
Targeting FGF19/FGFR4 Pathway: A Novel Therapeutic Strategy for Hepatocellular Carcinoma

Abstract
:1. Introduction
2. FGF Family
- Intracrine: they consist of the FGF11 subfamily (FGF11, 12, 13, 14). These factors are secreted intracellularly, they do not interact with FGFRs and their main role is regulation of the electrical excitability of neurons and other excitable cells like cardiomyocytes.
- Paracrine: they consist of the FGF1 subfamily (FGF1,2), the FGF4 subfamily (FGF4,5,6), the FGF7 subfamily (FGF3,7,10,22), the FGF9 subfamily (FGF9,17,18) and the FGF8 subfamily (FGF8,17,18). These are secreted proteins that bind to FGFRs and use heparin/heparan sulphate as a cofactor, although FGF1, 2, 3 can directly translocate to the nucleus and act as intracrine proteins. They are involved in embryogenesis and tissue repair by regulating cell proliferation, differentiation and survival.
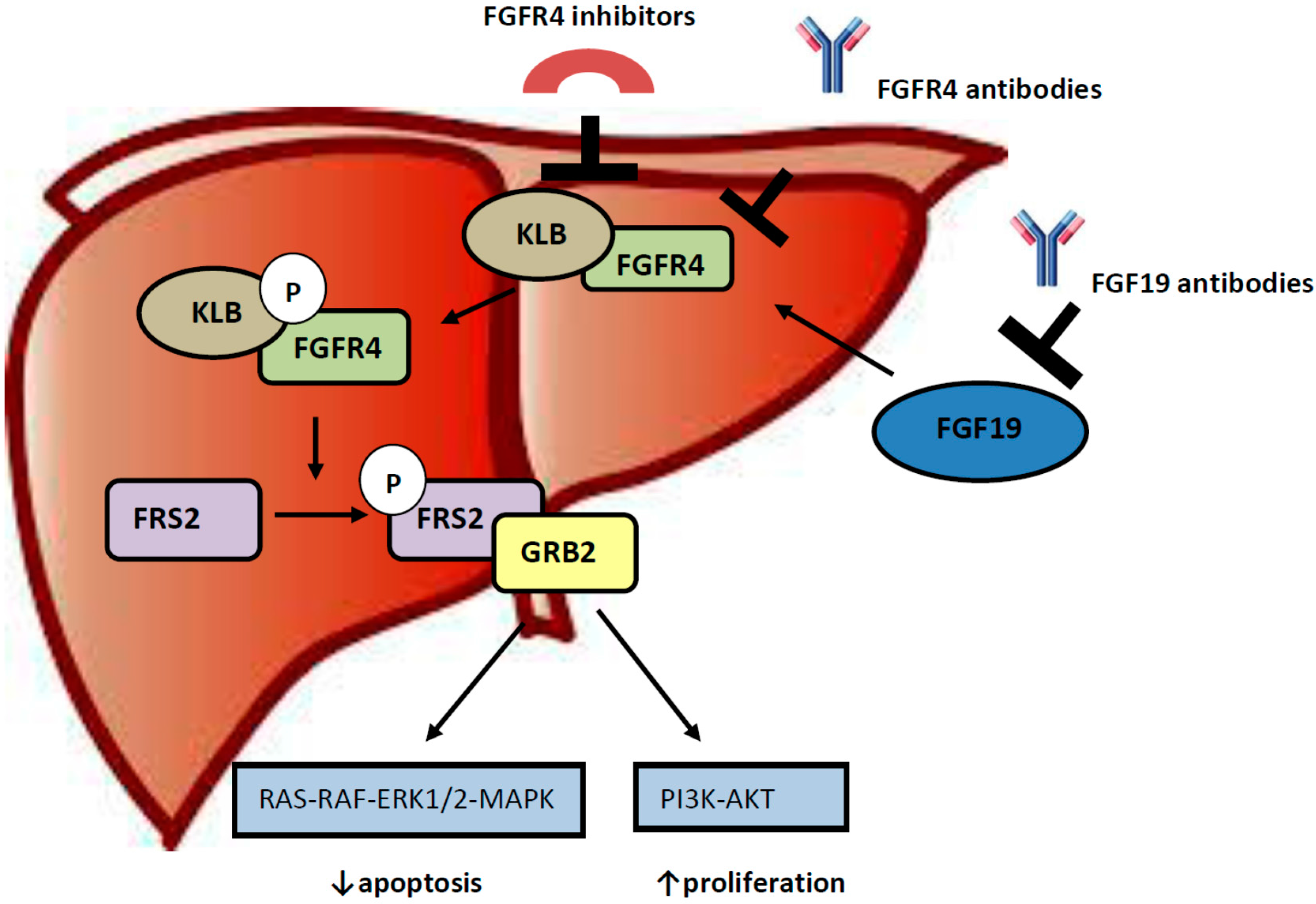
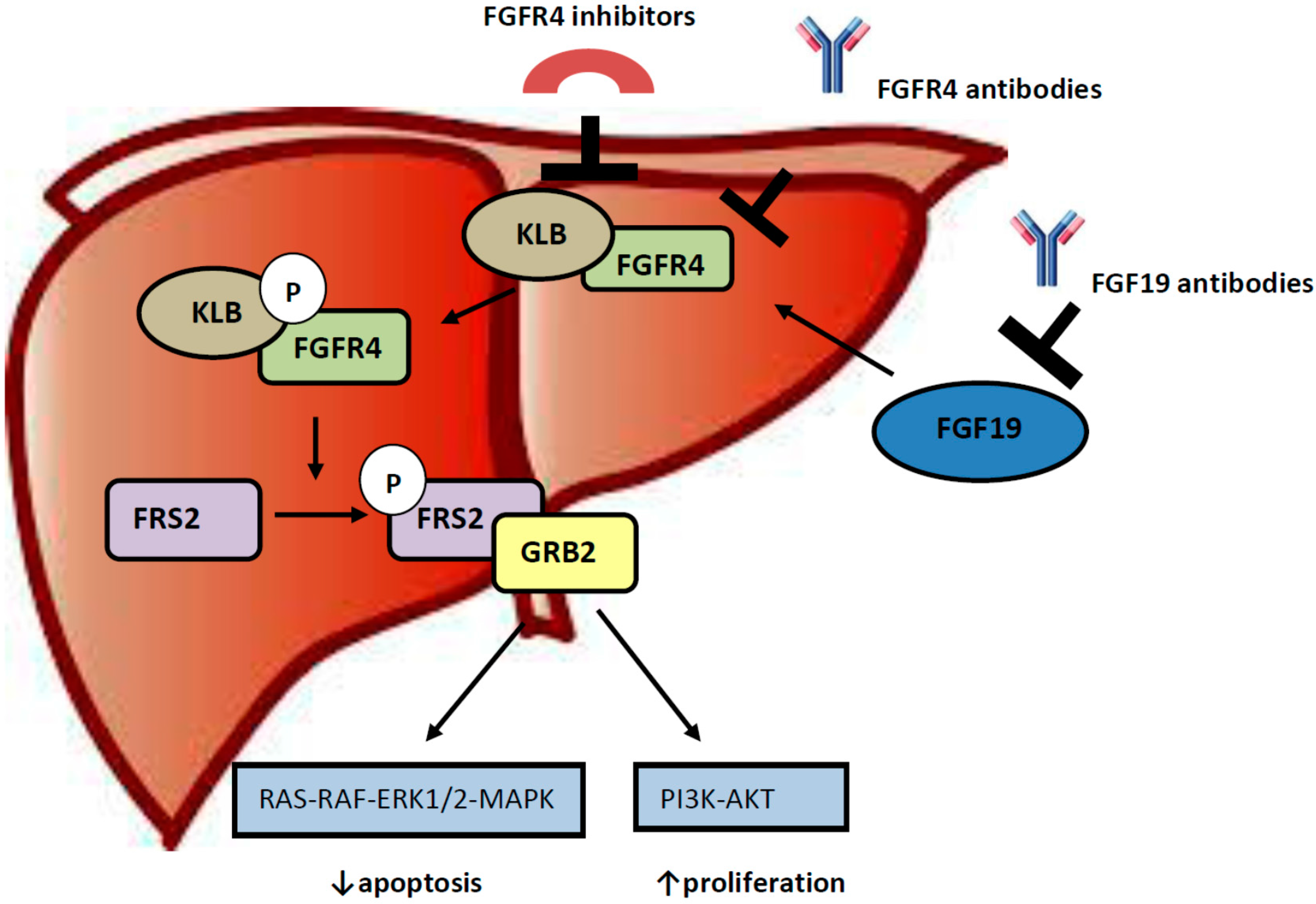
3. Role of FGF 19-FGFR4 Pathway in Hepatocellular Carcinogenesis
4. Targeting the FGF19/FGFR4 Pathway

{kind=link}
| Open Trials Targeting the FGF19/FGFR4 Pathway in Various Tumours | |
|---|---|
| AZD4547 | AZD4547 & Anastrozole or Letrozole in ER+ Breast Cancer Patients Who Have Progressed on NSAIs (RADICAL) |
| AZD4547 | Lung-MAP: S1400 Biomarker-Targeted Second-Line Therapy in Treating Patients with Recurrent Stage IIIB-IV Squamous Cell Lung Cancer |
