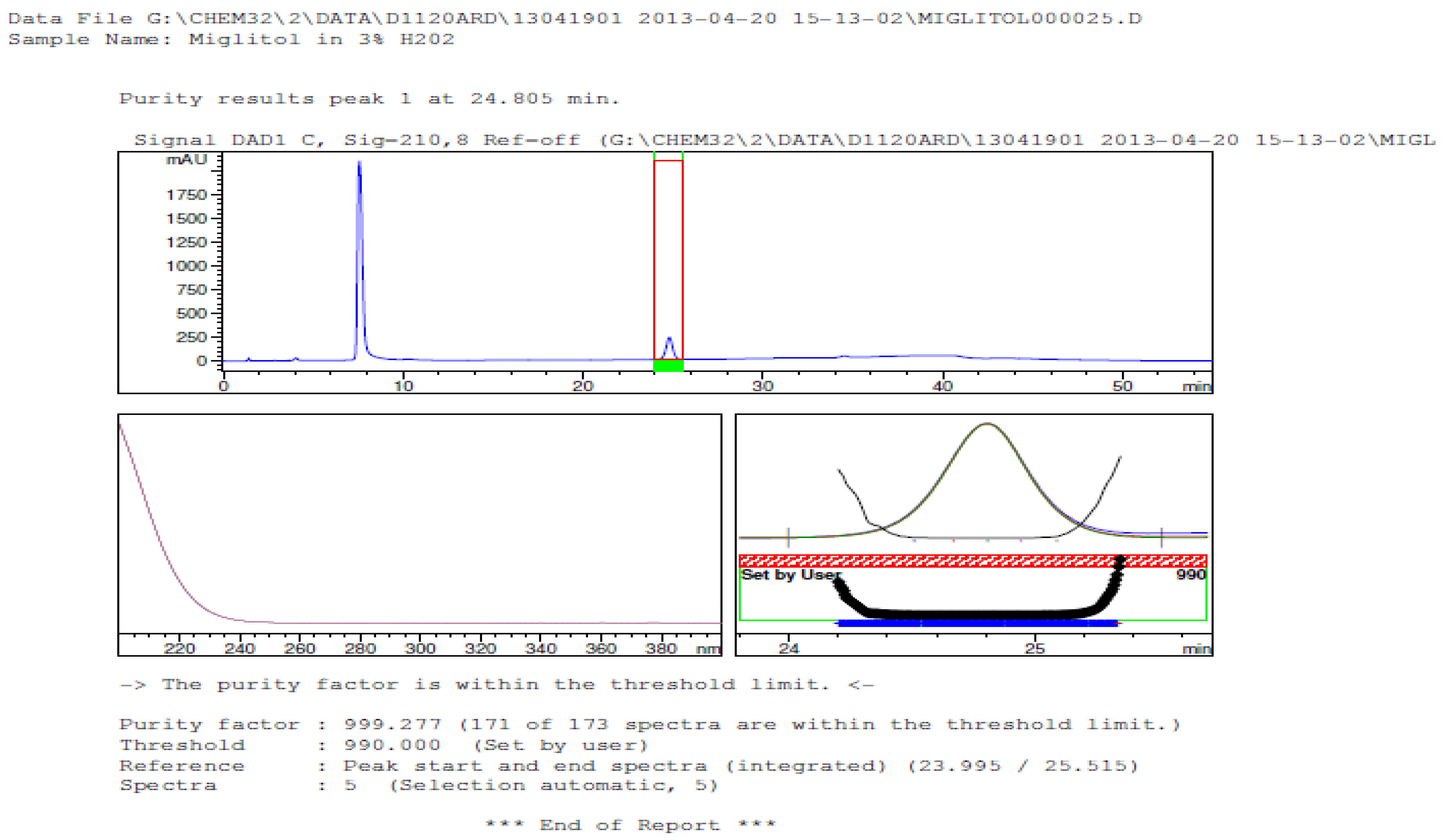
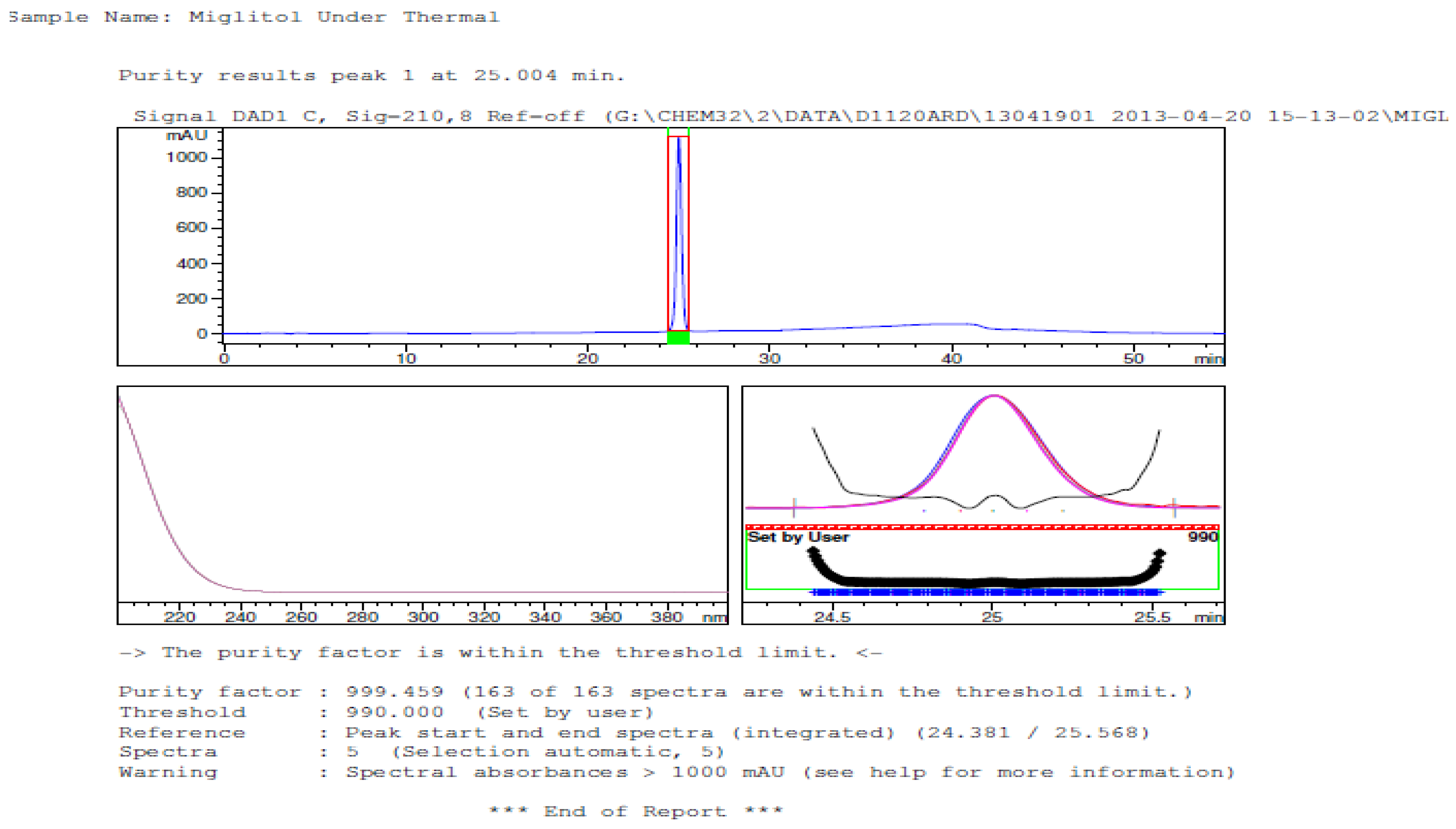
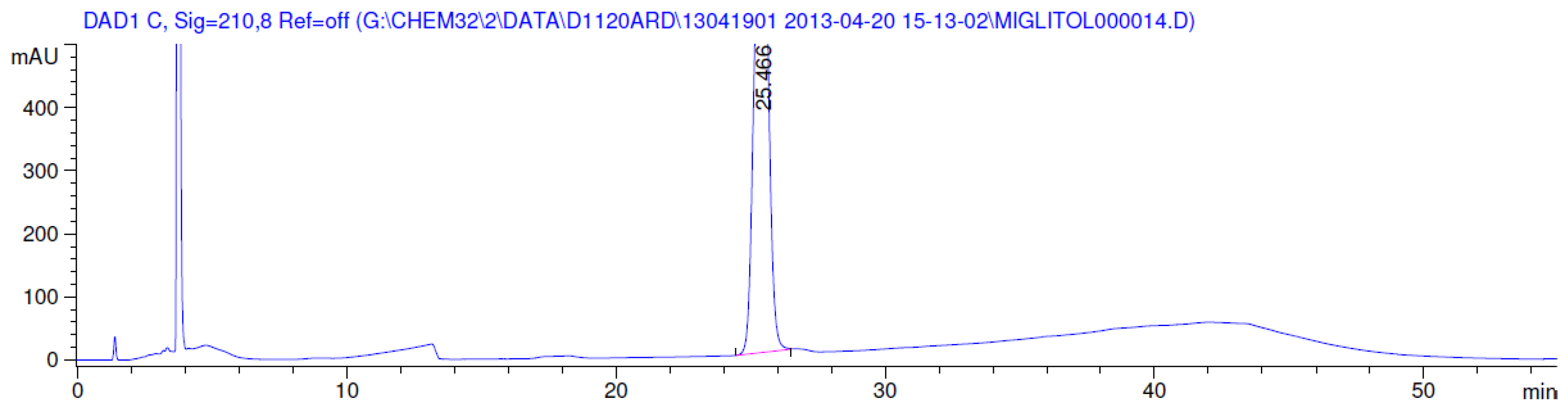
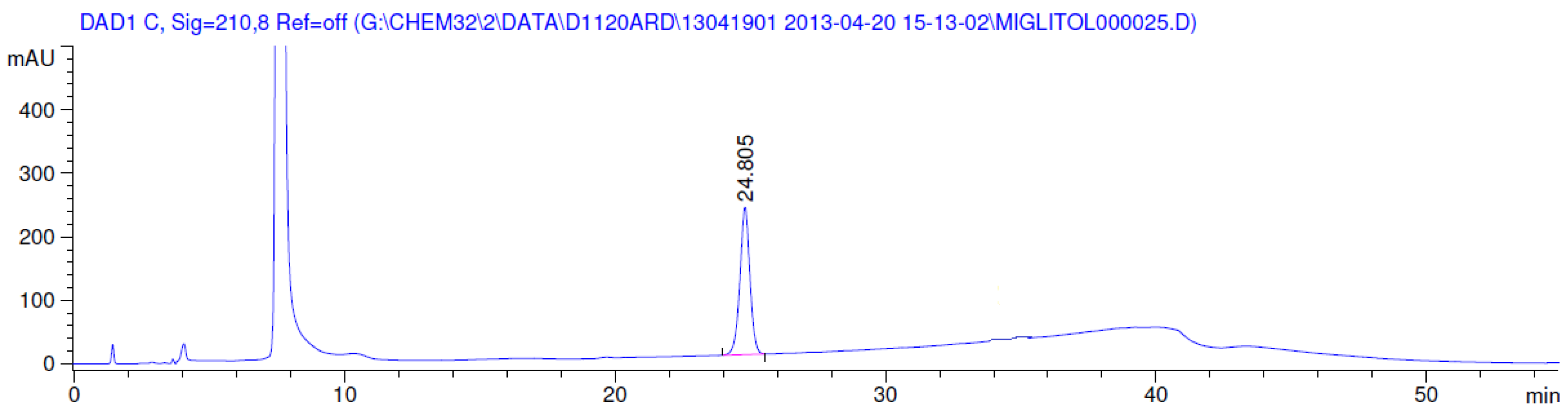
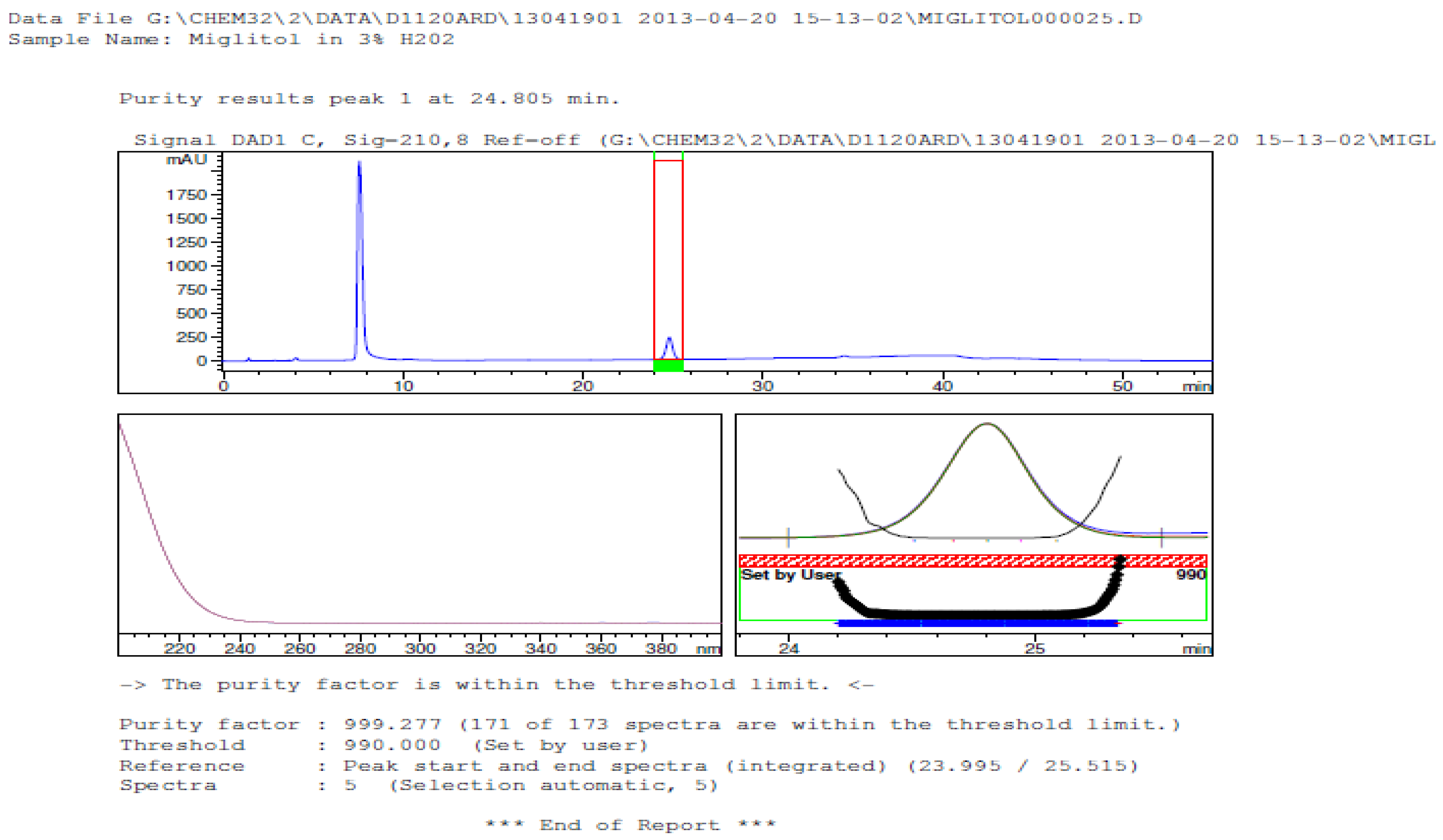
