The Evolving Understanding of the Molecular and Therapeutic Landscape of Pancreatic Ductal Adenocarcinoma


{kind=link}
Abstract
1. Introduction
2. Clinical Presentation
2.1. Clinicopathological Staging of PDAC
2.2. Current Treatment Approaches for Advanced PDAC
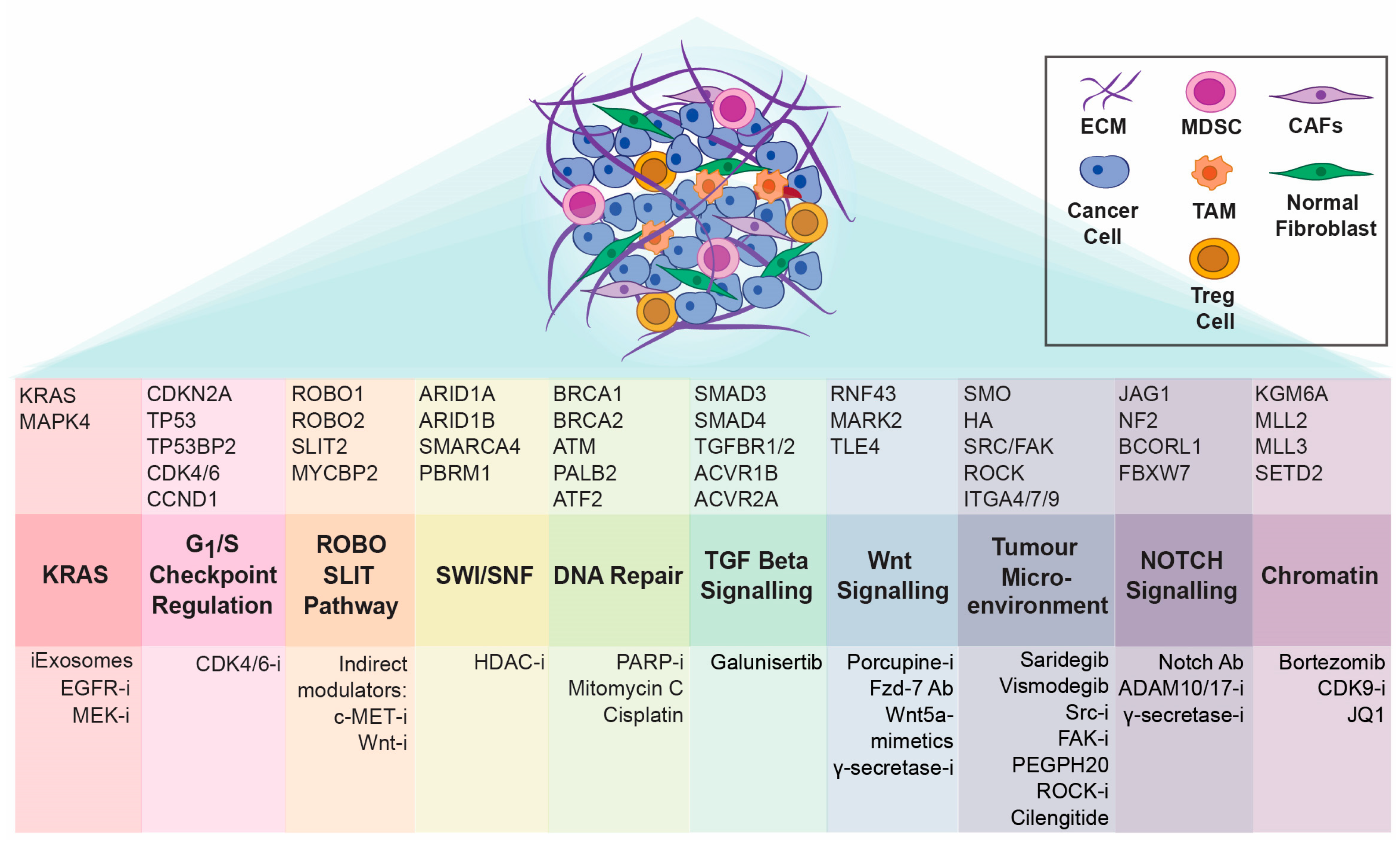
3. The “omic” Diversity of PDAC
- (1)
- “stable” subtype, present in 20% of all patients whose tumour genomes harboured fewer than 50 SV events;
- (2)
- “locally rearranged” subtype, detected in 30% of the cohort, characterised by a single focal event on one-two chromosomes, breakage-fusion-bridge events, chromothripsis or low prevalence alterations in known oncogenes and therapeutic targets (focal amplifications in KRAS, SOX9, GATA6, ERBB2, MET, CDK6);
- (3)
- the “scattered” subtype, present in 36% of tumours, showed a range of non-random chromosomal damage with less than 200 structural rearrangements;
- (4)
- the “unstable” or high SV subtype, present in 14% of PDAC, characterised by a large extent of SV (>200 events), suggesting major defects in DNA maintenance, with associated increased sensitivity to DNA-damaging agents [5]. Deleterious mutations in BRCA1, BRCA2 and PALB2 genes, essential components of homologous recombination-mediated DNA repair, were associated with the “unstable” PDAC subtype, and similarly, the top quintile of the previously identified BRCA mutational signature [40], was present in the majority (10/14) of unstable genomes [5].
