
Interaction of Positively Charged Oligopeptides with Blood Plasma Proteins

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Interactions with Albumin, α1-Acid Glycoprotein, and γ-Globulin—Spectroscopic Studies
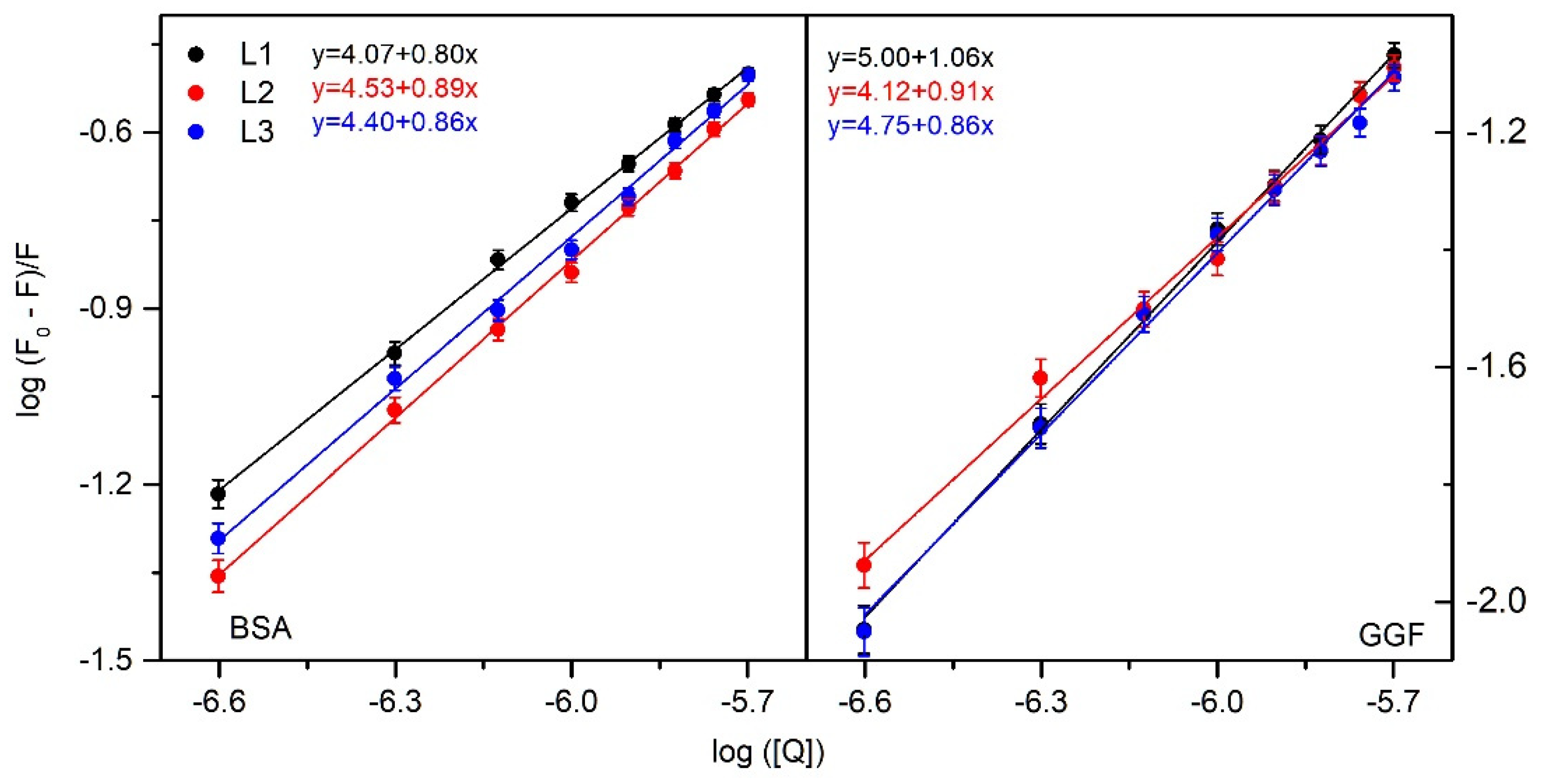
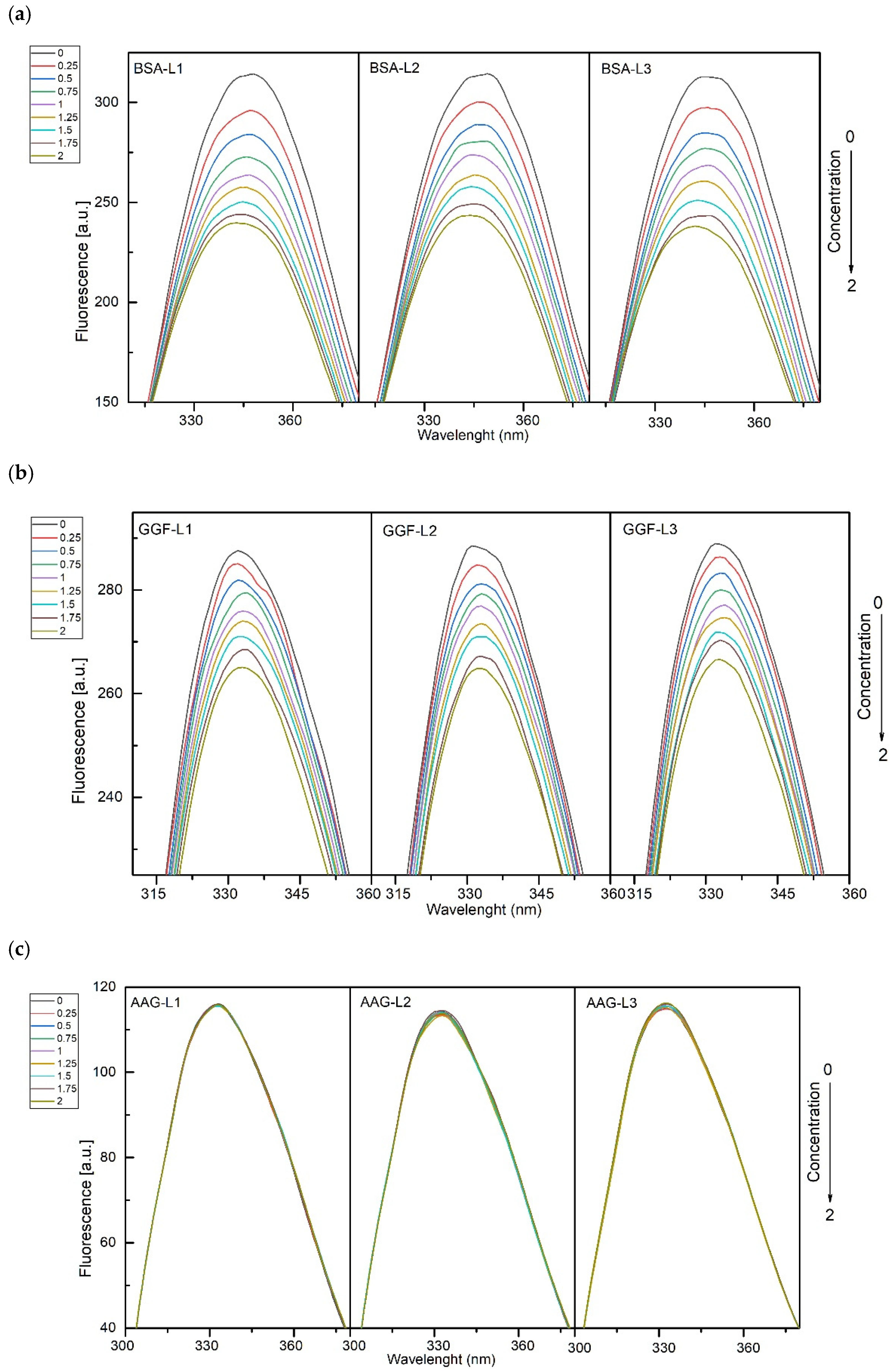
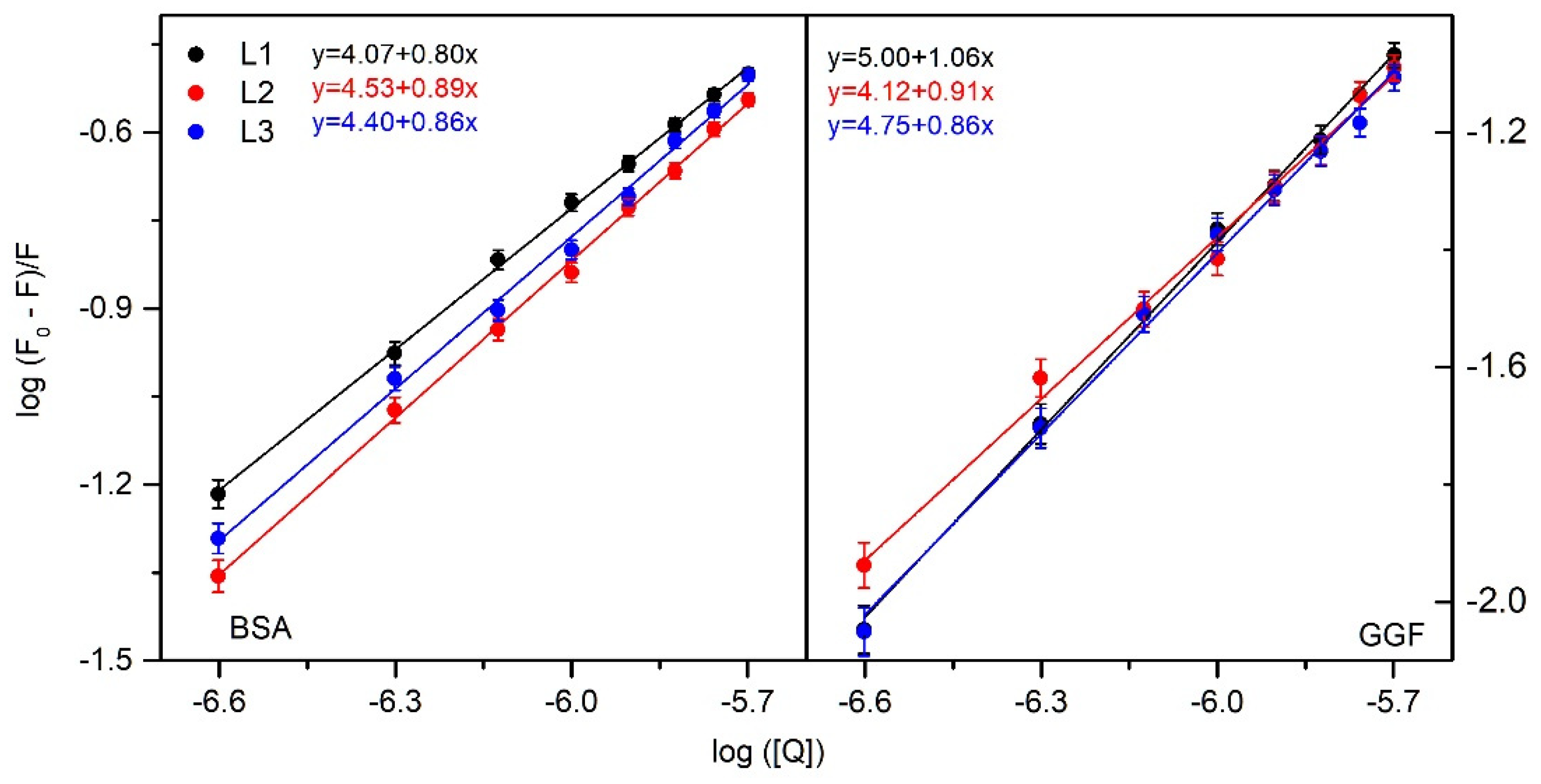
2.1.1. Fluorescence Spectroscopy
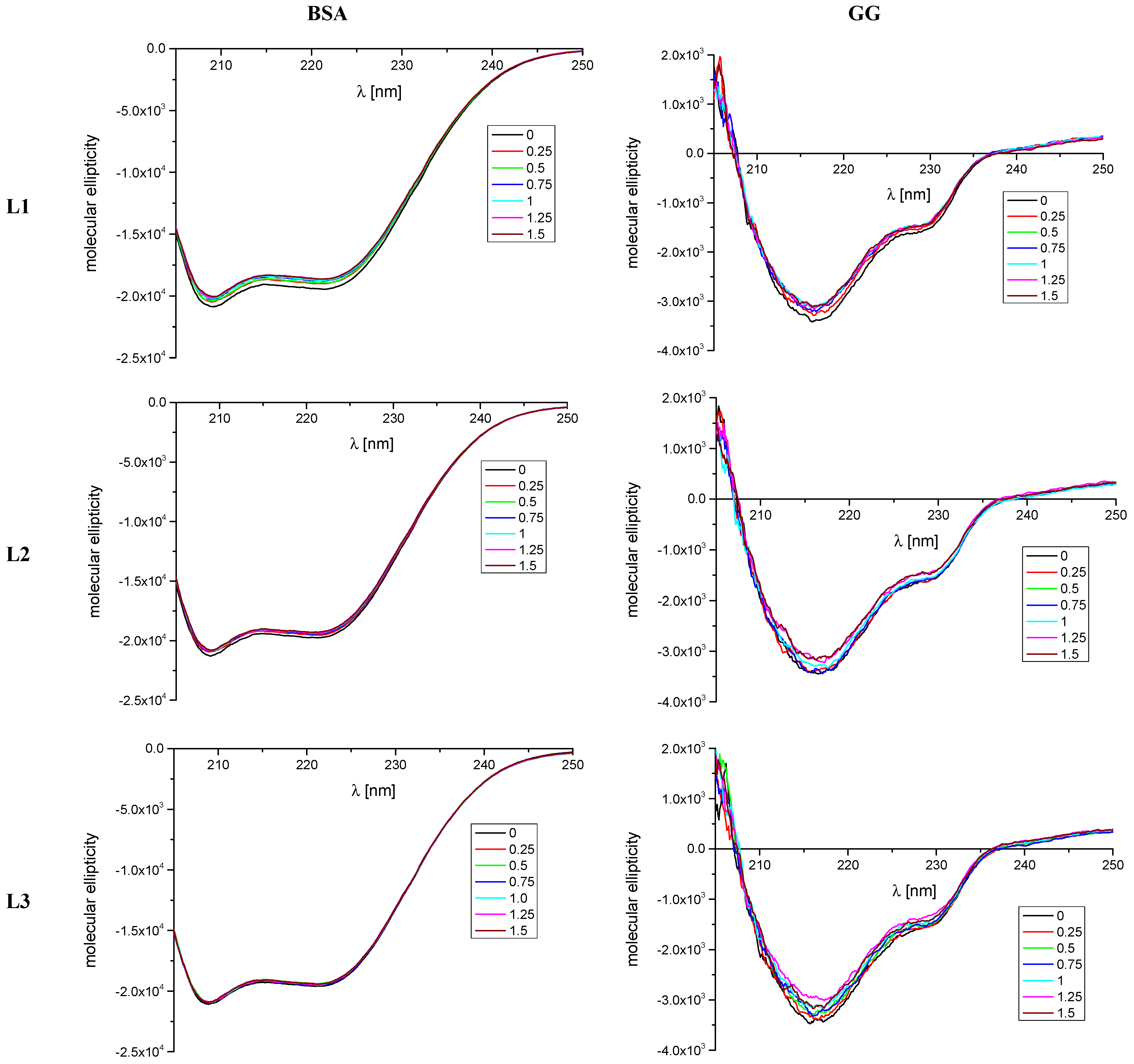
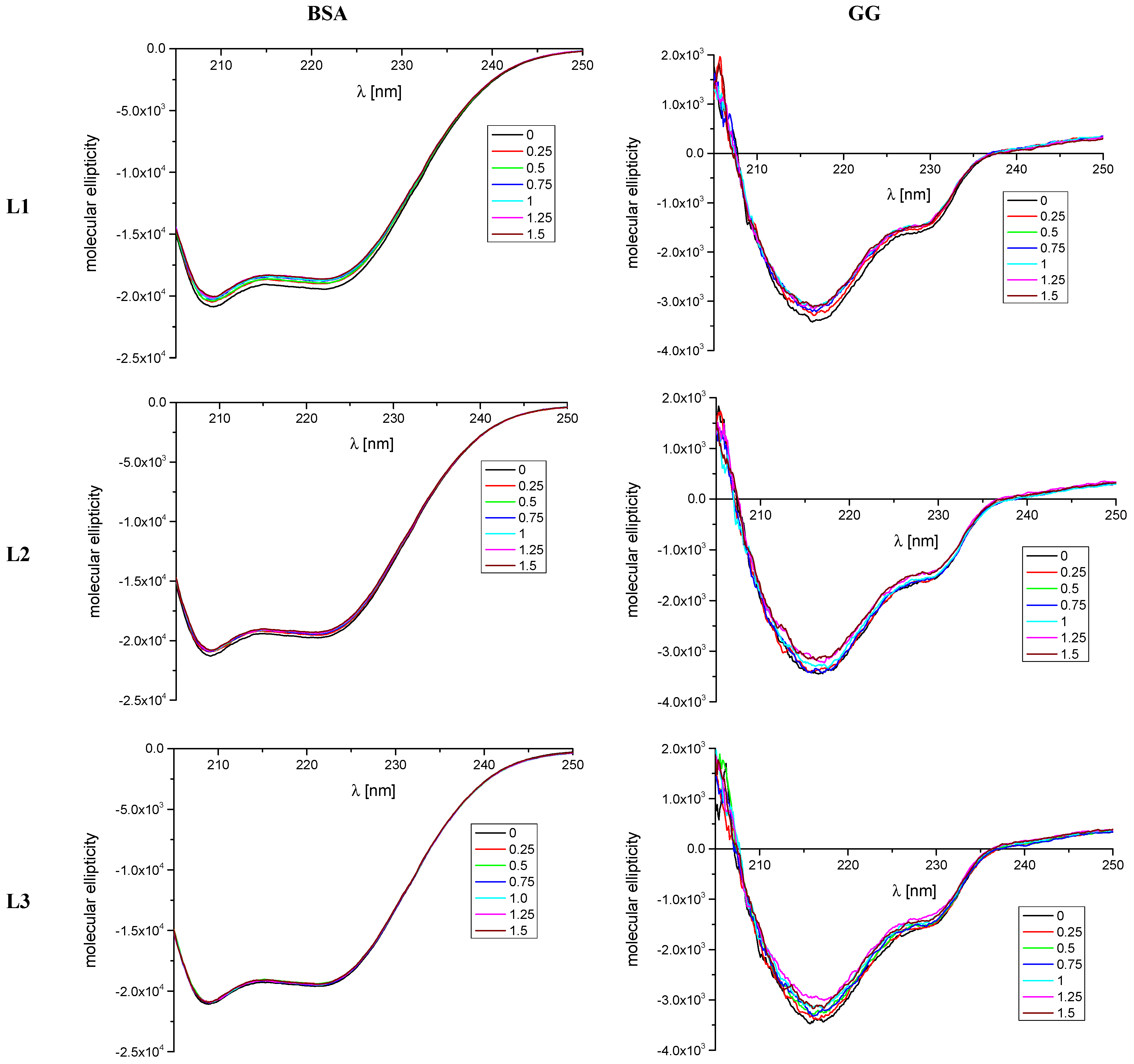
2.1.2. CD Spectroscopy
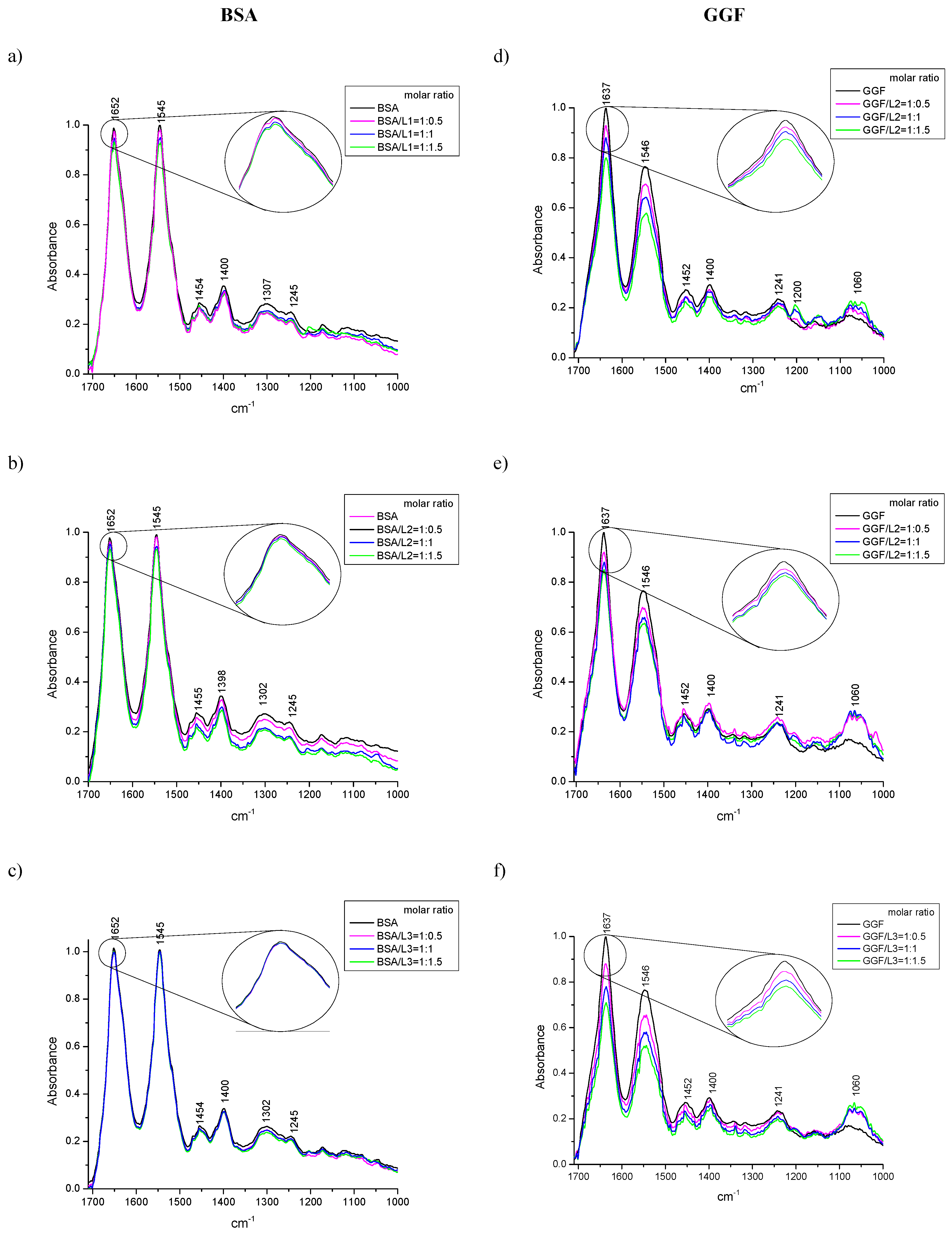
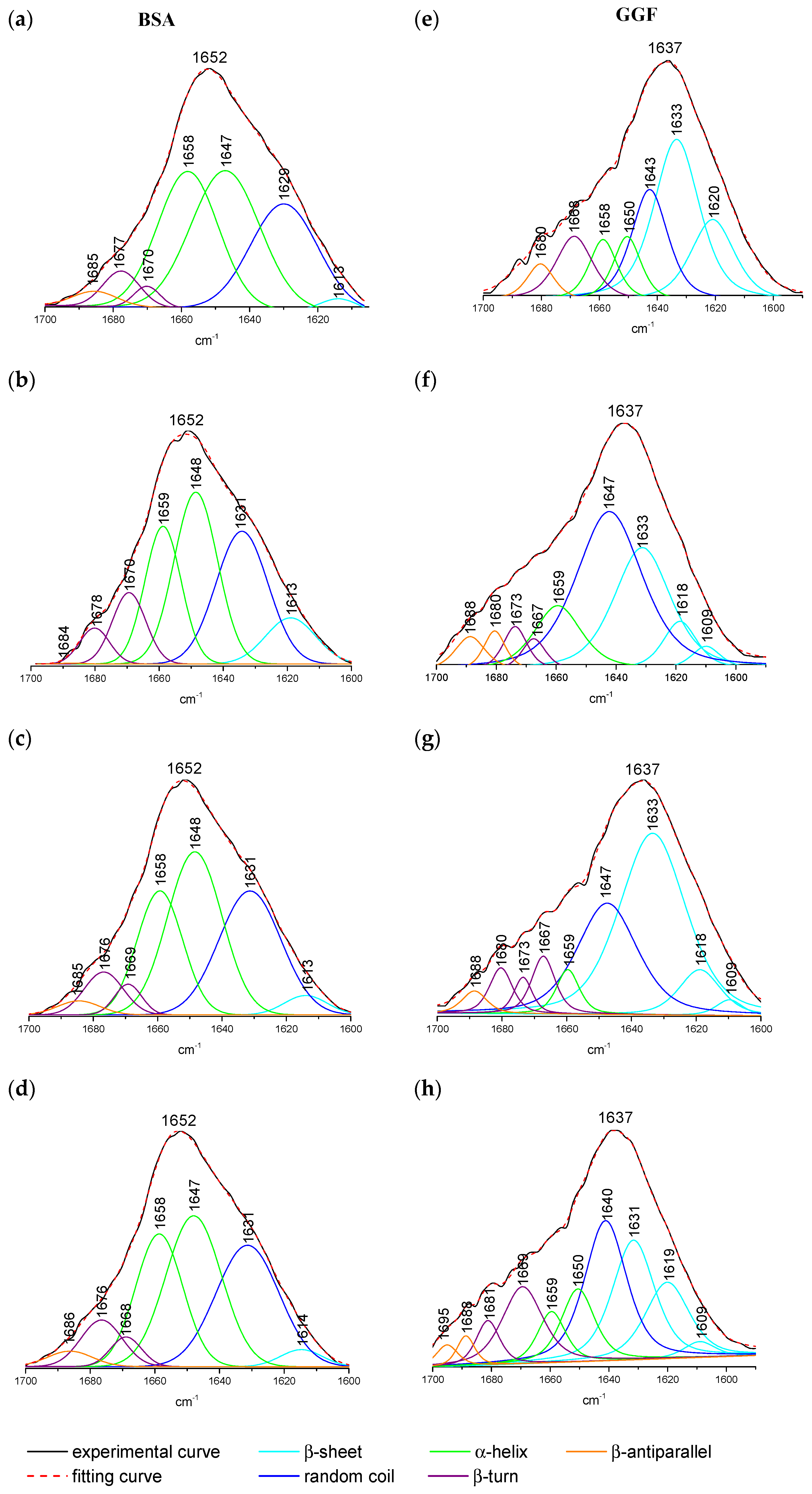
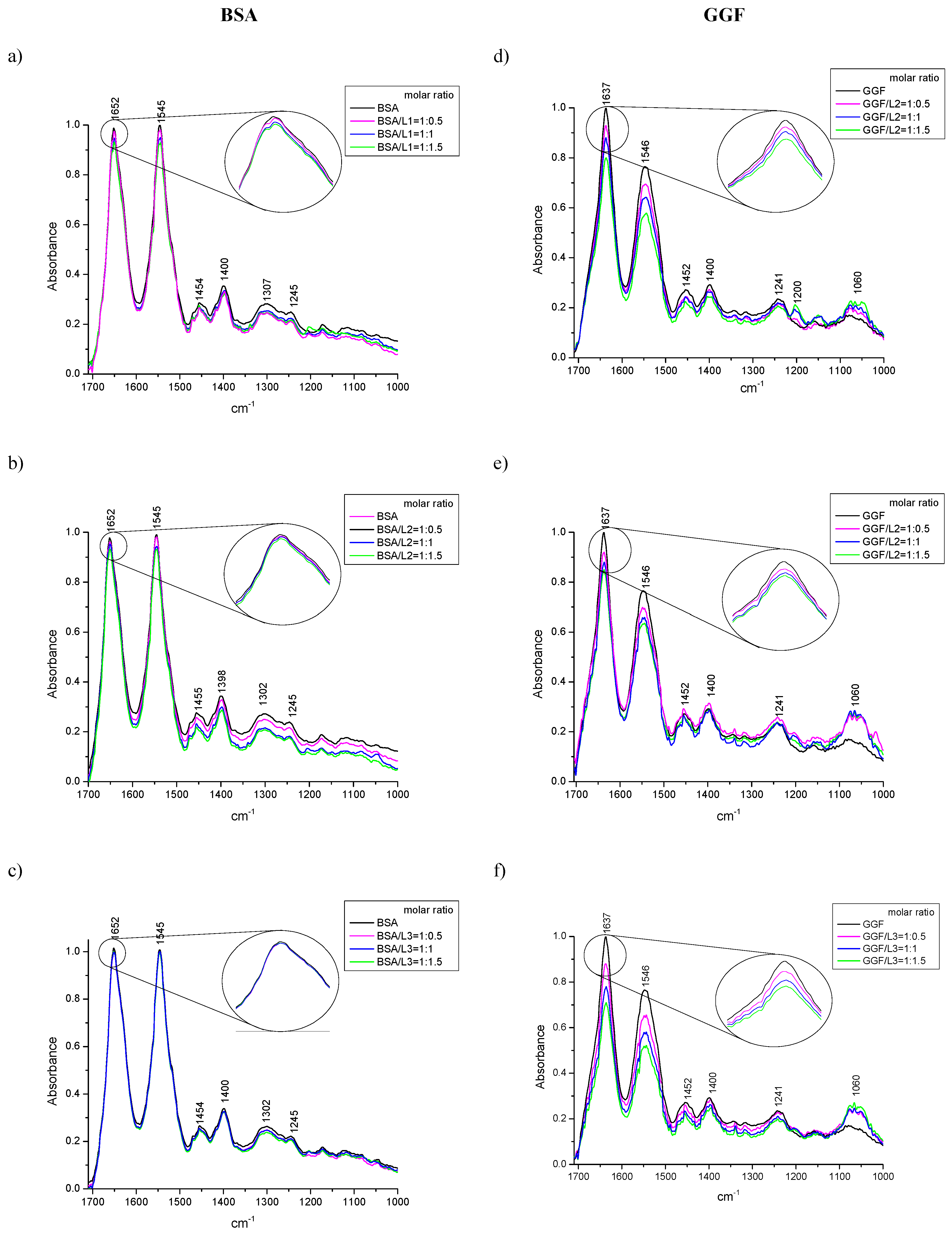
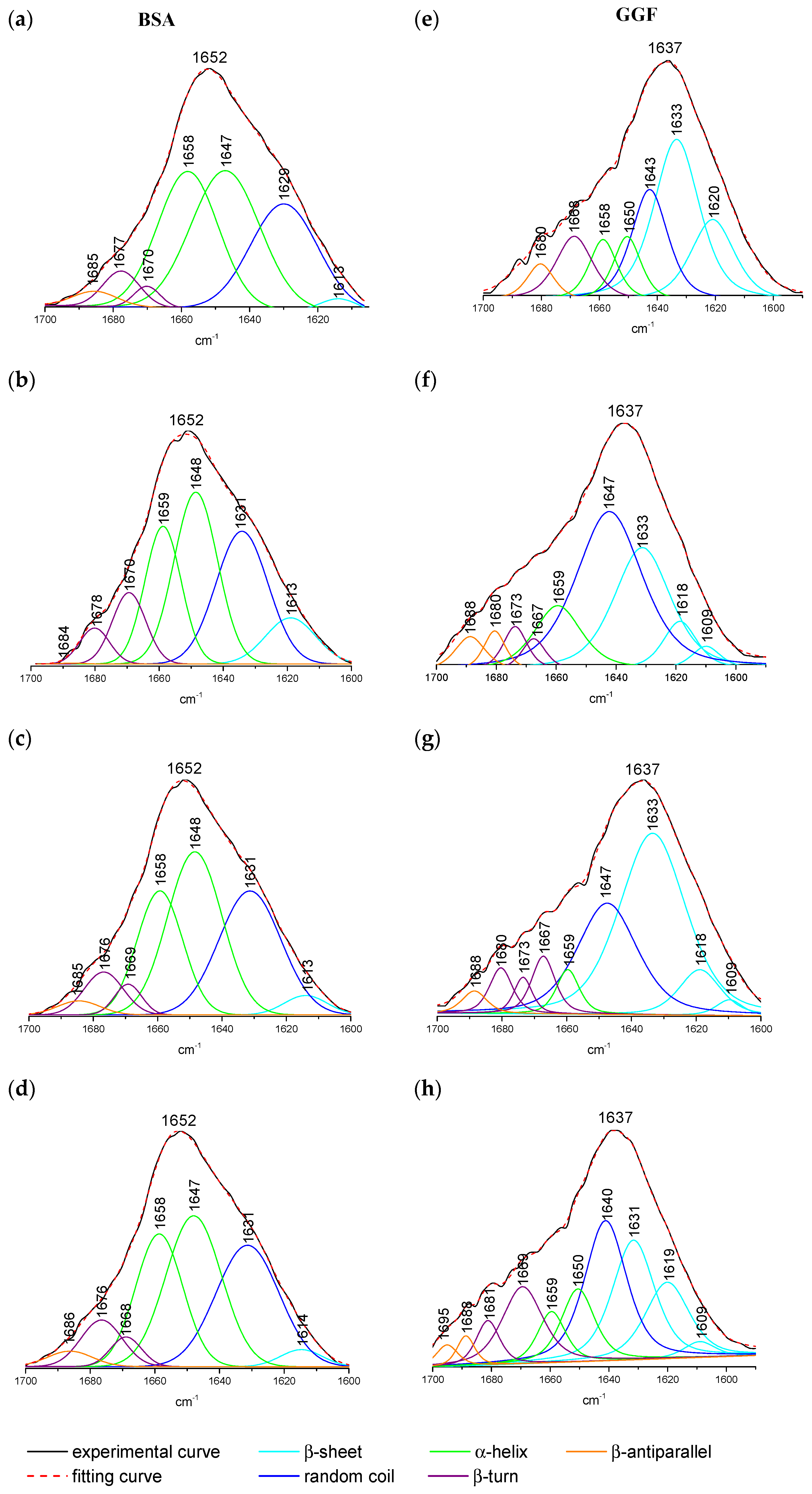
2.1.3. ATR-FTIR Spectroscopy
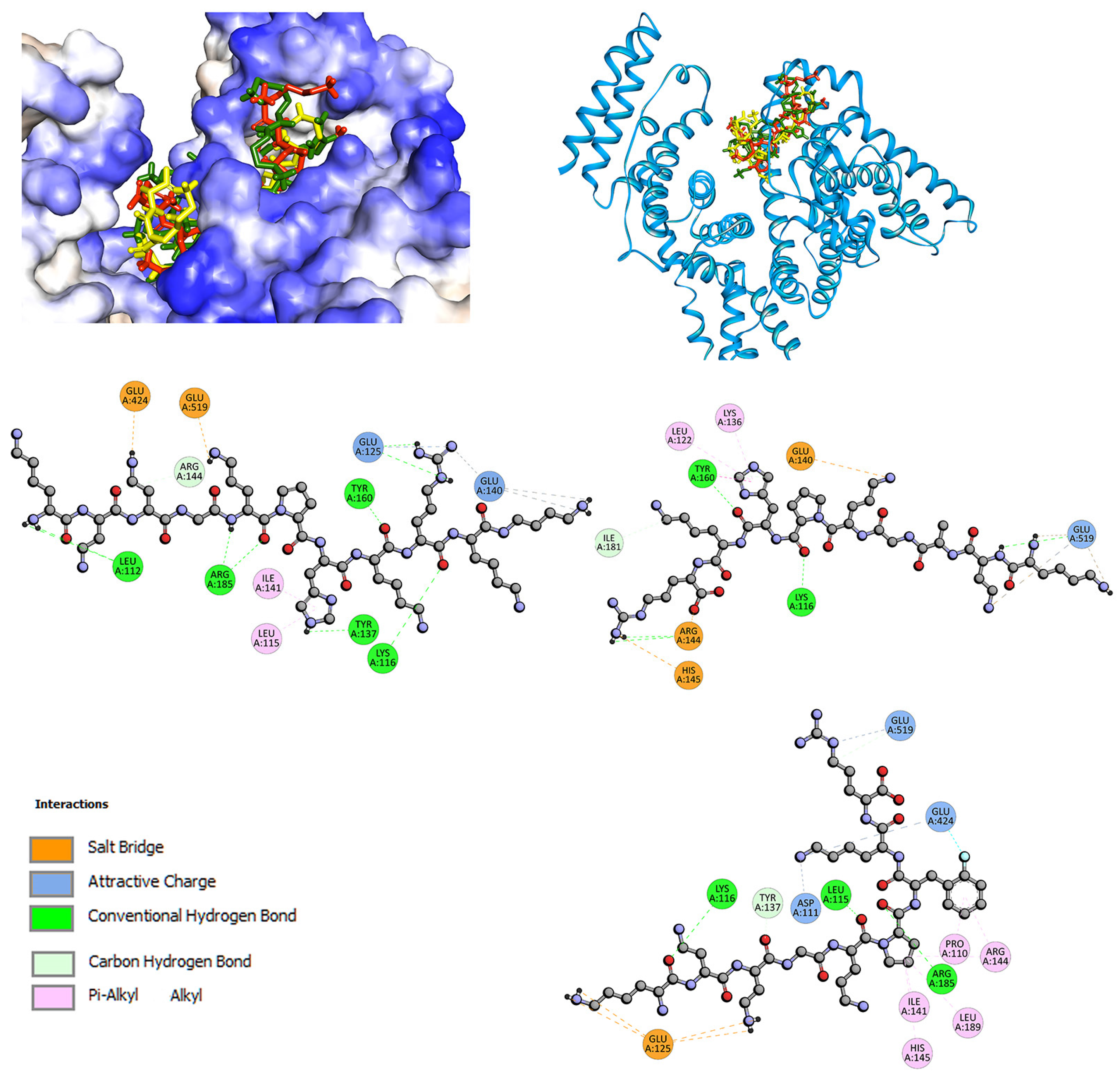
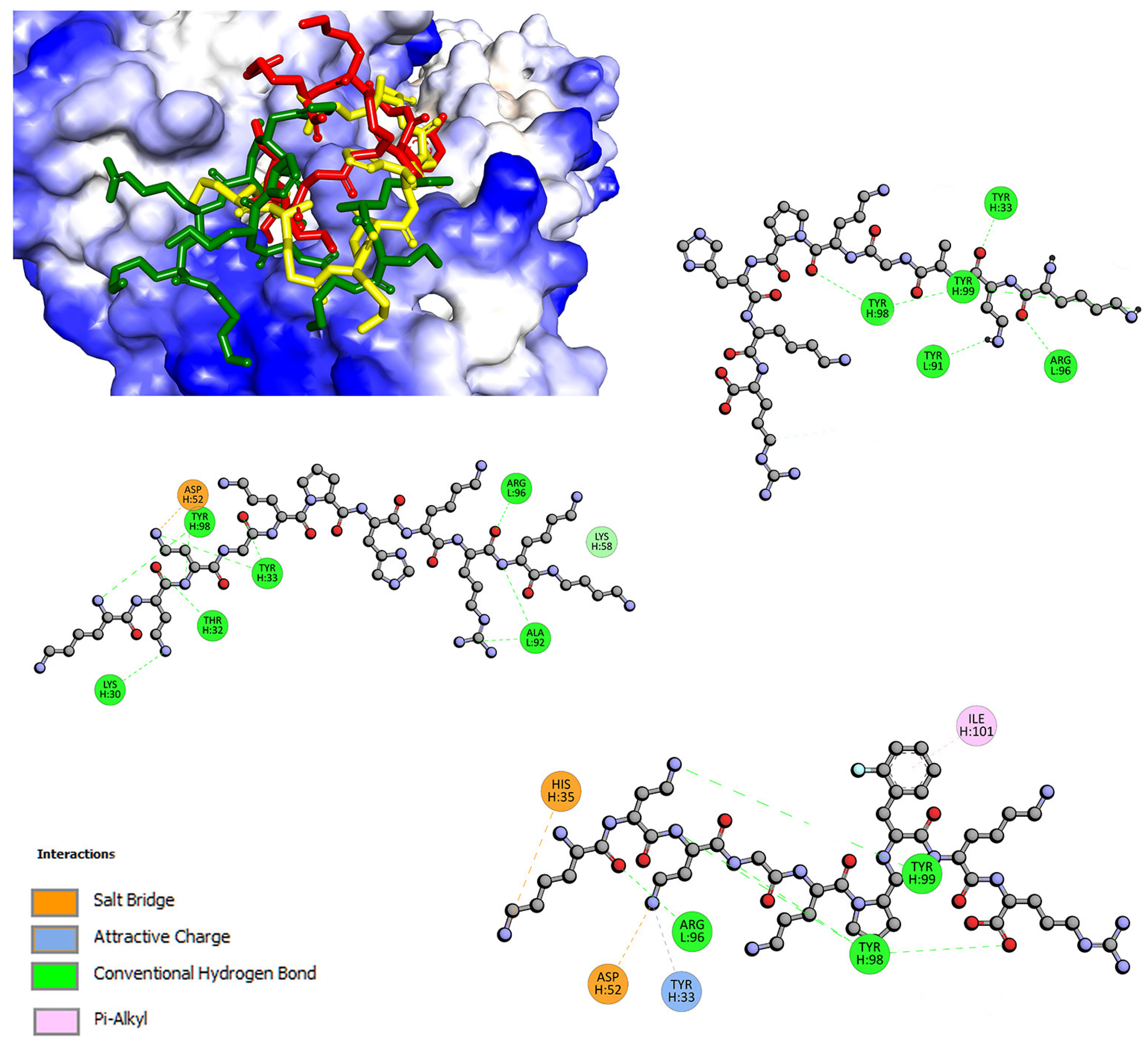
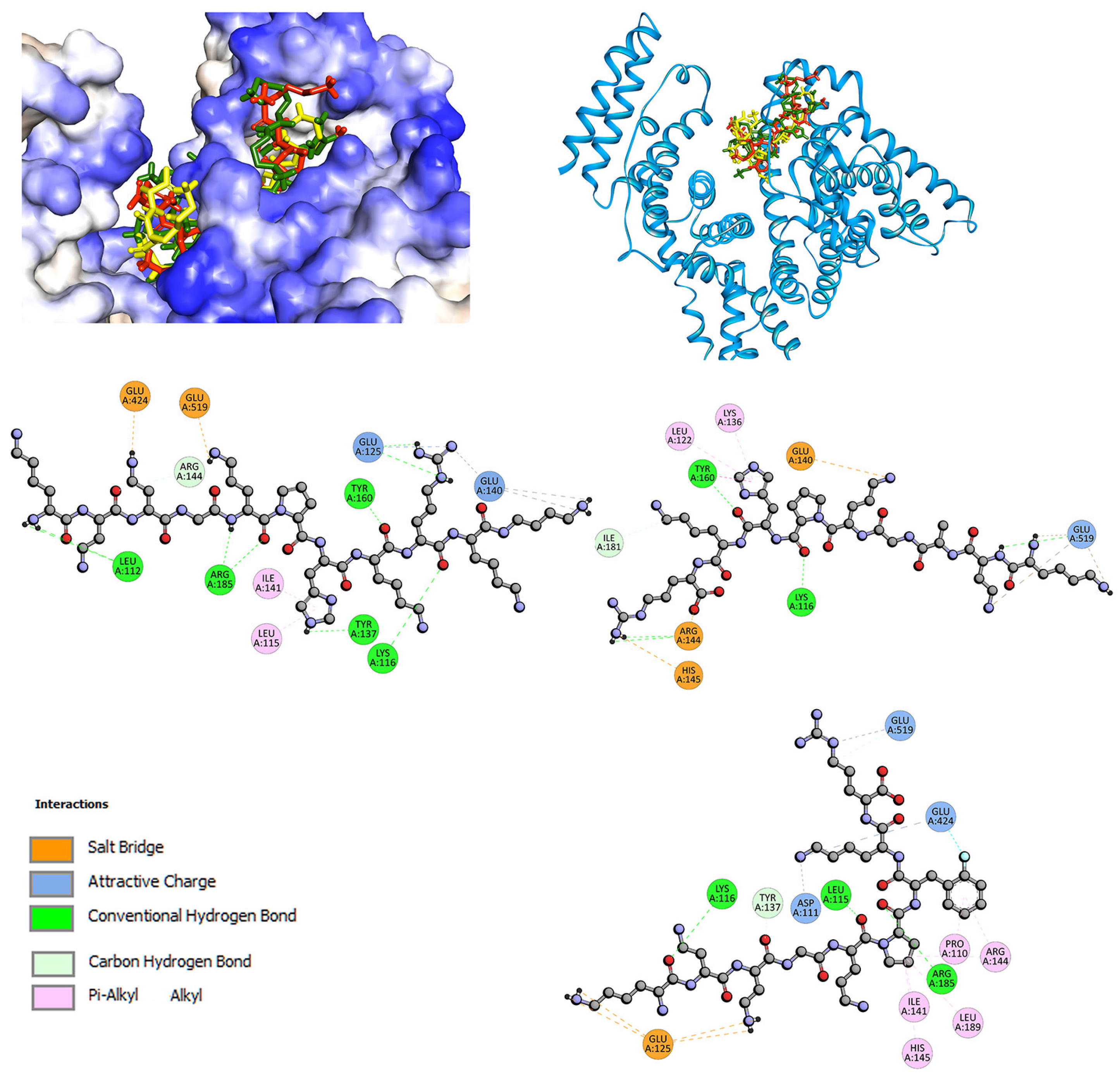
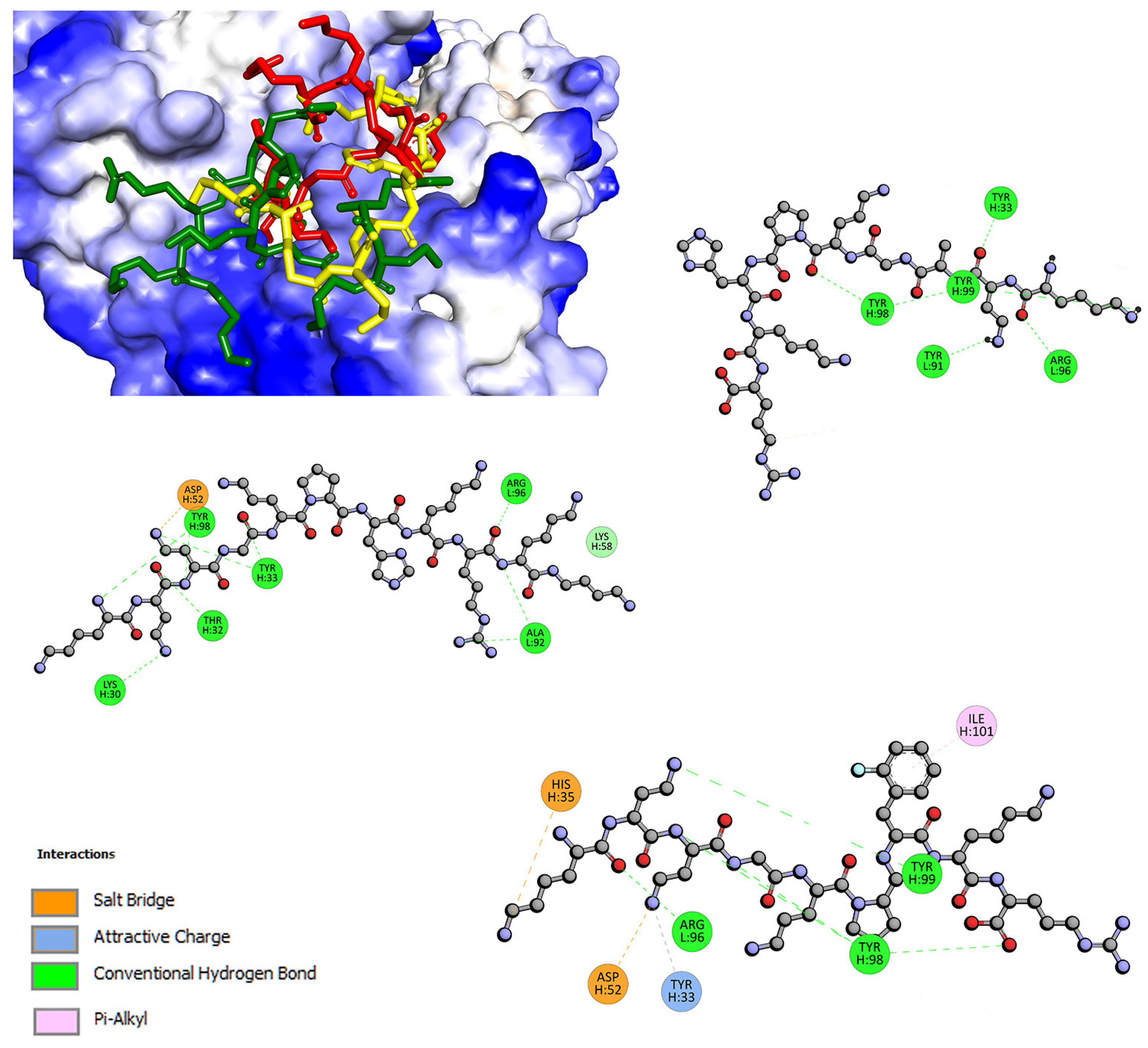
2.2. Interactions with Albumin and γ-Globulin—Molecular Docking Studies
3. Materials and Methods
3.1. Fluorescence Spectroscopy
3.2. CD Spectroscopy
3.3. ATR-FTIR Spectroscopy
3.4. Molecular Docking
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Banting, F.G.; Best, C.H.; Collip, J.B.; Campbell, W.R.; Fletcher, A.A. Pancreatic extracts in the treatment of diabetes mellitus. Can. Med. Assoc. J. 1922, 12, 141–146. [Google Scholar] [PubMed]
- Garber, K. Peptide leads new class of chronic pain drugs. Nat. Biotechnol. 2005, 23, 399. [Google Scholar] [CrossRef] [PubMed]
- Pernot, M.; Vanderesse, R.; Frochot, C.; Guillemin, F.; Barberi-Heyob, M. Stability of peptides and therapeutic success in cancer. Expert Opin. Drug Metab. Toxicol. 2011, 7, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Quiñones-Mateu, M.E.; Schols, D. Virus-inhibitory peptide: A natural HIV entry inhibitor in search for a formal target in the viral genome. AIDS 2011, 25, 1663–1664. [Google Scholar] [CrossRef] [PubMed]
- Rima, M.; Rima, M.; Fajloun, Z.; Sabatier, J.-M.; Bechinger, B.; Naas, T. Antimicrobial Peptides: A Potent Alternative to Antibiotics. Antibiotics 2021, 10, 1095. [Google Scholar] [CrossRef]
- Del Gatto, A.; Saviano, M.; Zaccaro, L. An Overview of Peptide-Based Molecules as Potential Drug Candidates for Multiple Sclerosis. Molecules 2021, 26, 5227. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.K.; Viswanathan, M.; Kent, R.B.; Wood, C.R. Therapeutic peptides: Technological advances driving peptides into development. Curr. Opin. Biotechnol. 2006, 17, 638–642. [Google Scholar] [CrossRef]
- Latham, P.W. Therapeutic peptides revisited. Nat. Biotechnol. 1999, 17, 755–757. [Google Scholar] [CrossRef]
- Martínez-Núñez, M.A.; López, V.E.L.Y. Nonribosomal peptides synthetases and their applications in industry. Sustain. Chem. Process. 2016, 4, 13. [Google Scholar] [CrossRef]