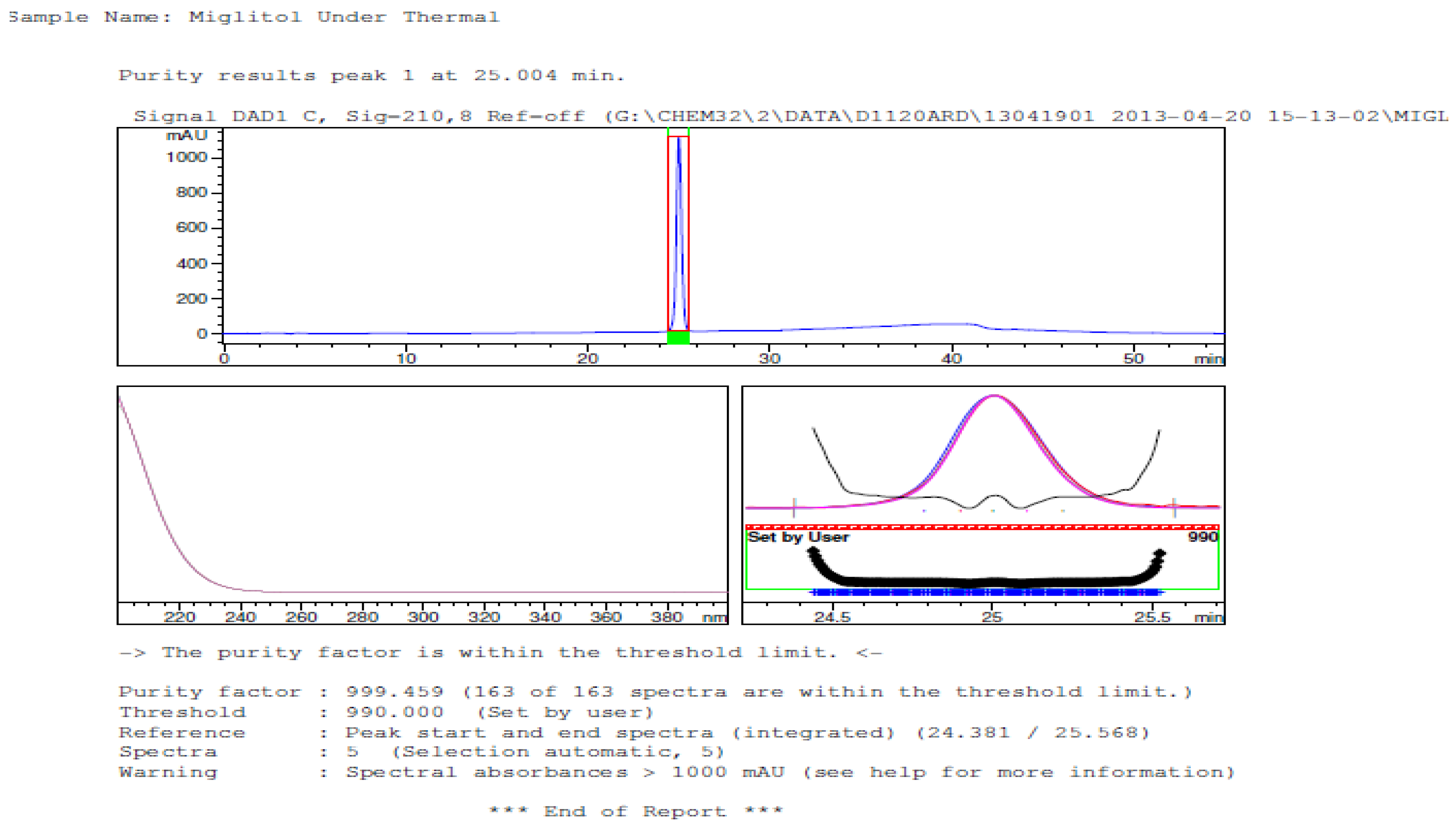
3.2. Results of Forced Degradation Studies
3.3. Precision
3.4. Limit of Detection and Limit of Quantification
3.5. Linearity
3.6. Accuracy
3.7. Solution Stability and Mobile Phase Stability
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aylward, G.W. Progressive changes in diabetics and their mangement. Eye (Lond.) 2005, 19, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Wild, S.; Roglic, G.; Green, A.; Sicree, R.; King, H. Global prevalance of diabetes; estimates for the year 2000 and projections for 2030. Diabetes Care 2004, 27, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Aronson, D. hyperglycemia and pathobiology of diabetic complications. Adv Cardiol. 2008, 45, 1–16. [Google Scholar] [PubMed]
- International Conference on Harmonization. Q3A (R2), Impurities in New Drug Substances, International Conference on Harmonization (ICH) Guidelines. 2006.