- (1)
- “squamous” subtype, associated with the worst patient prognosis;
- (2)
- “pancreatic progenitor” subtype, enriched for transcriptional networks containing PDX1, MNX1, HNF4G, HNF4A, HNF1B, HNF1A, FOXA2, FOXA3 and HES1 genes;
- (3)
- “aberrantly differentiated endocrine exocrine” (ADEX) subtype, a sub-class of the “pancreatic progenitor” group, defined by transcriptional networks that are essential in later stages of pancreatic development and differentiation. These include upregulation of NR5A2, MIST1, RBPJL and their downstream targets, which regulate acinar cell differentiation and pancreatitis/regeneration;
- (4)
- “immunogenic” subtype, associated with a significant immune infiltrate, with predominant expression profiles related to infiltrating B and T cells, upregulation of CTLA4 and PD1 immuno-suppressive pathways, inferring therapeutic opportunities with immune modulating agents for specific tumours in this class.
3.1. Targeting KRAS
3.2. G1/S Checkpoint as a Therapeutic Target in PDAC
3.3. Targeting DNA Damage Repair Signalling in PDAC
3.4. Mismatch Repair Deficiency in PDAC
4. Tumour Microenvironment Matters: Exploration of Stromal Components as Therapeutic Targets in Pancreatic Cancer
5. Concluding Remarks
Conflicts of Interest
References
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Hruban, R.H.; Adsay, N.V.; Albores-Saavedra, J.; Compton, C.; Garrett, E.S.; Goodman, S.N.; Kern, S.E.; Klimstra, D.S.; Kloppel, G.; Longnecker, D.S.; et al. Pancreatic intraepithelial neoplasia: A new nomenclature and classification system for pancreatic duct lesions. Am. J. Surg. Pathol. 2001, 25, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Hruban, R.H.; Goggins, M.; Parsons, J.; Kern, S.E. Progression model for pancreatic cancer. Clin. Cancer Res. 2000, 6, 2969–2972. [Google Scholar] [PubMed]
- Wilentz, R.E.; Iacobuzio-Donahue, C.A.; Argani, P.; McCarthy, D.M.; Parsons, J.L.; Yeo, C.J.; Kern, S.E.; Hruban, R.H. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: Evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000, 60, 2002–2006. [Google Scholar] [PubMed]
- Real, F.X. A “catastrophic hypothesis” for pancreas cancer progression. Gastroenterology 2003, 124, 1958–1964. [Google Scholar] [CrossRef]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.B.; Brown, A.M.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef] [PubMed]
- McCarroll, J.A.; Naim, S.; Sharbeen, G.; Russia, N.; Lee, J.; Kavallaris, M.; Goldstein, D.; Phillips, P.A. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front. Physiol. 2014, 5, 141. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Gaujoux, S.; Williet, N.; Bachet, J.B.; Bauguion, L.; Colson Durand, L.; Conroy, T.; Dahan, L.; Gilabert, M.; Huguet, F.; et al. Pancreatic cancer: French clinical practice guidelines for diagnosis, treatment and follow-up (SNFGE, FFCD, GERCOR, UNICANCER, SFCD, SFED, SFRO, ACHBT, AFC). Dig. Liver Dis. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Modolell, I.; Guarner, L.; Malagelada, J.R. Vagaries of clinical presentation of pancreatic and biliary tract cancer. Ann. Oncol. 1999, 10, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Bakkevold, K.E.; Arnesjo, B.; Kambestad, B. Carcinoma of the pancreas and papilla of Vater: Presenting symptoms, signs, and diagnosis related to stage and tumour site. A prospective multicentre trial in 472 patients. Norwegian Pancreatic Cancer Trial. Scand. J. Gastroenterol. 1992, 27, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, G.; Ma, Q.; Liu, H.; Li, W.; Han, L. The bidirectional interation between pancreatic cancer and diabetes. World J. Surg. Oncol. 2012, 10, 171. [Google Scholar] [CrossRef] [PubMed]
