
Involvement of Oxidative Stress and the Innate Immune System in SARS-CoV-2 Infection

 , , , and
, , , and 
{kind=link}
Abstract
1. Introduction
2. Structural Characteristics of SARS-CoV-2
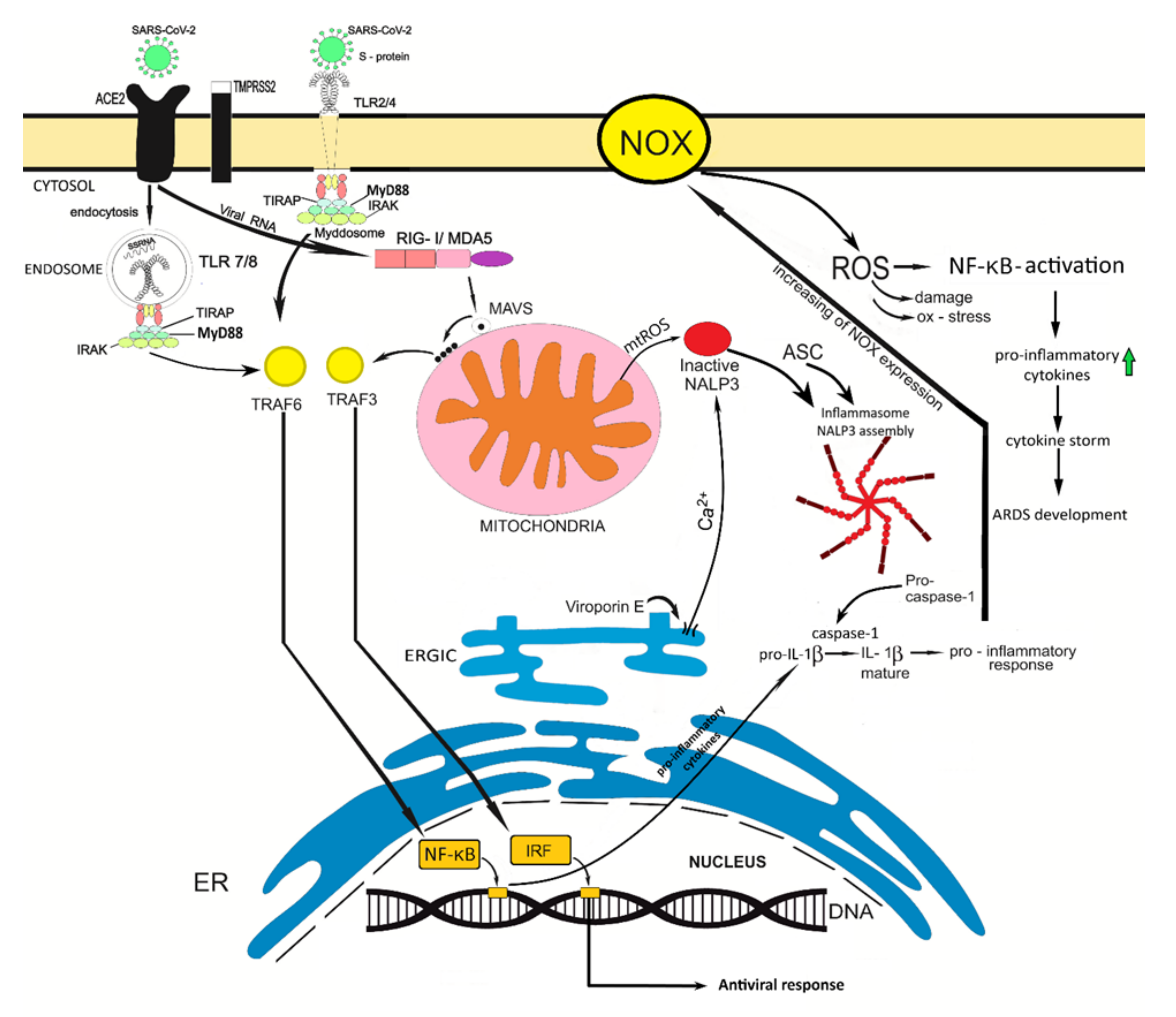
3. SARS-CoV-2 Infection Is Associated with Oxidative Stress
4. The Role of the Innate Immunity in Coronavirus Infection
5. Can Oxidative Stress Contribute to the Development of a More Severe SARS-Cov-2 Infection Course?
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Yeager, C.L.; Ashmun, R.A.; Williams, R.K.; Cardellichio, C.B.; Shapiro, L.H.; Look, A.T.; Holmes, K.V. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 1992, 357, 420–422. [Google Scholar] [CrossRef]
- Abdul-Rasool, S.; Fielding, B.C. Understanding human coronavirus HCoV-NL63. Open Virol. J. 2010, 4, 76–84. [Google Scholar] [CrossRef]
- Anderson, R.M.; Fraser, C.; Ghani, A.C.; Donnelly, C.A.; Riley, S.; Ferguson, N.M.; Leung, G.M.; Lam, T.H.; Hedley, A.J. Epidemiology, transmission dynamics and control of SARS: The 2002–2003 epidemic. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004, 359, 1091–1105. [Google Scholar] [CrossRef] [PubMed]
- Tong, T.R. Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV). Perspect. Med. Virol. 2006, 16, 43–95. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D. The chronology of the 2002–2003 SARS mini pandemic. Paediatr. Respir. Rev. 2004, 5, 262–269. [Google Scholar] [CrossRef]
- Chafekar, A.; Fielding, B.C. MERS-CoV: Understanding the latest human coronavirus threat. Viruses 2018, 10, 93. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282. [Google Scholar] [CrossRef]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Coronaviridae study group of the international committee on taxonomy of viruses. The species severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nyodu, R.; Maurya, V.K.; Saxena, S.K. Morphology, genome organization, replication, and pathogenesis of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). In Coronavirus Disease 2019 (COVID-19). Medical Virology: From Pathogenesis to Disease Control; Saxena, S., Ed.; Springer: Singapore, 2020. [Google Scholar]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Yoshimoto, F.K. The proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS CoV-2 or n-COV19), the cause of COVID-19. Protein J. 2020, 39, 198–216. [Google Scholar] [CrossRef]
- Ysrafil, A.I. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. 2020, 14, 407–412. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Rabaan, A.A.; Al-Ahmed, S.H.; Haque, S.; Sah, R.; Tiwari, R.; Malik, Y.S.; Dhama, K.; Yatoo, M.I.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. SARS-CoV-2, SARS-CoV, and MERS-COV: A comparative overview. Infez. Med. 2020, 28, 174–184. [Google Scholar] [PubMed]
- Jaimes, J.A.; Millet, J.K.; Whittaker, G.R. Proteolytic cleavage of the SARS-CoV-2 spike protein and the role of the novel S1/S2 site. iScience 2020, 23, 101212. [Google Scholar] [CrossRef]