- Le Govic, Y.; Papon, N.; Le Gal, S.; Bouchara, J.P.; Vandeputte, P. Non-ribosomal Peptide Synthetase Gene Clusters in the Human Pathogenic Fungus Scedosporium apiospermum. Front. Microbiol. 2019, 10, 2062. [Google Scholar] [CrossRef]
- Vazquez-Lombardi, R.; Phan, T.; Today, C.Z.-D. Challenges and opportunities for non-antibody scaffold drugs. Drug Discov. Today 2015, 20, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Kintzing, J.; Interrante, M.; Pharmacological, J.C.-T. Emerging strategies for developing next-generation protein therapeutics for cancer treatment. Trends Pharmacol. Sci. 2016, 37, 993–1008. [Google Scholar] [CrossRef] [PubMed]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef] [PubMed]
- Houston, M.E.; Campbell, A.P.; Lix, B.; Kay, C.M.; Sykes, B.D.; Hodges, R.S. Lactam Bridge Stabilization of R-Helices: The Role of Hydrophobicity in Controlling Dimeric versus Monomeric R-Helices. Biochemistry 1996, 35, 10041–10050. [Google Scholar] [CrossRef]
- Trabi, M.; Craik, D.J. Circular proteins—No end in sight. Trends Biochem. Sci. 2002, 27, 132–138. [Google Scholar] [CrossRef]
- Rozek, A.; Powers, J.P.S.; Friedrich, C.L.; Hancock, R.E.W. Structure-based design of an indolicidin peptide analogue with increased protease stability. Biochemistry 2003, 42, 14130–14138. [Google Scholar] [CrossRef] [PubMed]
- Kessler, H. Conformation and Biological Activity of Cyclic Peptides. Angew. Chemie Int. Ed. 1982, 21, 512–523. [Google Scholar] [CrossRef]
- Chatterjee, J.; Rechenmacher, F.; Kessler, H. N-Methylation of Peptides and Proteins: An Important Element for Modulating Biological Functions. Angew. Chemie Int. Ed. 2013, 52, 254–269. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.M.; Cabalteja, C.C.; Horne, W.S. Peptide Backbone Composition and Protease Susceptibility: Impact of Modification Type, Position, and Tandem Substitution. ChemBioChem 2016, 17, 712–718. [Google Scholar] [CrossRef]
- Gao, W.; Cho, E.; Liu, Y.; Lu, Y. Advances and challenges in cell-free incorporation of unnatural amino acids into proteins. Front. Pharmacol. 2019, 10, 611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velkov, T.; Roberts, K.D.; Nation, R.L.; Thompson, P.E.; Li, J. Pharmacology of polymyxins: New insights into an ’old class of antibiotics. Future Microbiol. 2013, 8, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Martini, F.; Welch, K.; Newsome, M. Fundamentals of Anatomy and Physiology: Applications Manual; Prentice Hall: Denver, CO, USA, 1998; ISBN 0137518684. [Google Scholar]