- Guillén-Ponce, C.; Blázquez, J.; González, I.; de-Madaria, E.; Montáns, J.; Carrato, A. Diagnosis and staging of pancreatic ductal adenocarcinoma. Clin. Transl. Oncol. 2017, 19, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Javed, A.A.; Thoburn, C.; Wong, F.; Tie, J.; Gibbs, P.; Schmidt, C.M.; Yip-Schneider, M.T.; Allen, P.J.; Schattner, M. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 10202–10207. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Thery, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Che, S.P.; Kurywchak, P.; Tavormina, J.L.; Gansmo, L.B.; Correa de Sampaio, P.; Tachezy, M.; Bockhorn, M.; Gebauer, F.; Haltom, A.R.; et al. Detection of mutant KRAS and TP53 DNA in circulating exosomes from healthy individuals and patients with pancreatic cancer. Cancer Biol. Ther. 2017, 18, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Allen, P.J.; Kuk, D.; Castillo, C.F.; Basturk, O.; Wolfgang, C.L.; Cameron, J.L.; Lillemoe, K.D.; Ferrone, C.R.; Morales-Oyarvide, V.; He, J.; et al. Multi-institutional Validation Study of the American Joint Commission on Cancer (8th Edition) Changes for T and N Staging in Patients With Pancreatic Adenocarcinoma. Ann. Surg. 2017, 265, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Kamarajah, S.K.; Burns, W.R.; Frankel, T.L.; Cho, C.S.; Nathan, H. Validation of the American Joint Commission on Cancer (AJCC) 8th Edition Staging System for Patients with Pancreatic Adenocarcinoma: A Surveillance, Epidemiology and End Results (SEER) Analysis. Ann. Surg. Oncol. 2017, 24, 2023–2030. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Poplin, E.; Feng, Y.; Berlin, J.; Rothenberg, M.L.; Hochster, H.; Mitchell, E.; Alberts, S.; O’Dwyer, P.; Haller, D.; Catalano, P.; et al. Phase III, randomized study of gemcitabine and oxaliplatin versus gemcitabine (fixed-dose rate infusion) compared with gemcitabine (30-min infusion) in patients with pancreatic carcinoma E6201: A trial of the Eastern Cooperative Oncology Group. J. Clin. Oncol. 2009, 27, 3778–3785. [Google Scholar] [CrossRef] [PubMed]
- Bria, E.; Milella, M.; Gelibter, A.; Cuppone, F.; Pino, M.S.; Ruggeri, E.M.; Carlini, P.; Nistico, C.; Terzoli, E.; Cognetti, F.; et al. Gemcitabine-based combinations for inoperable pancreatic cancer: Have we made real progress? A meta-analysis of 20 phase 3 trials. Cancer 2007, 110, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Chin, V.; Nagrial, A.; Sjoquist, K.; O’Connor, C.A.; Chantrill, L.; Biankin, A.V.; Scholten, R.; Yip, D. Chemotherapy and radiotherapy for advanced pancreatic cancer. Cochrane Data. System. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sultana, A.; Smith, C.T.; Cunningham, D.; Starling, N.; Neoptolemos, J.P.; Ghaneh, P. Meta-analyses of chemotherapy for locally advanced and metastatic pancreatic cancer. J. Clin. Oncol. 2007, 25, 2607–2615. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Signorovitch, J.E.; Yang, H.; Patterson-Lomba, O.; Xiang, C.Q.; Ung, B.; Parisi, M.; Marshall, J.L. Comparative Effectiveness of nab-Paclitaxel Plus Gemcitabine vs. FOLFIRINOX in Metastatic Pancreatic Cancer: A Retrospective Nationwide Chart Review in the United States. Adv. Ther. 2018, 35, 1564–1577. [Google Scholar] [CrossRef] [PubMed]
- Sohal, D.P.; Mangu, P.B.; Laheru, D. Metastatic Pancreatic Cancer: American Society of Clinical Oncology Clinical Practice Guideline Summary. J. Oncol. Pract. 2017, 13, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Sohal, D.P.S.; Kennedy, E.B.; Khorana, A.; Copur, M.S.; Crane, C.H.; Garrido-Laguna, I.; Krishnamurthi, S.; Moravek, C.; O’Reilly, E.M.; Philip, P.A.; et al. Metastatic Pancreatic Cancer: ASCO Clinical Practice Guideline Update. J. Clin. Oncol. 2018, 36, 2545–2556. [Google Scholar] [CrossRef] [PubMed]