- Tilocca, B.; Soggiu, A.; Sanguinetti, M.; Musella, V.; Britti, D.; Bonizzi, L.; Urbani, A.; Roncada, P. Comparative computational analysis of SARS-CoV-2 nucleocapsid protein epitopes in taxonomically related coronaviruses. Microbes Infect. 2020, 22, 188–194. [Google Scholar] [CrossRef]
- Gralinski, L.E.; Menachery, V.D. Return of the coronavirus: 2019-nCoV. Viruses 2020, 12, 135. [Google Scholar] [CrossRef]
- McBride, R.; van Zyl, M.; Fielding, B.C. The coronavirus nucleocapsid is a multifunctional protein. Viruses 2014, 6, 2991–3018. [Google Scholar] [CrossRef]
- Hurst, K.R.; Koetzner, C.A.; Masters, P.S. Characterization of a critical interaction between the coronavirus nucleocapsid protein and nonstructural protein 3 of the viral replicase-transcriptase complex. J. Virol. 2013, 87, 9159–9172. [Google Scholar] [CrossRef]
- Kang, S.; Yang, M.; Hong, Z.; Zhang, L.; Huang, Z.; Chen, X.; He, S.; Zhou, Z.; Zhou, Z.; Chen, Q.; et al. Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm. Sin. B 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Udugama, B.; Kadhiresan, P.; Kozlowski, H.N.; Malekjahani, A.; Osborne, M.; Li, V.Y.C.; Chen, H.; Mubareka, S.; Gubbay, J.B.; Chan, W.C.W. Diagnosing COVID-19: The disease and tools for detection. ACS Nano 2020, 14, 3822–3835. [Google Scholar] [CrossRef]
- Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses 2020, 12, 254. [Google Scholar] [CrossRef] [PubMed]
- Che, X.Y.; Hao, W.; Wang, Y.; Di, B.; Yin, K.; Xu, Y.C.; Feng, C.S.; Wan, Z.Y.; Cheng, V.C.; Yuen, K.Y. Nucleocapsid protein as early diagnostic marker for SARS. Emerg. Infect. Dis. 2004, 10, 1947–1949. [Google Scholar] [CrossRef]
- Satarker, S.; Nampoothiri, M. Structural proteins in Severe Acute Respiratory Syndrome Coronavirus-2. Arch. Med. Res. 2020, 51, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wen, J.; Tang, L.; Zhang, H.; Zhang, X.; Li, Y.; Wang, J.; Han, Y.; Li, G.; Shi, J.; et al. The M protein of SARS-CoV: Basic structural and immunological properties. Genom. Proteom. Bioinform. 2003, 1, 118–130. [Google Scholar] [CrossRef]
- Hasöksüz, M.; Kiliç, S.; Saraç, F. Coronaviruses and SARS-COV-2. Turk. J. Med. Sci. 2020, 50, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Gao, J.; Zheng, H.; Li, B.; Kong, L.; Zhang, Y.; Wang, W.; Zeng, Y.; Ye, L. The membrane protein of SARS-CoV suppresses NF-kappaB activation. J. Med. Virol. 2007, 79, 1431–1439. [Google Scholar] [CrossRef]
- Fung, T.S.; Liu, D.X. Post-translational modifications of coronavirus proteins: Roles and function. Future Virol. 2018, 13, 405–430. [Google Scholar] [CrossRef]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef] [PubMed]
- Castaño-Rodriguez, C.; Honrubia, J.M.; Gutiérrez-Álvarez, J.; DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Verdia-Baguena, C.; Queralt-Martin, M.; et al. Role of Severe Acute Respiratory Syndrome Coronavirus Viroporins E, 3a, and 8a in replication and pathogenesis. mBio 2018, 9, e02325-e17. [Google Scholar] [CrossRef]
- Nieva, J.; Madan, V.; Carrasco, L. Viroporins: Structure and biological functions. Nat. Rev. Microbiol. 2012, 10, 563–574. [Google Scholar] [CrossRef]