- Sharma, A.; Ghosh, K.S.; Singh, B.P.; Gathania, A.K. Spectroscopic investigation of interaction between bovine gamma globulin and gold nanoparticles. Methods Appl. Fluoresc. 2015, 3, 25002. [Google Scholar] [CrossRef] [PubMed]
- Barrett, K.E.; Brooks, H.; Boitano, S.B.S. Review of Medical Phsiology Twenty; A LANGE medical book; McGraw Hill: New York, NY, USA, 2010; ISBN 978-0-07-160568-7. [Google Scholar]
- He, X.M.; Carter, D.C. Atomic structure and chemistry of human serum albumin. Nature 1992, 358, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Sugio, S.; Kashima, A.; Mochizuki, S.; Noda, M.; Kobayashi, K. Crystal structure of human serum albumin at 2.5 A resolution. Protein Eng. 1999, 12, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.X.; Kim, H.Y.; Dass, C. Probing three-dimensional structure of bovine serum albumin by chemical cross-linking and mass spectrometry. J. Am. Soc. Mass Spectrom. 2004, 15, 1237–1247. [Google Scholar] [CrossRef]
- Ketrat, S.; Japrung, D.; Pongprayoon, P. Exploring how structural and dynamic properties of bovine and canine serum albumins differ from human serum albumin. J. Mol. Graph. Model. 2020, 98, 107601. [Google Scholar] [CrossRef]
- Bujacz, A. Structures of bovine, equine and leporine serum albumin. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 1278–1289. [Google Scholar] [CrossRef]
- Neault, J.F.; Tajmir-Riahi, H.A. Interaction of cisplatin with human serum albumin. Drug binding mode and protein secondary structure. Biochim. Biophys. Acta—Protein Struct. Mol. Enzymol. 1998, 1384, 153–159. [Google Scholar] [CrossRef]
- Tajmir-Riahi, H.A. An Overview of Drug Binding to Human Serum Albumin: Protein Folding and Unfolding. Sci. Iran. 2007, 14, 87–95. [Google Scholar]
- Jiao, Q.; Wang, R.; Jiang, Y.; Liu, B. Study on the interaction between active components from traditional Chinese medicine and plasma proteins. Chem. Cent. J. 2018, 12, 48. [Google Scholar] [CrossRef] [PubMed]
- Hadichegeni, S.; Goliaei, B.; Taghizadeh, M.; Davoodmanesh, S.; Taghavi, F.; Hashemi, M. Characterization of the interaction between human serum albumin and diazinon via spectroscopic and molecular docking methods. Hum. Exp. Toxicol. 2018, 37, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. The Characterization of Two Specific Drug Binding Sites on Human Serum Albumin. Mol. Pharmacol. 1975, 11, 824–832. [Google Scholar] [PubMed]
- Zsila, F. Subdomain IB is the third major drug binding region of human serum albumin: Toward the three-sites model. Mol. Pharm. 2013, 10, 1668–1682. [Google Scholar] [CrossRef] [PubMed]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Waters, N.J. Pharmacokinetic and Pharmacodynamic Considerations for Drugs Binding to Alpha-1-Acid Glycoprotein. Pharm. Res. 2019, 36, 30. [Google Scholar] [CrossRef]