- Nageswara Rao, R.; Nagaraju, V. An over view of the recent trends in development of HPLC methods for determination of impurities in drugs. J. Pharm. Biomed. Anal. 2003, 33, 335–377. [Google Scholar] [CrossRef]
- Singh, S.; Handa, T.; Narayanam, M.; Sahu, A.; Junwal, M.; Shah, R.P. A critical review on the use of modern sophisticated hyphenated tools in the characterization of impurities and degradation products. J. Pharm. Biomed. Anal. 2012, 69, 148–173. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.N.; Granner, D.K. Insulin, Oral hypoglycemic agents and the pharmacology of endocrine Pancreas. In Goodman and Gilman’s the Pharmacological Basis of Therapeutics, 10th ed.; Hardman, J.G., Limbird, L.E., Gilman, A.G., Eds.; MacGraw-Hill Companies Inc.: New York, NY, USA, 2001; pp. 1679–1714. [Google Scholar]
- Asamoto, H.; Nobushi, Y.; Oi, T.; Uchikura, K. Determination of Miglitol by Column-Switching Ion-Pair HPLC with Tris(2,2′-bipyridine)ruthenium(II)-Electrogenerated Chemiluminescence Detection. Chem. Pharm. Bull. (Tokyo) 2015, 63, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Lukram, O.; Dwivedi, A. Ultra-performance liquid chromatography electrospray ionization–tandem mass spectrometry method for the estimation of miglitol in human plasma using metformin as the internal standard. Drug Test. Anal. 2011, 3, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Cahours, X.; Daali, Y.; Cherkaoui, S.; Veuthey, J.L. Simultaneous analysis of polyhydroxylated alkaloids by capillary electrophoresis using borate complexation and evaluation of sweeping technique for sensitivity improvement. Chromatographia 2002, 55, 211–216. [Google Scholar] [CrossRef]
- Ibrahim, F.A.; Ali, F.A. Kinetic determination of acarbose and miglitol in bulk and pharmaceutical formulations using alkaline potassium permanganate. Int. J. Biomed. Sci. 2007, 3, 20–30. [Google Scholar] [PubMed]
- Li, X.; Wang, Y.; Wang, J.; Fawcett, J.P.; Zhao, L.; Gu, J. Determination of miglitol in human plasma by liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Nirogi, R.V.; Kandikere, V.N.; Shukla, M.; Mudigonda, K.; Maurya, S.; Boosi, R.K.; Yerramilli, A. Liquid chromatographic tandem mass spectrometry method for the quantification of miglitol in human plasma. Arzneimittelforschung 2006, 56, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Jin, X.Z.; Xiu-mei, D. Determination of miglitol in human plasma by HPLC-MS/ESI. Chin. Pharm. J. 2005, 40, 51–53. [Google Scholar]
- Chittora, N.C.; Shrivastava, A.; Jain, A. New RP-HPLC Method of Miglitol in Tablet Dosage form Including Forced Degradation Studies and Estimation in Spiked Rabbit Plasma. J. Young Pharm. 2009, 1, 364–370. [Google Scholar] [CrossRef]
- Snyder, L.R.; Glajch, J.L.; Kirkland, J.J. Practical HPLC Method Development, 2nd ed.; Wiley Interscience: New York, NY, USA, 1997. [Google Scholar]
- O’Neil, M.J. The Merck Index: An Encyclopedia of Chemicals, Drugs and Biologicals, 14th ed.; Merck Research Lab.: Whitehouse Station, NJ, USA, 2006; p. 1067. [Google Scholar]
- International Conference on Harmonization. Q2 (R1), Validation of Analytical Procedures: Text and Methodology, ICH Guidelines. 2005.
- International Conference on Harmonization. Q1B, Photostability Testing of New Drug Substances and Products, ICH Guidelines. 1996.
- The United States Pharmacopoeia 39 and National Formulary 34, Asian ed.; United States Pharmacopoeia Convection: Rockville, MD, USA, 2016.
- World Health Organization (WHO). Fact Sheet 275 [EB/OL]. 2003–09, 2016.