- Reni, M.; Zanon, S.; Peretti, U.; Chiaravalli, M.; Barone, D.; Pircher, C.; Balzano, G.; Macchini, M.; Romi, S.; Gritti, E.; et al. Nab-paclitaxel plus gemcitabine with or without capecitabine and cisplatin in metastatic pancreatic adenocarcinoma (PACT-19): A randomised phase 2 trial. Lancet Gastroenterol. Hepatol. 2018, 3, 691–697. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Timms, L.; Kalimuthu, S.N.; Selander, I.; McPherson, T.; Wilson, G.W.; Chan-Seng-Yue, M.A.; Borozan, I.; et al. Association of Distinct Mutational Signatures With Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2017, 3, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Gara, R.K.; Kumari, S.; Ganju, A.; Yallapu, M.M.; Jaggi, M.; Chauhan, S.C. Slit/Robo pathway: A promising therapeutic target for cancer. Drug Discov. Today 2015, 20, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Nones, K.; Waddell, N.; Song, S.; Patch, A.M.; Miller, D.; Johns, A.; Wu, J.; Kassahn, K.S.; Wood, D.; Bailey, P.; et al. Genome-wide DNA methylation patterns in pancreatic ductal adenocarcinoma reveal epigenetic deregulation of SLIT-ROBO, ITGA2 and MET signaling. Int. J. Cancer 2014, 135, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Huang, Y.H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massague, J. TGF-beta Tumor Suppression through a Lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef] [PubMed]
- Vivekanandhan, S.; Mukhopadhyay, D. Genetic status of KRAS influences Transforming Growth Factor-beta (TGF-beta) signaling: An insight into Neuropilin-1 (NRP1) mediated tumorigenesis. Semin. Cancer Biol. 2018; [Epub ahead of print]. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Mishra, L.; Deng, C.X. The role of TGF-beta/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2018, 62, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.R.W.; Herrmann, D.; Evans, T.R.J.; Morton, J.P.; Timpson, P. Combating pancreatic cancer with PI3K pathway inhibitors in the era of personalized medicine. Gut 2018. [Google Scholar] [CrossRef] [PubMed]
- Boyiadzis, M.M.; Kirkwood, J.M.; Marshall, J.L.; Pritchard, C.C.; Azad, N.S.; Gulley, J.L. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J. Immunother. Cancer 2018, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Mendt, M.; Kamerkar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.C.; Gagea, M.; Yang, S.; Blanko, E.V.R.; Peng, Q.; Ma, X.; et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Ihle, N.T.; Byers, L.A.; Kim, E.S.; Saintigny, P.; Lee, J.J.; Blumenschein, G.R.; Tsao, A.; Liu, S.; Larsen, J.E.; Wang, J.; et al. Effect of KRAS oncogene substitutions on protein behavior: Implications for signaling and clinical outcome. J. Natl. Cancer Inst. 2012, 104, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Der, C.J.; Philips, M.R. Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery? Clin. Cancer Res. 2015, 21, 1819–1827. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.H.; Cochrane, C.G.; Bourne, J.R.; Solski, P.A.; Buss, J.E.; Der, C.J. Farnesol modification of Kirsten-ras exon 4B protein is essential for transformation. Proc. Natl. Acad. Sci. USA 1990, 87, 3042–3046. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, J.S.; McCoy, S.; Whitehead, R.P.; Iqbal, S.; Wade, J.L., 3rd; Giguere, J.K.; Abbruzzese, J.L. A phase II study of farnesyl transferase inhibitor R115777 in pancreatic cancer: A Southwest oncology group (SWOG 9924) study. Investig. New Drugs 2005, 23, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; van de Velde, H.; Karasek, P.; Oettle, H.; Vervenne, W.L.; Szawlowski, A.; Schoffski, P.; Post, S.; Verslype, C.; Neumann, H.; et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J. Clin. Oncol. 2004, 22, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Ruess, D.A.; Heynen, G.J.; Ciecielski, K.J.; Ai, J.; Berninger, A.; Kabacaoglu, D.; Gorgulu, K.; Dantes, Z.; Wormann, S.M.; Diakopoulos, K.N.; et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med. 2018, 24, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. CSH Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, H.; Counter, C.M. Interrogating the protein interactomes of RAS isoforms identifies PIP5K1A as a KRAS-specific vulnerability. Nat. Commun. 2018, 9, 3646. [Google Scholar] [CrossRef] [PubMed]