- Otagiri, M. A Molecular Functional Study on the Interactions of Drugs with Plasma Proteins. Drug Metab. Pharmacokinet. 2005, 20, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Kopecký, V.; Ettrich, R.; Hofbauerová, K.; Baumruk, V. Structure of human α1-acid glycoprotein and its high-affinity binding site. Biochem. Biophys. Res. Commun. 2003, 300, 41–46. [Google Scholar] [CrossRef]
- Taguchi, K.; Nishi, K.; Giam Chuang, V.T.; Maruyama, T.; Otagiri, M. Molecular Aspects of Human Alpha-1 Acid Glycoprotein—Structure and Function. In Acute Phase Proteins; IntechOpen: London, UK, 2013. [Google Scholar] [CrossRef]
- Zsila, F.; Iwao, Y. The drug binding site of human α1-acid glycoprotein: Insight from induced circular dichroism and electronic absorption spectra. Biochim. Biophys. Acta—Gen. Subj. 2007, 1770, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Zsila, F.; Bikádi, Z.; Simonyi, M. Induced circular dichroism spectra reveal binding of the antiinflammatory curcumin to human α1-acid glycoprotein. Bioorganic Med. Chem. 2004, 12, 3239–3245. [Google Scholar] [CrossRef]
- Schmid, K.; Emura, J.; Schmid, M.F.; Stevens, R.L.; Nimberg, R.B. Sequence comparison of human plasma alpha1-acid glycoprotein and the immunoglobulins. Int. J. Pept. Protein Res. 1978, 11, 42–48. [Google Scholar] [CrossRef]
- Kremer, J.M.H.; Wilting, J.; Janssen, L.H.M. Drug binding to human alpha-1-acid glycoprotein in health and disease. Pharmacol. Rev. 1988, 40, 1–47. [Google Scholar] [PubMed]
- Müller, W.E. Drug binding sites on human alpha-1-acid glycoprotein. Prog. Clin. Biol. Res. 1989, 300, 363–378. [Google Scholar] [PubMed]
- Schley, J.; MÜLler-Oerlinghausen, B. Investigation of the binding of various tricyclic neuroleptics and antidepressants to alpha 1-acid glycoprotein. J. Pharm. Pharmacol. 1986, 38, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lomakin, A.; Latypov, R.F.; Laubach, J.P.; Hideshima, T.; Richardson, P.G.; Munshi, N.C.; Anderson, K.C.; Benedek, G.B. Phase transitions in human IgG solutions. J. Chem. Phys. 2013, 139, 121904. [Google Scholar] [CrossRef] [PubMed]
- Jian, J.; Wu, Z. Influences of traditional Chinese medicine on non-specific immunity of Jian Carp (Cyprinus carpio var. Jian). Fish Shellfish Immunol. 2004, 16, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Ying, L.; Chao, W.; Guanghua, L. Interaction of Jatrorrhizine with human gamma globulin in membrane mimetic environments: Probing of the binding mechanism and binding site by spectroscopic and molecular modeling methods. J. Mol. Struct. 2010, 980, 108–113. [Google Scholar] [CrossRef]