- Farag, N.S.; Breitinger, U.; Breitinger, H.G.; El Azizi, M.A. Viroporins and inflammasomes: A key to understand virus-induced inflammation. Int. J. Biochem. Cell Biol. 2020, 122, 105738. [Google Scholar] [CrossRef]
- Ueda, M.; Daidoji, T.; Du, A.; Yang, C.S.; Ibrahim, M.S.; Ikuta, K. Highly pathogenic H5N1 avian influenza virus induces extracellular Ca2+ influx, leading to apoptosis in avian cells. J. Virol. 2010, 84, 3068–3078. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Roche, L.; Mesta, F. Oxidative stress as key player in Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) infection. Arch. Med. Res. 2020, 51, 384–387. [Google Scholar] [CrossRef]
- Sarkar, K.; Sil, P.C. Infseases and endogenous oxidative stress. Oxid. Stress Lung Dis. 2019, 2, 125–148. [Google Scholar]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.B.; Rahu, N. Oxidative stress and inflammation: What polyphenols can do for us? Oxid. Med. Cell Longev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Laurindo, F.R.M. Redox cellular signaling pathways in endothelial dysfunction and vascular disease. Endothel. Cardiovasc. Dis. 2018, 127–145. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Liu, M.; Chen, F.; Liu, T.; Chen, F.; Liu, S.; Yang, J. The role of oxidative stress in influenza virus infection. Microbes Infect. 2017, 19, 580–586. [Google Scholar] [CrossRef]
- Kellner, M.; Noonepalle, S.; Lu, Q.; Srivastava, A.; Zemskov, E.; Black, S.M. ROS signaling in the pathogenesis of Acute Lung Injury (ALI) and Acute Respiratory Distress Syndrome (ARDS). Adv. Exp. Med. Biol. 2017, 967, 105–137. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet Lond. Engl. 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Oran, D.P.; Topol, E.J. Prevalence of asymptomatic SARS-CoV-2 infection: A narrative review. Ann. Intern. Med. 2020, 173, 362–367. [Google Scholar] [CrossRef]
- Li, X.; Geng, M.; Peng, Y.; Meng, L.; Lu, S. Molecular immune pathogenesis and diagnosis of COVID-19. J. Pharm. Anal. 2020, 10, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Elner, S.G.; Bian, Z.M.; Till, G.O.; Petty, H.R.; Elner, V.M. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp. Eye Res. 2007, 85, 462–472. [Google Scholar] [CrossRef]
- Leisman, D.E.; Ronner, L.; Pinotti, R.; Taylor, M.D.; Sinha, P.; Calfee, C.S.; Hirayama, A.V.; Mastroiani, F.; Turtle, C.J.; Harhay, M.O.; et al. Cytokine elevation in severe and critical COVID-19: A rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 2020, 12, 1233–1244. [Google Scholar] [CrossRef]
- Golonka, R.M.; Saha, P.; Yeoh, B.S.; Chattopadhyay, S.; Gewirtz, A.T.; Joe, B.; Vijay-Kumar, M. Harnessing innate immunity to eliminate SARS-CoV-2 and ameliorate COVID-19 disease. Physiol. Genom. 2020, 52, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Laforge, M.; Elbim, C.; Frère, C.; Hémadi, M.; Massaud, C.; Nuss, P.; Benoliel, J.; Becker, C. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat. Rev. Immunol. 2020, 20, 515–516. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol Toxxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Cecchini, R.; Cecchini, A.L. SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med. Hypotheses 2020, 143, 110102. [Google Scholar] [CrossRef]