- Bournet, B.; Buscail, C.; Muscari, F.; Cordelier, P.; Buscail, L. Targeting KRAS for diagnosis, prognosis, and treatment of pancreatic cancer: Hopes and realities. Eur. J. Cancer 2016, 54, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Sinn, M.; Bahra, M.; Liersch, T.; Gellert, K.; Messmann, H.; Bechstein, W.; Waldschmidt, D.; Jacobasch, L.; Wilhelm, M.; Rau, B.M.; et al. CONKO-005: Adjuvant Chemotherapy With Gemcitabine Plus Erlotinib Versus Gemcitabine Alone in Patients After R0 Resection of Pancreatic Cancer: A Multicenter Randomized Phase III Trial. J. Clin. Oncol. 2017, 35, 3330–3337. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Vervenne, W.L.; Bennouna, J.; Humblet, Y.; Gill, S.; Van Laethem, J.L.; Verslype, C.; Scheithauer, W.; Shang, A.; Cosaert, J.; et al. Phase III trial of bevacizumab in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. J. Clin. Oncol. 2009, 27, 2231–2237. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, V.; Vehling-Kaiser, U.; Waldschmidt, D.; Kettner, E.; Marten, A.; Winkelmann, C.; Klein, S.; Kojouharoff, G.; Gauler, T.C.; von Weikersthal, L.F.; et al. Gemcitabine plus erlotinib followed by capecitabine versus capecitabine plus erlotinib followed by gemcitabine in advanced pancreatic cancer: Final results of a randomised phase 3 trial of the ‘Arbeitsgemeinschaft Internistische Onkologie’ (AIO-PK0104). Gut 2013, 62, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Siveke, J.T.; Schenk, M.; Lerch, M.M.; Caca, K.; Freiberg-Richter, J.; Fischer von Weikersthal, L.; Kullmann, F.; Reinacher-Schick, A.; Fuchs, M.; et al. Efficacy of gemcitabine plus erlotinib in rash-positive patients with metastatic pancreatic cancer selected according to eligibility for FOLFIRINOX: A prospective phase II study of the ‘Arbeitsgemeinschaft Internistische Onkologie’. Eur. J. Cancer 2018, 94, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Boeck, S.; Jung, A.; Laubender, R.P.; Neumann, J.; Egg, R.; Goritschan, C.; Vehling-Kaiser, U.; Winkelmann, C.; Fischer von Weikersthal, L.; Clemens, M.R.; et al. EGFR pathway biomarkers in erlotinib-treated patients with advanced pancreatic cancer: Translational results from the randomised, crossover phase 3 trial AIO-PK0104. Br. J. Cancer 2013, 108, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Schultheis, B.; Reuter, D.; Ebert, M.P.; Siveke, J.; Kerkhoff, A.; Berdel, W.E.; Hofheinz, R.; Behringer, D.M.; Schmidt, W.E.; Goker, E.; et al. Gemcitabine combined with the monoclonal antibody nimotuzumab is an active first-line regimen in KRAS wildtype patients with locally advanced or metastatic pancreatic cancer: A multicenter, randomized phase IIb study. Ann. Oncol. 2017, 28, 2429–2435. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Schutte, M.; Hruban, R.H.; Geradts, J.; Maynard, R.; Hilgers, W.; Rabindran, S.K.; Moskaluk, C.A.; Hahn, S.A.; Schwarte-Waldhoff, I.; Schmiegel, W.; et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997, 57, 3126–3130. [Google Scholar] [PubMed]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar] [PubMed]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. Thirty-Fifth Annual CTRC-AACR San Antonio Breast Cancer Symposium; Cancer Research: San Antonio, TX, USA, 2012; Volume 72. [Google Scholar]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.; Balaji, U.; Freinkman, E.; Witkiewicz, A.K.; Knudsen, E.S. Metabolic Reprogramming of Pancreatic Cancer Mediated by CDK4/6 Inhibition Elicits Unique Vulnerabilities. Cell Rep. 2016, 14, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.; Witkiewicz, A.K.; Knudsen, E.S. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget 2014, 5, 6512–6525. [Google Scholar] [CrossRef] [PubMed]