- Kotynia, A.; Wiatrak, B.; Kamysz, W.; Neubauer, D.; Jawień, P.; Marciniak, A. Cationic peptides and their cu(Ii) and ni(ii) complexes: Coordination and biological characteristics. Int. J. Mol. Sci. 2021, 22, 2028. [Google Scholar] [CrossRef]
- Yin, L.M.; Edwards, M.A.; Li, J.; Yip, C.M.; Deber, C.M. Roles of Hydrophobicity and Charge Distribution of Cationic Antimicrobial Peptides in Peptide-Membrane Interactions. J. Biol. Chem. 2012, 287, 7738–7745. [Google Scholar] [CrossRef]
- Huang, Y.; Huang, J.; Chen, Y. Alpha-helical cationic antimicrobial peptides: Relationships of structure and function. Protein Cell 2010, 1, 143–152. [Google Scholar] [CrossRef]
- Chen, G.Z.; Huang, X.Z.; Xu, J.H.; Zneng, Z.Z.; Wang, Z.B. The Methods of Fluorescence Analysis, 2nd ed.; Science: Beijing, China, 1990. [Google Scholar]
- Lakowicz, J.R.; Weber, G. Quenching of fluorescence by oxygen. Probe for structural fluctuations in macromolecules. Biochemistry 1973, 12, 4161–4170. [Google Scholar] [CrossRef]
- Lakowicz, J.R. (Ed.) Principles of Fluorescence Spectroscopy, 3rd ed.; Springer US: Boston, MA, USA, 2006; ISBN 978-0-387-31278-1. [Google Scholar]
- Ware, W.R. Oxygen quenching of fluorescence in solution: An experimental study of the diffusion process. J. Phys. Chem. 1962, 66, 455–458. [Google Scholar] [CrossRef]
- Ross, P.D.; Subramanian, S. Thermodynamics of Protein Association Reactions: Forces Contributing to Stability. Biochemistry 1981, 20, 3096–3102. [Google Scholar] [CrossRef] [PubMed]
- Szczukowski, Ł.; Krzyżak, E.; Wiatrak, B.; Jawień, P.; Marciniak, A.; Kotynia, A.; Świątek, P. New n-substituted-1,2,4-triazole derivatives of pyrrolo [3,4-d]pyridazinone with significant anti-inflammatory activity—Design, synthesis and complementary in vitro, computational and spectroscopic studies. Int. J. Mol. Sci. 2021, 22, 11235. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.F.; Liu, T.T.; Wang, Q.; Jiang, M.; Shi, J.H. Spectroscopic and molecular docking studies of binding interaction of gefitinib, lapatinib and sunitinib with bovine serum albumin (BSA). J. Photochem. Photobiol. B Biol. 2015, 153, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. Further Characterization of Specific Drug Binding Sites on Human Serum Albumin. Mol. Pharmacol. 1976, 12, 1052–1061. [Google Scholar] [PubMed]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to study proteins by circular dichroism. Biochim. Biophys. Acta—Proteins Proteom. 2005, 1751, 119–139. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, W.; Liu, J.; Sheng, F.; Hu, Z.; Chen, X. Binding of the bioactive component Jatrorrhizine to human serum albumin. Biochim. Biophys. Acta—Gen. Subj. 2005, 1722, 15–21. [Google Scholar] [CrossRef]