- Health Ministry’s Disease Control Division, Chinese Medical Association Diabetes Society. China Guideline for Diabetes; Peking University Press: Beijing, China, 2015. [Google Scholar]
- Council of Europe. European Pharmacopoeia (Ph. Eur.), 8th ed.; Council of Europe: Strasbourg, France, 2015. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Miglitol | Imp A | Imp B | Imp C | Imp D | Imp E | Imp F |
|---|---|---|---|---|---|---|---|
| LOD % (w/w) w.r.t analyte concentration | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 |
| LOQ % (w/w) w.r.t analyte concentration | 0.05 | 0.05 | 0.05 | 0.05 | 0.05 | 0.05 | 0.05 |
| Slope | 385.7 | 540.08 | 365.7 | 305.3 | 676.3 | 612.5 | 164.9 |
| Intercept | −0.271 | 0.833 | 0.875 | 0.121 | 0.124 | −2.669 | −1.22 |
| Correlation coefficient | 0.999 | 0.999 | 1.000 | 0.998 | 0.999 | 0.999 | 0.999 |
| Method precision (% RSD) | 5.21 | 3.54 | 1.76 | 3.44 | 1.49 | 5.28 | 6.91 |
| Intermediate precision (% RSD) | 2.28 | 0.87 | 0.54 | 2.62 | 4.90 | 3.56 | 3.43 |
| Name of the Compound | Structure | Chemical Name | Emprical Formula |
|---|---|---|---|
| Miglitol |  | (2R,3R,4R,5S)-1-(2-hydroxyethyl)-2-(hydroxymethyl)piperidine-3,4,5-triol | C8H17NO5 |
| Impurity A (Dialkylated Miglitol) | 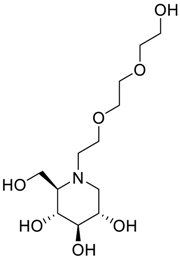 | (2R,3R,4R,5S)-1-(2-(2-(2-hydroxyethoxy)ethoxy)ethyl)-2-(hydroxymethyl)piperidine-3,4,5-triol | C12H25NO7 |
| Impurity B (Ido) | 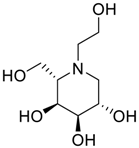 | (2S,3S,4R,5S)-1-(2-hydroxyethyl)-2-(hydroxymethyl)piperidine-3,4,5-triol | C8H17NO5 |
| Impurity C (Monoalkyl Miglitol) |  | (2R,3R,4R,5S)-1-(2-(2-hydroxyethoxy)ethyl)-2-(hydroxymethyl)piperidine-3,4,5-triol | C10H21NO6 |
| Impurity D (Taro) | 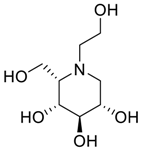 | (2S,3R,4R,5S)-1-(2-hydroxyethyl)-2-(hydroxymethyl)piperidine-3,4,5-triol | C8H17NO5 |
| Impurity E (Galacto) |  | (2R,3S,4R,5S)-1-(2-hydroxyethyl)-2-(hydroxymethyl)piperidine-3,4,5-triol | C8H17NO5 |
| Impurity F (Deoxynirijomycin) |  | (2R,3R,4R,5S)-2-(hydroxymethyl)piperidine-3,4,5-triol | C6H13NO4 |
| Miglitol N-oxide | 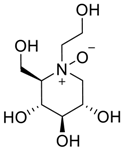 | -- | C8H17NO6 |
| LCMS-MS/MS | |||
|---|---|---|---|
| [M + H]+ | Collision energy (eV) | Fragmentation pattern (m/z) | |
| Miglitol | 208.2 | 20 | 190.10,172.10,154.10,146.10,74.10 |
| 50 | 140.10,96.10,80.10,56.20 | ||
| Imp-A | 296.2 | 20 | 146.10,128.10,110.10,102.10,69.20,60.20 |
| Imp-B | 208.2 | 20 | 190.10,172.10,154.10,146.10,74.10 |
| 50 | 140.10,96.10,80.10,56.20 | ||
| Imp-C | 252.2 | 20 | 234.10,190.10,172.10,118.10,86.10,58.20 |
| Imp-D | 208.2 | 20 | 190.10,172.10,154.10,146.10,74.10 |
| 50 | 140.10,94.10,80.10,56.20 | ||
| Imp-E | 208.0 | 20 | 190.10,172.10,154.10,146.10,74.10 |
| 50 | 140.10,94.10,80.10,56.20 | ||
| Imp-F | 164.2 | 20 | 146.10,128.10,110.10,69.20,60.20 |
| MIG N-Oxide | 224.2 | 20 | 176.10,146.10,74.20 |
| Name of Compound | HRMS Data | |||
|---|---|---|---|---|
| [M + H]+ | ppm | Double Bond Equivalance | Elemental Composition | |