- Hosakote, Y.M.; Liu, T.; Castro, S.M.; Garofalo, R.P.; Casola, A. Respiratory syncytial virus induces oxidative stress by modulating antioxidant enzymes. Am. J. Respir Cell Mol. Biol. 2009, 41, 348–357. [Google Scholar] [CrossRef]
- Abouhashem, A.S.; Singh, K.; Azzazy, H.M.; Sen, C.K. Is low alveolar type II Cell SOD3 in the lungs of elderly linked to the observed severity of COVID-19? Antioxid. Redox Signal. 2020, 33, 59–65. [Google Scholar] [CrossRef]
- Lazarian, G.; Quinquenel, A.; Bellal, M.; Siavellis, J.; Jacquy, C.; Re, D.; Merabet, F.; Mekinian, A.; Braun, T.; Damaj, G.; et al. Autoimmune haemolytic anaemia associated with COVID-19 infection. Br. J. Haematol. 2020, 190, 29–31. [Google Scholar] [CrossRef]
- Chow, C.; Herrera Abreu, M.T.; Suzuki, T.; Downey, G.P. Oxidative stress and acute lung injury. Am. J. Respir. Cell Mol. Biol. 2003, 29, 427–431. [Google Scholar] [CrossRef]
- Mellman, I. Dendritic cells: Master regulators of the immune response. Cancer Immunol. Res. 2013, 1, 145–149. [Google Scholar] [CrossRef]
- Hu, G.; Christman, J.W. Editorial: Alveolar macrophages in lung inflammation and resolution. Front. Immunol. 2019, 10, 2275. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Fan, Y.; Lai, Y.; Han, T.; Li, Z.; Zhou, P. Coronavirus infections and immune responses. J. Med. Virol. 2020, 92, 424–432. [Google Scholar] [CrossRef]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Catanzaro, M.; Fagiani, F.; Racchi, M.; Corsini, E.; Govoni, S.; Lanni, C. Immune response in COVID-19: Addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2. Signal Transduct. Target Ther. 2020, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef]
- Saxena, M.; Yeretssian, G. NOD-like receptors: Master regulators of inflammation and cancer. Front. Immunol. 2014, 5, 327. [Google Scholar] [CrossRef]
- Abadía-Molina, F.; Morón-Calvente, V.; Baird, S.D.; Shamim, F.; Martin, F.; MacKenzie, A. Neuronal apoptosis inhibitory protein (NAIP) localizes to the cytokinetic machinery during cell division. Sci. Rep. 2017, 7, 39981. [Google Scholar] [CrossRef]
- MacDonald, J.A.; Wijekoon, C.P.; Liao, K.C.; Muruve, D.A. Biochemical and structural aspects of the ATP-binding domain in inflammasome-forming human NLRP proteins. IUBMB Life 2013, 65, 851–862. [Google Scholar] [CrossRef]
- Velloso, F.J.; Trombetta-Lima, M.; Anschau, V.; Sogayar, M.C.; Correa, R.G. NOD-like receptors: Major players (and targets) in the interface between innate immunity and cancer. Biosci. Rep. 2019, 39, BSR20181709. [Google Scholar] [CrossRef]
- Motta, V.; Soares, F.; Sun, T.; Philpott, D.J. NOD-like receptors: Versatile cytosolic sentinels. Physiol. Rev. 2015, 95, 149–178. [Google Scholar] [CrossRef] [PubMed]
- Correa, R.G.; Milutinovic, S.; Reed, J.C. Roles of NOD1 (NLRC1) and NOD2 (NLRC2) in innate immunity and inflammatory diseases. Biosci. Rep. 2012, 32, 597–608. [Google Scholar] [CrossRef]