- Maciążek-Jurczyk, M.; Morak-Młodawska, B.; Jeleń, M.; Kopeć, W.; Szkudlarek, A.; Owczarzy, A.; Kulig, K.; Rogóż, W.; Pożycka, J. The influence of oxidative stress on serum albumin structure as a carrier of selected diazaphenothiazine with potential anticancer activity. Pharmaceuticals 2021, 14, 285. [Google Scholar] [CrossRef]
- Svenson, J.; Brandsdal, B.O.; Stensen, W.; Svendsen, J.S. Albumin binding of short cationic antimicrobial micropeptides and its influence on the in vitro bactericidal effect. J. Med. Chem. 2007, 50, 3334–3339. [Google Scholar] [CrossRef]
- Zorzi, A.; Linciano, S.; Angelini, A. Non-covalent albumin-binding ligands for extending the circulating half-life of small biotherapeutics. Medchemcomm 2019, 10, 1068. [Google Scholar] [CrossRef]
- Khalid, I.M.; Sharkh, S.E.A.; Samamarh, H.; Alfaqeeh, R.; Abuteir, M.M.; Darwish, S.M. Spectroscopic Characterization of the Interaction between Dopamine and Human Serum Albumin. Open J. Biophys. 2019, 9, 110–130. [Google Scholar] [CrossRef]
- Pakbin, B.; Zolghadr, L.; Rafiei, S.; Brück, W.M.; Brück, T.B. FTIR differentiation based on genomic DNA for species identification of Shigella isolates from stool samples. Sci. Rep. 2022, 12, 2780. [Google Scholar] [CrossRef] [PubMed]
- Byler, D.M.; Susi, H. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolymers 1986, 25, 469–487. [Google Scholar] [CrossRef]
- Lu, X.; Al-Qadiri, H.M.; Lin, M.; Rasco, B.A. Application of Mid-infrared and Raman Spectroscopy to the Study of Bacteria. Food Bioprocess Technol. 2011, 4, 919–935. [Google Scholar] [CrossRef]
- Barth, A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta—Bioenerg. 2007, 1767, 1073–1101. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wang, F.; Liu, S. Radiolabeled cyclic RGD peptides as radiotracers for tumor imaging. Biophys. Reports 2016, 2, 1–20. [Google Scholar] [CrossRef]
- Ahmed, A.; Tajmir-Riahi, H.A.; Carpentier, R. A quantitative secondary structure analysis of the 33 kDa extrinsic polypeptide of photosystem II by FTIR spectroscopy. FEBS Lett. 1995, 363, 65–68. [Google Scholar] [CrossRef]
- Liu, Y.; Xie, M.-X.; Kang, J.; Zheng, D. Studies on the interaction of total saponins of panax notoginseng and human serum albumin by Fourier transform infrared spectroscopy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2003, 59, 2747–2758. [Google Scholar] [CrossRef]
- Kong, J.; Yu, S. Fourier Transform Infrared Spectroscopic Analysis of Protein Secondary Structures. Acta Biochim. Biophys. Sin. 2007, 39, 549–559. [Google Scholar] [CrossRef]