- Chiorean, E.G.; Hochster, H.S.; Nanda, S.; Thornton, D.; Klise, S.A. A phase II study of abemaciclib as a monotherapy and in combination with other agents in patients with previously treated metastatic pancreatic ductal adenocarcinoma (PDAC). J. Clin. Oncol. 2017. [Google Scholar] [CrossRef]
- Chou, A.; Froio, D.; Nagrial, A.M.; Parkin, A.; Murphy, K.J.; Chin, V.T.; Wohl, D.; Steinmann, A.; Stark, R.; Drury, A.; et al. Tailored first-line and second-line CDK4-targeting treatment combinations in mouse models of pancreatic cancer. Gut 2018, 67, 2142–2155. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Yu, J.; Deng, M.; Yin, Y.; Zhang, H.; Luo, K.; Qin, B.; Li, Y.; Wu, C.; Ren, T.; et al. CDK4/6-dependent activation of DUB3 regulates cancer metastasis through SNAIL1. Nature commun. 2017, 8, 13923. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Ford, D.; Easton, D.F.; Bishop, D.T.; Narod, S.A.; Goldgar, D.E. Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet 1994, 343, 692–695. [Google Scholar] [CrossRef]
- Greer, J.B.; Whitcomb, D.C. Role of BRCA1 and BRCA2 mutations in pancreatic cancer. Gut 2007, 56, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Sehdev, A.; Gbolahan, O.; Hancock, B.A.; Stanley, M.; Shahda, S.; Wan, J.; Wu, H.H.; Radovich, M.; O’Neil, B.H. Germline and Somatic DNA Damage Repair Gene Mutations and Overall Survival in Metastatic Pancreatic Adenocarcinoma Patients Treated with FOLFIRINOX. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Villarroel, M.C.; Rajeshkumar, N.V.; Garrido-Laguna, I.; De Jesus-Acosta, A.; Jones, S.; Maitra, A.; Hruban, R.H.; Eshleman, J.R.; Klein, A.; Laheru, D.; et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol. Cancer Ther. 2011, 10, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, V.; Quietzsch, D.; Gieseler, F.; Gonnermann, M.; Schonekas, H.; Rost, A.; Neuhaus, H.; Haag, C.; Clemens, M.; Heinrich, B.; et al. Randomized phase III trial of gemcitabine plus cisplatin compared with gemcitabine alone in advanced pancreatic cancer. J. Clin. Oncol. 2006, 24, 3946–3952. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, V.; Wilke, H.; Mergenthaler, H.G.; Clemens, M.; Konig, H.; Illiger, H.J.; Arning, M.; Schalhorn, A.; Possinger, K.; Fink, U. Gemcitabine and cisplatin in the treatment of advanced or metastatic pancreatic cancer. Ann. Oncol. 2000, 11, 1399–1403. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, V.; Labianca, R.; Hinke, A.; Louvet, C. Increased survival using platinum analog combined with gemcitabine as compared to single-agent gemcitabine in advanced pancreatic cancer: Pooled analysis of two randomized trials, the GERCOR/GISCAD intergroup study and a German multicenter study. Ann. Oncol. 2007, 18, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Sonnenblick, A.; Kadouri, L.; Appelbaum, L.; Peretz, T.; Sagi, M.; Goldberg, Y.; Hubert, A. Complete remission, in BRCA2 mutation carrier with metastatic pancreatic adenocarcinoma, treated with cisplatin based therapy. Cancer Biol. Ther. 2011, 12, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Kanji, Z.S.; Epelbaum, R.; Devaud, N.; Dagan, E.; Holter, S.; Aderka, D.; Paluch-Shimon, S.; Kaufman, B.; Gershoni-Baruch, R.; et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br. J. Cancer 2014, 111, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.T.; Hendifar, A.; McWilliams, R.R.; Geva, R.; Epelbaum, R.; Rolfe, L.; Goble, S.; Lin, K.K.; Biankin, A.V.; Giordano, H.; et al. Rucaparib Monotherapy in Patients With Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis. Oncol. 2018, 2, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lowery, M.A.; Kelsen, D.P.; Capanu, M.; Smith, S.C.; Lee, J.W.; Stadler, Z.K.; Moore, M.J.; Kindler, H.L.; Golan, T.; Segal, A.; et al. Phase II trial of veliparib in patients with previously treated BRCA-mutated pancreas ductal adenocarcinoma. Eur. J. Cancer 2018, 89, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Tempero, M.A.; Malafa, M.P.; Al-Hawary, M.; Asbun, H.; Bain, A.; Behrman, S.W.; Benson, A.B., 3rd; Binder, E.; Cardin, D.B.; Cha, C.; et al. Pancreatic Adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J. Nat. Compr. Cancer Net. JNCCN 2017, 15, 1028–1061. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crino, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Aglietta, M.; Barone, C.; Sawyer, M.B.; Moore, M.J.; Miller, W.H., Jr.; Bagala, C.; Colombi, F.; Cagnazzo, C.; Gioeni, L.; Wang, E.; et al. A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann. Oncol. 2014, 25, 1750–1755. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Baretti, M.; Le, D.T. DNA mismatch repair in cancer. Pharmacol. Ther. 2018, 189, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.I.; Shia, J.; Stadler, Z.K.; Varghese, A.M.; Capanu, M.; Salo-Mullen, E.; Lowery, M.A.; Diaz, L.A., Jr.; Mandelker, D.; Yu, K.H.; et al. Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clin. Cancer Res. 2018, 24, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Vennin, C.; Murphy, K.J.; Morton, J.P.; Cox, T.R.; Pajic, M.; Timpson, P. Reshaping the Tumor Stroma for Treatment of Pancreatic Cancer. Gastroenterology 2018, 154, 820–838. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Samuel, M.S.; Lopez, J.I.; McGhee, E.J.; Croft, D.R.; Strachan, D.; Timpson, P.; Munro, J.; Schroder, E.; Zhou, J.; Brunton, V.G.; et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and beta-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell 2011, 19, 776–791. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Ibbetson, S.J.; Pyne, N.T.; Pollard, A.N.; Olson, M.F.; Samuel, M.S. Mechanotransduction pathways promoting tumor progression are activated in invasive human squamous cell carcinoma. Am. J. Pathol. 2013, 183, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, G.E.; Barrett, A.S.; Hill, R.C.; Lakins, J.N.; Schlaepfer, D.D.; Mouw, J.K.; et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Chin, V.T.; Nagrial, A.M.; Chou, A.; Biankin, A.V.; Gill, A.J.; Timpson, P.; Pajic, M. Rho-associated kinase signalling and the cancer microenvironment: Novel biological implications and therapeutic opportunities. Expert Rev. Mol. Med. 2015, 17, e17. [Google Scholar] [CrossRef] [PubMed]
- Pajic, M.; Herrmann, D.; Vennin, C.; Conway, J.R.; Chin, V.T.; Johnsson, A.E.; Welch, H.C.; Timpson, P. The dynamics of Rho GTPase signaling and implications for targeting cancer and the tumor microenvironment. Small GTPases 2015, 6, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Sabbineni, H.; Clarke, A.; Somanath, P.R. Novel roles of Src in cancer cell epithelial-to-mesenchymal transition, vascular permeability, microinvasion and metastasis. Life Sci. 2016, 157, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Frankson, R.; Yu, Z.H.; Bai, Y.; Li, Q.; Zhang, R.Y.; Zhang, Z.Y. Therapeutic Targeting of Oncogenic Tyrosine Phosphatases. Cancer Res. 2017, 77, 5701–5705. [Google Scholar] [CrossRef] [PubMed]
- Kanteti, R.; Mirzapoiazova, T.; Riehm, J.J.; Dhanasingh, I.; Mambetsariev, B.; Wang, J.; Kulkarni, P.; Kaushik, G.; Seshacharyulu, P.; Ponnusamy, M.P.; et al. Focal adhesion kinase a potential therapeutic target for pancreatic cancer and malignant pleural mesothelioma. Cancer Biol. Ther. 2018, 19, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Begum, A.; Ewachiw, T.; Jung, C.; Huang, A.; Norberg, K.J.; Marchionni, L.; McMillan, R.; Penchev, V.; Rajeshkumar, N.V.; Maitra, A.; et al. The extracellular matrix and focal adhesion kinase signaling regulate cancer stem cell function in pancreatic ductal adenocarcinoma. PLoS ONE 2017, 12, e0180181. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, I.M.; Kolev, V.N.; Vidal, C.M.; Kadariya, Y.; Ring, J.E.; Wright, Q.; Weaver, D.T.; Menges, C.; Padval, M.; McClatchey, A.I.; et al. Merlin deficiency predicts FAK inhibitor sensitivity: A synthetic lethal relationship. Sci. Transl. Med. 2014, 6, 237ra68. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Gan, H.K.; Blagden, S.P.; Plummer, R.; Arkenau, H.T.; Ranson, M.; Evans, T.R.; Zalcman, G.; Bahleda, R.; Hollebecque, A.; et al. A phase I, pharmacokinetic and pharmacodynamic study of GSK2256098, a focal adhesion kinase inhibitor, in patients with advanced solid tumors. Ann. Oncol. 2016, 27, 2268–2274. [Google Scholar] [CrossRef] [PubMed]