- Philpott, D.J.; Sorbara, M.T.; Robertson, S.J.; Croitoru, K.; Girardin, S.E. NOD proteins: Regulators of inflammation in health and disease. Nat. Rev. Immunol. 2013, 14, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Le, H.T.; Harton, J.A. Pyrin- and CARD-only proteins as regulators of NLR functions. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef]
- Stutz, A.; Kolbe, C.C.; Stahl, R.; Horvath, G.L.; Franklin, B.S.; van Ray, O.; Brinkschulte, R.; Geyer, M.; Meissner, F.; Latz, E. NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J. Exp. Med. 2017, 214, 1725–1736. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Castano-Rodriguez, C.; Fernandez-Delgado, R.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 2015, 485, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Kucia, M. SARS-CoV-2 infection and overactivation of Nlrp3 inflammasome as a trigger of cytokine “storm” and risk factor for damage of hematopoietic stem cells. Leukemia 2020. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.D. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Kanneganti, T.D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Shah, A. Novel coronavirus-induced NLRP3 inflammasome activation: A potential drug target in the treatment of COVID-19. Front. Immunol. 2020, 11, 1021. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Liu, Y.; Olagnier, D.; Lin, R. Host and viral modulation of RIG-I-mediated antiviral immunity. Front. Immunol. 2017, 7. [Google Scholar] [CrossRef]
- Brisse, M.; Ly, H. Comparative structure and function analysis of the RIG-I-like receptors: RIG-I and MDA5. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Gack, M.U. Mechanisms of RIG-I-like receptor activation and manipulation by viral pathogens. J. Virol. 2014, 88, 5213–5216. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Fujita, T. Cytoplasmic viral RNA sensors: RIG-I-like receptors. Encycl. Immunobiol. 2016, 352–359. [Google Scholar] [CrossRef]
- Totura, A.L.; Baric, R.S. SARS coronavirus pathogenesis: Host innate immune responses and viral antagonism of interferon. Curr. Opin. Virol. 2012, 2, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Vabret, N.; Britton, G.J.; Gruber, C.; Hegde, S.; Kim, J.; Kuksin, M.; Levantovsky, R.; Malle, L.; Moreira, A.; Park, M.D.; et al. Sinai immunology review project. Immunology of COVID-19: Current state of the science. Immunity 2020, 52, 910–941. [Google Scholar] [CrossRef]
- Samuel, C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar] [CrossRef]
- Matsuyama, T.; Kubli, S.P.; Yoshinaga, S.K.; Pfeffer, K.; Mak, T.W. An aberrant STAT pathway is central to COVID-19. Cell Death Differ. 2020, 27, 3209–3225. [Google Scholar] [CrossRef]
- Miorin, L.; Kehrer, T.; Sanchez-Aparici, M.T.; Zhang, K.; Cohen, P.; Patel, R.S.; Cupic, A.; Makio, T.; Mei, M.; Moreno, E.; et al. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 28344–28354. [Google Scholar] [CrossRef]