- Majorek, K.A.; Porebski, P.J.; Dayal, A.; Zimmerman, M.D.; Jablonska, K.; Stewart, A.J.; Chruszcz, M.; Minor, W. Structural and immunologic characterization of bovine, horse, and rabbit serum albumins. Mol. Immunol. 2012, 52, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Wedemayer, G.J.; Patten, P.A.; Wang, L.H.; Schultz, P.G.; Stevens, R.C. Structural insights into the evolution of an antibody combining site. Science 1997, 276, 1665–1669. [Google Scholar] [CrossRef] [PubMed]
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life (Int. Union Biochem. Mol. Biol. Life) 2005, 57, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S.I.; Amisaki, T. Molecular dynamics study of conformational changes in human serum albumin by binding of fatty acids. Proteins Struct. Funct. Bioinforma. 2006, 64, 730–739. [Google Scholar] [CrossRef]
- Curry, S.; Brick, P.; Franks, N.P. Fatty acid binding to human serum albumin: New insights from crystallographic studies. Biochim. Biophys. Acta—Mol. Cell Biol. Lipids 1999, 1441, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ho, J.X.; Ruble, J.R.; Rose, J.; Rüker, F.; Ellenburg, M.; Murphy, R.; Click, J.; Soistman, E.; Wilkerson, L.; et al. Structural studies of several clinically important oncology drugs in complex with human serum albumin. Biochim. Biophys. Acta—Gen. Subj. 2013, 1830, 5356–5374. [Google Scholar] [CrossRef] [PubMed]
- Sivertsen, A.; Isaksson, J.; Leiros, H.K.S.; Svenson, J.; Svendsen, J.S.; Brandsdal, B.O. Synthetic cationic antimicrobial peptides bind with their hydrophobic parts to drug site II of human serum albumin. BMC Struct. Biol. 2014, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Windshügel, B. LEADS-PEP: A Benchmark Data Set for Assessment of Peptide Docking Performance. J. Chem. Inf. Model. 2016, 56, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Krüger, D.M.; Glas, A.; Bier, D.; Pospiech, N.; Wallraven, K.; Dietrich, L.; Ottmann, C.; Koch, O.; Hennig, S.; Grossmann, T.N. Structure-Based Design of Non-natural Macrocyclic Peptides That Inhibit Protein-Protein Interactions. J. Med. Chem. 2017, 60, 8982–8988. [Google Scholar] [CrossRef]
- Li, Y.; Liu, X.; Dong, X.; Zhang, L.; Sun, Y. Biomimetic design of affinity peptide ligand for capsomere of virus-like particle. Langmuir 2014, 30, 8500–8508. [Google Scholar] [CrossRef]
- Rentzsch, R.; Renard, B.Y. Docking small peptides remains a great challenge: An assessment using AutoDock Vina. Brief. Bioinform. 2015, 16, 1045–1056. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian~16 {R}evision {A}.03 2016. Available online: https://gaussian.com/citation/ (accessed on 23 January 2023).