- Quan, M.; Cui, J.; Xia, T.; Jia, Z.; Xie, D.; Wei, D.; Huang, S.; Huang, Q.; Zheng, S.; Xie, K.; et al. Merlin/NF2 Suppresses Pancreatic Tumor Growth and Metastasis by Attenuating the FOXM1-Mediated Wnt/beta-Catenin Signaling. Cancer Res. 2015, 75, 4778–4789. [Google Scholar] [CrossRef] [PubMed]
- Rath, N.; Morton, J.P.; Julian, L.; Helbig, L.; Kadir, S.; McGhee, E.J.; Anderson, K.I.; Kalna, G.; Mullin, M.; Pinho, A.V.; et al. ROCK signaling promotes collagen remodeling to facilitate invasive pancreatic ductal adenocarcinoma tumor cell growth. EMBO Mol. Med. 2017, 9, 198–218. [Google Scholar] [CrossRef] [PubMed]
- Rath, N.; Munro, J.; Cutiongco, M.F.; Jagiello, A.; Gadegaard, N.; McGarry, L.; Unbekandt, M.; Michalopoulou, E.; Kamphorst, J.J.; Sumpton, D.; et al. Rho Kinase Inhibition by AT13148 Blocks Pancreatic Ductal Adenocarcinoma Invasion and Tumor Growth. Cancer Res. 2018, 78, 3321–3336. [Google Scholar] [PubMed]
- Vennin, C.; Chin, V.T.; Warren, S.C.; Lucas, M.C.; Herrmann, D.; Magenau, A.; Melenec, P.; Walters, S.N.; Del Monte-Nieto, G.; Conway, J.R.; et al. Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Satoh, S.; Takayasu, M.; Kawasaki, K.; kegaki, I.; Hitomi, A.; Yano, K.; Shibuya, M.; Asano, T. Antivasospastic effects of hydroxyfasudil, a Rho-kinase inhibitor, after subarachnoid hemorrhage. J. Pharmacol. Sci. 2012, 118, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M.; Hirai, S.; Seto, M.; Satoh, S.; Ohtomo, E. Effects of fasudil in acute ischemic stroke: Results of a prospective placebo-controlled double-blind trial. J. Neurol. Sci. 2005, 238, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.M.; Horton, K.J.; Coveler, A.L.; Hingorani, S.R.; Harris, W.P. Targeting the Tumor Stroma: The Biology and Clinical Development of Pegylated Recombinant Human Hyaluronidase (PEGPH20). Curr. Oncol. Rep. 2017, 19, 47. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Harris, W.P.; Beck, J.T.; Berdov, B.A.; Wagner, S.A.; Pshevlotsky, E.M.; Tjulandin, S.A.; Gladkov, O.A.; Holcombe, R.F.; Korn, R.; et al. Phase Ib Study of PEGylated Recombinant Human Hyaluronidase and Gemcitabine in Patients with Advanced Pancreatic Cancer. Clin. Cancer Res. 2016, 22, 2848–2854. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parkin, A.; Man, J.; Chou, A.; Nagrial, A.M.; Samra, J.; Gill, A.J.; Timpson, P.; Pajic, M. The Evolving Understanding of the Molecular and Therapeutic Landscape of Pancreatic Ductal Adenocarcinoma. Diseases 2018, 6, 103. https://doi.org/10.3390/diseases6040103
Parkin A, Man J, Chou A, Nagrial AM, Samra J, Gill AJ, Timpson P, Pajic M. The Evolving Understanding of the Molecular and Therapeutic Landscape of Pancreatic Ductal Adenocarcinoma. Diseases. 2018; 6(4):103. https://doi.org/10.3390/diseases6040103
Chicago/Turabian StyleParkin, Ashleigh, Jennifer Man, Angela Chou, Adnan M Nagrial, Jaswinder Samra, Anthony J Gill, Paul Timpson, and Marina Pajic. 2018. "The Evolving Understanding of the Molecular and Therapeutic Landscape of Pancreatic Ductal Adenocarcinoma" Diseases 6, no. 4: 103. https://doi.org/10.3390/diseases6040103
APA StyleParkin, A., Man, J., Chou, A., Nagrial, A. M., Samra, J., Gill, A. J., Timpson, P., & Pajic, M. (2018). The Evolving Understanding of the Molecular and Therapeutic Landscape of Pancreatic Ductal Adenocarcinoma. Diseases, 6(4), 103. https://doi.org/10.3390/diseases6040103