- Collin, F. Chemical basis of reactive oxygen species reactivity and involvement in neurodegenerative diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef]
- Touyz, R.M.; Briones, A.M.; Sedeek, M.; Burger, D.; Montezano, A.C. NOX isoforms and reactive oxygen species in vascular health. Mol. Interv. 2011, 11, 27–35. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef]
- Rastogi, R.; Geng, X.; Li, F.; Ding, Y. NOX activation by subunit interaction and underlying mechanisms in disease. Front. Cell Neurosci. 2017, 10. [Google Scholar] [CrossRef]
- Haslund-Vinding, J.; McBean, G.; Jaquet, V.; Vilhardt, F. NADPH oxidases in oxidant production by microglia: Activating receptors, pharmacology and association with disease. Br. J. Pharmacol. 2016, 174, 1733–1749. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Li, W.; Xie, J.; Hou, Y.; You, C. Cytokine storm induced by SARS-CoV-2. Clin. Chim. Acta 2020, 509, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH oxidase activation and bacterial resistance. Front. Cell Infect. Microbiol. 2017, 7. [Google Scholar] [CrossRef]
- Lingappan, K. NF-κB in Oxidative stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Chelombitko, M.A. Role of reactive oxygen species in inflammation: A minireview. Mosc. Univ. Biol. Sci. Bull. 2018, 73, 199–202. [Google Scholar] [CrossRef]
- Leach Ingram, S.; Diotallevi, M. Reactive oxygen species: Rapid fire in inflammation. Biochemist 2017, 39, 30–33. [Google Scholar] [CrossRef]
- Yuki, K.; Fujiogi, M.; Koutsogiannaki, S. COVID-19 pathophysiology: A review. Clin. Immunol. 2020, 215, 108427. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-mediated cellular signaling. Oxid. Med. Cell Longev. 2016. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Yang, F.; Zheng, Q.; Tang, W.; Li, J. The potential role of the NLRP3 inflammasome activation as a link between mitochondria ROS generation and neuroinflammation in postoperative cognitive dysfunction. Front. Cell Neurosci. 2019, 13, 73. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Poznyak, A.V.; Sobenin, I.A.; Nikiforov, N.N.; Ivanova, E.A. Mitochondrion as a selective target for treatment of atherosclerosis, role of mitochondrial DNA mutations and defective mitophagy in the pathogenesis of atherosclerosis and chronic inflammation. Curr. Neuropharmacol. 2019, 18. [Google Scholar] [CrossRef]
- Michalakis, K.; Ilias, I. SARS-CoV-2 infection and obesity: Common inflammatory and metabolic aspects. Diabetes Metab. Syndr. 2020, 14, 469–471. [Google Scholar] [CrossRef]
- Marseglia, L.; Manti, S.; D’Angelo, G.; Nicotera, A.; Parisi, E.; Di Rosa, G.; Gitto, E.; Arrigo, T. Oxidative stress in obesity: A critical component in human diseases. Int. J. Mol. Sci. 2014, 16, 378–400. [Google Scholar] [CrossRef]
- Carnevale, R.; Cammisotto, V.; Pagano, F.; Nocella, C. Effects of smoking on oxidative stress and vascular function. Smok. Prev. Cessat. 2018. [Google Scholar] [CrossRef]