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Quenching | Binding | Thermodynamic | |||||||
|---|---|---|---|---|---|---|---|---|---|
| T (K) | Ksv × 105 (dm3mol−1) | kq × 1013 (dm3mol−1s−1) | logKb | Kb × 104 (dm3mol−1) | n | ΔG° (kJmol−1) | ΔH° (kJmol−1) | ΔS° (Jmol−1K−1) | |
| L1 | 297 303 308 | 1.55 ± 0.07 1.21 ± 0.03 0.70 ± 0.06 | 1.55 ± 0.07 1.21 ± 0.03 0.70 ± 0.06 | 4.07 ± 0.07 3.95 ± 0.09 3.72 ± 0.15 | 1.17 ± 0.02 0.89 ± 0.02 0.53 ± 0.02 | 0.80 ± 0.02 0.80 ± 0.04 0.80 ± 0.02 | −23 ± 1 | −54 ± 2 | −106 ± 3 |
| L2 | 297 303 308 | 1.41 ± 0.02 1.39 ± 0.06 1.12 ± 0.07 | 1.41 ± 0.02 1.39 ± 0.06 1.12 ± 0.07 | 4.53 ± 0.08 4.04 ± 0.17 3.74 ± 0.15 | 3.39 ± 0.06 1.10 ± 0.05 0.55 ± 0.02 | 0.89 ± 0.02 0.81 ± 0.04 0.77 ± 0.04 | −25 ± 1 | −126 ± 1 | −339 ± 3 |
| L3 | 297 303 308 | 1.53 ± 0.03 1.22 ± 0.05 1.13 ± 0.06 | 1.53 ± 0.03 1.22 ± 0.05 1.13 ± 0.06 | 4.40 ± 0.12 4.10 ± 0.15 3.88 ± 0.16 | 2.52 ± 0.07 1.13 ± 0.04 0.76 ± 0.03 | 0.86 ± 0.02 0.83 ± 0.04 0.80 ± 0.03 | −25 ± 1 | −83 ± 1 | −195 ± 2 |
| Quenching | Binding | Thermodynamic | |||||||
|---|---|---|---|---|---|---|---|---|---|
| T (K) | Ksv × 104 (dm3mol−1) | kq × 1012 (dm3mol−1s−1) | logKb | Kb × 104 (dm3mol−1) | n | ΔG° (kJmol−1) | ΔH° (kJmol−1) | ΔS° (Jmol−1K−1) | |
| L1 | 297 303 308 | 4.24 ± 0.06 4.03 ± 0.13 3.60 ± 0.06 | 4.24 ± 0.06 4.03 ± 0.13 3.60 ± 0.06 | 5.00 ± 0.12 4.79 ± 0.20 4.39 ± 0.21 | 10.10 ± 0.24 6.17 ± 0.25 2.51 ± 0.12 | 1.06 ± 0.02 1.03 ± 0.06 0.97 ± 0.04 | −29 ± 1 | −94 ± 2 | −220 ± 7 |
| L2 | 297 303 308 | 3.99 ± 0.10 2.11 ± 0.12 1.00 ± 0.10 | 3.99 ± 0.06 2.11 ± 0.12 1.00 ± 0.10 | 4.12 ± 0.17 3.50 ± 0.20 3.31 ± 0.30 | 1.32 ± 0.15 0.32 ± 0.02 0.21 ± 0.02 | 0.89 ± 0.02 0.85 ± 0.05 0.88 ± 0.05 | −23 ± 1 | −130 ± 4 | −363 ± 11 |
| L3 | 297 303 308 | 3.87 ± 0.10 3.12 ± 0.14 2.65 ± 0.11 | 3.87 ± 0.10 3.12 ± 0.14 2.65 ± 0.11 | 4.75 ± 0.17 4.45 ± 0.19 3.83 ± 0.30 | 5.63 ± 0.20 2.82 ± 0.08 0.68 ± 0.01 | 0.86 ± 0.02 0.83 ± 0.04 0.80 ± 0.03 | −27 ± 1 | −144 ± 6 | −393 ± 15 |
| Site Marker | logKb | ||
|---|---|---|---|
| L1 | L2 | L3 | |
| - | 4.07 ± 0.07 | 4.53 ± 0.08 | 4.40 ± 0.12 |
| BSA + PHB (site I) | 4.00 ± 0.12 | 4.30 ± 0.20 | 4.30 ± 0.26 |
| BSA + IBP (site II) | 4.03 ± 0.15 | 4.42 ± 0.30 | 4.07 ± 0.31 |
| BSA: Analyzed. Peptide Molar Ratio | % α-Helix | % β-Sheet | % β-Turn | % Other |
|---|---|---|---|---|
| L1 | ||||
| 1:0 | 60.7% | 5.2% | 9.9% | 24.1% |
| 1:0.25 | 59.4% | 6.0% | 10.0% | 24.6% |
| 1:0.5 | 59.2% | 5.9% | 10.1% | 24.8% |
| 1:0.75 | 58.6% | 6.3% | 10.1% | 25.0% |
| 1:1 | 58.5% | 6.4% | 10.1% | 25.0% |
| 1:1.5 | 58.2% | 6.6% | 10.1% | 25.1% |
| L2 | ||||
| 1:0 | 61.9% | 4.7% | 9.9% | 23.5% |
| 1:0.25 | 61.2% | 5.1% | 9.9% | 23.7% |
| 1:0.5 | 60.9% | 5.5% | 9.9% | 23.7% |
| 1:0.75 | 60.9% | 5.5% | 9.9% | 23.6% |
| 1:1 | 60.7% | 5.8% | 9.9% | 23.6% |
| 1:1.5 | 60.7% | 5.9% | 9.9% | 23.5% |
| L3 | ||||
| 1:0 | 61.3% | 4.9% | 9.9% | 23.9% |
| 1:0.25 | 60.9% | 5.3% | 10.0% | 23.8% |
| 1:0.5 | 60.9% | 5.4% | 10.0% | 23.8% |
| 1:0.75 | 61.1% | 5.1% | 9.9% | 23.9% |
| 1:1 | 61.0% | 5.3% | 9.9% | 23.9% |
| 1:1.5 | 60.9% | 5.2% | 9.9% | 23.9% |
| GGF: Analyzed Peptide Molar Ratio | % α-Helix | % β-Sheet | % β-Turn | % Other |
|---|---|---|---|---|
| L1 | ||||
| 1:0 | 8.3% | 37.9% | 13.2% | 40.6% |
| 1:0.25 | 7.9% | 38.0% | 13.2% | 40.9% |
| 1:0.5 | 7.9% | 38.0% | 13.2% | 40.9% |
| 1:0.75 | 7.6% | 37.8% | 13.3% | 41.3% |
| 1:1 | 7.4% | 37.9% | 13.3% | 41.4% |
| 1:1.5 | 7.5% | 37.7% | 13.3% | 41.5% |
| L2 | ||||
| 1:0 | 8.4% | 37.6% | 13.2% | 40.7% |
| 1:0.25 | 8.3% | 37.7% | 13.2% | 40.8% |
| 1:0.5 | 8.3% | 37.6% | 13.2% | 40.8% |
| 1:0.75 | 8.3% | 37.7% | 13.2% | 40.7% |
| 1:1 | 8.1% | 37.9% | 13.2% | 40.8% |
| 1:1.5 | 7.5% | 37.8% | 13.3% | 41.4% |
| L3 | ||||
| 1:0 | 8.2% | 37.6% | 13.2% | 41.0% |
| 1:0.25 | 8.0% | 37.7% | 13.2% | 41.1% |
| 1:0.5 | 7.8% | 38.0% | 13.2% | 40.9% |
| 1:0.75 | 7.8% | 38.0% | 13.2% | 41.0% |
| 1:1 | 7.4% | 37.9% | 13.3% | 41.5% |
| 1:1.5 | 7.0% | 38.0% | 13.3% | 41.7% |
| Protein/Complex | (%) | ||||
|---|---|---|---|---|---|
| α-Helix | β-Sheet | β-Turn | β-Anti | Random Coil | |
| free BSA | 63.71 | 1.64 | 6.65 | 2.38 | 25.62 |
| BSA/L1 | |||||
| 1:0.5 | 61.36 | 3.87 | 7.47 | 0.77 | 26.54 |
| 1:1.0 | 49.45 | 8.28 | 13.86 | 0.49 | 27.93 |
| 1:1.5 | 49.40 | 8.69 | 13.99 | 0.31 | 27.61 |
| BSA/L2 | |||||
| 1:0.5 | 55.96 | 3.31 | 8.42 | 2.54 | 29.77 |
| 1:1.0 | 55.19 | 3.17 | 9.59 | 2.56 | 29.49 |
| 1:1.5 | 54.12 | 3.00 | 10.15 | 2.81 | 29.93 |
| BSA/L3 | |||||
| 1:0.5 | 55.00 | 3.31 | 9.86 | 2.35 | 29.47 |
| 1:1.0 | 55.31 | 3.19 | 9.40 | 2.68 | 29.43 |
| 1:1.5 | 54.40 | 3.52 | 9.71 | 2.69 | 29.69 |
| free GGF | 14.17 | 47.7 | 14.55 | 5.73 | 17.69 |
| GGF/L1 | |||||
| 1:0.5 | 11.67 | 34.54 | 10.12 | 3.85 | 39.82 |
| 1:1.0 | 9.41 | 35.36 | 11.38 | 4.13 | 39.71 |
| 1:1.5 | 9.37 | 35.72 | 10.12 | 4.12 | 40.67 |
| GGF/L2 | |||||
| 1:0.5 | 10.54 | 43.42 | 8.55 | 3.22 | 34.28 |
| 1:1.0 | 9.59 | 44.29 | 8.48 | 4.31 | 33.33 |
| 1:1.5 | 9.54 | 43.35 | 8.35 | 4.86 | 33.91 |
| GGF/L3 | |||||
| 1:0.5 | 13.84 | 40.58 | 17.16 | 6.23 | 22.19 |
| 1:1.0 | 12.52 | 40.11 | 18.63 | 5.53 | 23.22 |
| 1:1.5 | 12.08 | 40.70 | 18.16 | 5.01 | 24.05 |
| Serum Albumin | γ-Globulin Fraction | |||
|---|---|---|---|---|
| Domain I (Site 3) | Domain II (Site 1) | Domain III (Site 2) | ||
| L1 | −5.5 | −4.6 | −5.3 | −5.5 |
| L2 | −6.1 | −4.4 | −5.9 | −6.1 |
| L3 | −6.2 | −4.2 | −5.8 | −5.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotynia, A.; Marciniak, A.; Kamysz, W.; Neubauer, D.; Krzyżak, E. Interaction of Positively Charged Oligopeptides with Blood Plasma Proteins. Int. J. Mol. Sci. 2023, 24, 2836. https://doi.org/10.3390/ijms24032836
Kotynia A, Marciniak A, Kamysz W, Neubauer D, Krzyżak E. Interaction of Positively Charged Oligopeptides with Blood Plasma Proteins. International Journal of Molecular Sciences. 2023; 24(3):2836. https://doi.org/10.3390/ijms24032836
Chicago/Turabian StyleKotynia, Aleksandra, Aleksandra Marciniak, Wojciech Kamysz, Damian Neubauer, and Edward Krzyżak. 2023. "Interaction of Positively Charged Oligopeptides with Blood Plasma Proteins" International Journal of Molecular Sciences 24, no. 3: 2836. https://doi.org/10.3390/ijms24032836
APA StyleKotynia, A., Marciniak, A., Kamysz, W., Neubauer, D., & Krzyżak, E. (2023). Interaction of Positively Charged Oligopeptides with Blood Plasma Proteins. International Journal of Molecular Sciences, 24(3), 2836. https://doi.org/10.3390/ijms24032836