| Miglitol | 208.1190 | 2.4 | 0.5 | C8H18NO5 |
| Imp-A | 296.1696 | −4.4 | 0.5 | C12H26NO7 |
| Imp-B | 208.1185 | −1.9 | 0.5 | C8H18NO5 |
| Imp-C | 252.1435 | −4.8 | 0.5 | C10H22NO6 |
| Imp-D | 208.1177 | −3.8 | 0.5 | C8H18NO5 |
| Imp-E | 208.1189 | 1.9 | 0.5 | C8H18NO5 |
| Imp-F | 164.0919 | −2.4 | 0.5 | C6H14NO4 |
| MIG N-Oxide | 224.1129 | −2.2 | 0.5 | C8H18NO6 |
| Stress condition | Duration | Purity of miglitol after forced degradation (%) | Content of major degradant (%) | Remarks |
|---|---|---|---|---|
| Acid hydrolysis | 10 days | 100 | - | No degradation products formed |
| Base hydrolysis | 10 days | 100 | - | No degradation products formed |
| Oxidation | 1 h | 0.0 | 100 | Significant degradation product formed |
| Thermal (105 °C) | 10 days | 100 | - | No degradation products formed |
| Photolytic as per ICH | 11 days | 100 | - | No degradation products formed |
| Compound | Level | Concentration (% w/w) | Recovery in % | |
|---|---|---|---|---|
| Individual | Mean | |||
| Imp-A | LOQ | 0.05 | 99.2 | 95.65 |
| 50% | 0.075 | 96.3 | ||
| 100% | 0.15 | 97.9 | ||
| 150% | 0.225 | 89.2 | ||
| Imp-B | LOQ | 0.05 | 100.2 | 96.03 |
| 50% | 0.075 | 97.6 | ||
| 100% | 0.15 | 97.5 | ||
| 150% | 0.225 | 88.8 | ||
| Imp-C | LOQ | 0.05 | 101.3 | 99.95 |
| 50% | 0.075 | 101.7 | ||
| 100% | 0.15 | 100.9 | ||
| 150% | 0.225 | 95.9 | ||
| Imp-D | LOQ | 0.05 | 93.0 | 97.58 |
| 50% | 0.075 | 99.1 | ||
| 100% | 0.15 | 102.0 | ||
| 150% | 0.225 | 96.2 | ||
| Imp-E | LOQ | 0.05 | 94.0 | 96.63 |
| 50% | 0.075 | 94.5 | ||
| 100% | 0.15 | 103.1 | ||
| 150% | 0.225 | 94.9 | ||
| Imp-F | LOQ | 0.05 | 91.0 | 95.25 |
| 50% | 0.075 | 99.3 | ||
| 100% | 0.15 | 103.1 | ||
| 150% | 0.225 | 87.6 | ||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balakumaran, K.; Janagili, M.; Rajana, N.; Papureddy, S.; Anireddy, J. Development and Validation of Miglitol and Its Impurities by RP-HPLC and Characterization Using Mass Spectrometry Techniques. Sci. Pharm. 2016, 84, 654-670. https://doi.org/10.3390/scipharm84040654
Balakumaran K, Janagili M, Rajana N, Papureddy S, Anireddy J. Development and Validation of Miglitol and Its Impurities by RP-HPLC and Characterization Using Mass Spectrometry Techniques. Scientia Pharmaceutica. 2016; 84(4):654-670. https://doi.org/10.3390/scipharm84040654
Chicago/Turabian StyleBalakumaran, Kesavan, Mosesbabu Janagili, Nagaraju Rajana, Sureshbabu Papureddy, and Jayashree Anireddy. 2016. "Development and Validation of Miglitol and Its Impurities by RP-HPLC and Characterization Using Mass Spectrometry Techniques" Scientia Pharmaceutica 84, no. 4: 654-670. https://doi.org/10.3390/scipharm84040654
APA StyleBalakumaran, K., Janagili, M., Rajana, N., Papureddy, S., & Anireddy, J. (2016). Development and Validation of Miglitol and Its Impurities by RP-HPLC and Characterization Using Mass Spectrometry Techniques. Scientia Pharmaceutica, 84(4), 654-670. https://doi.org/10.3390/scipharm84040654