- Smith, J.C.; Sausville, E.L.; Girish, V.; Yuan, M.L.; Vasudevan, A.; John, K.M.; Sheltzer, J.M. Cigarette smoke exposure and inflammatory signaling increase the expression of the SARS-CoV-2 receptor ACE2 in the Respiratory Tract. Dev. Cell 2020, 53, 514–529.e3. [Google Scholar] [CrossRef]
- Meini, S.; Fortini, A.; Andreini, R.; Sechi, L.A.; Tascini, C. The paradox of the low prevalence of current smokers among COVID-19 patients hospitalized in non-intensive care wards: Results from an italian multicenter case-control study. Nicotine Tob. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Usman, M.S.; Siddiqi, T.J.; Khan, M.S.; Patel, U.K.; Shahid, I.; Ahmed, J.; Kalra, A.; Michos, E.D. Is there a smoker’s paradox in COVID-19? BMJ Evid. Based Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kozakiewicz, M.; Kornatowski, M.; Krzywińska, O.; Kędziora-Kornatowska, K. Changes in the blood antioxidant defense of advanced age people. Clin. Interv. Aging 2019, 14, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Keles, E.S. Mild SARS-CoV-2 infections in children might be based on evolutionary biology and linked with host reactive oxidative stress and antioxidant capabilities. New Microbes New Infect. 2020, 36, 100723. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Evidence Used to Update the List of Underlying Medical Conditions that Increase a Person’s Risk of Severe Illness from COVID-19. Available online: https://www.cdc.gov/coronavirus/2019-ncov/need-extra-precautions/evidence-table.html (accessed on 13 August 2020).
- Poe, F.L.; Corn, J. N-Acetylcysteine: A potential therapeutic agent for SARS-CoV-2. Med. Hypotheses 2020, 143, 109862. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, X.; Ni, L.; Di, X.; Ma, B.; Niu, S.; Liu, C.; Reiter, R.J. COVID-19: Melatonin as a potential adjuvant treatment. Life Sci. 2020, 250, 117583. [Google Scholar] [CrossRef] [PubMed]
- Erol, N.; Saglam, L.; Saglam, Y.S.; Erol, H.S.; Altun, S.; Aktas, M.S.; Halici, M.B. The protection potential of antioxidant vitamins against acute respiratory distress syndrome: A rat trial. Inflammation 2019, 42, 1585–1594. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozlov, E.M.; Ivanova, E.; Grechko, A.V.; Wu, W.-K.; Starodubova, A.V.; Orekhov, A.N. Involvement of Oxidative Stress and the Innate Immune System in SARS-CoV-2 Infection. Diseases 2021, 9, 17. https://doi.org/10.3390/diseases9010017
Kozlov EM, Ivanova E, Grechko AV, Wu W-K, Starodubova AV, Orekhov AN. Involvement of Oxidative Stress and the Innate Immune System in SARS-CoV-2 Infection. Diseases. 2021; 9(1):17. https://doi.org/10.3390/diseases9010017
Chicago/Turabian StyleKozlov, Evgenii M., Ekaterina Ivanova, Andrey V. Grechko, Wei-Kai Wu, Antonina V. Starodubova, and Alexander N. Orekhov. 2021. "Involvement of Oxidative Stress and the Innate Immune System in SARS-CoV-2 Infection" Diseases 9, no. 1: 17. https://doi.org/10.3390/diseases9010017
APA StyleKozlov, E. M., Ivanova, E., Grechko, A. V., Wu, W.-K., Starodubova, A. V., & Orekhov, A. N. (2021). Involvement of Oxidative Stress and the Innate Immune System in SARS-CoV-2 Infection. Diseases, 9(1), 17. https://doi.org/10.3390/diseases9010017