| AZD4547 | Intergroup Trial UNICANCER UC 0105-1305/ IFCT 1301: Efficacy of Targeted Drugs Guided by Genomic Profiles in Metastatic NSCLC Patients (SAFIR02_Lung) |
| AZD4547 | Evaluation of the Efficacy of High Throughput Genome Analysis as a Therapeutic Decision Tool for Patients with Metastatic Breast Cancer (SAFIR02_Breast) |
| AZD4547 | Open-Label, Randomised, Multi-Drug, Biomarker-Directed, Phase 1b Study in Pts w/Muscle Invasive Bladder Cancer (BISCAY) |
| JNJ-42756493 | A Study to Evaluate the Safety, Pharmacokinetics, and Pharmacodynamics of JNJ-42756493 in Adult Participants with Advanced or Refractory Solid Tumors or Lymphoma |
| JNJ-42756493 | An Efficacy and Safety Study of JNJ-42756493 in Participants with Urothelial Cancer |
| JNJ-42756493 | Study to Evaluate the Safety, Pharmacokinetics, and Pharmacodynamics of JNJ-42756493 in Participants with Advanced Hepatocellular Carcinoma |
| FGF401 | Safety and Efficacy of FGF401 in Patients with Solid Malignancies |
| BLU554 | A Phase 1 Study of BLU-554 in Patients with Hepatocellular Carcinoma and Cholangiocarcinoma |
5. Discussion
6. Conclusions
Conflicts of Interest
References
- Ferlay, J.S.I.; Ervik, M.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [Internet]. International Agency for Research on Cancer: Lyon, France, 2013. Available online: http://globocan.iarc.fr (accessed on 20 July 2015).
- Simonetti, R.G.; Camma, C.; Fiorello, F.; Politi, F.; D’Amico, G.; Pagliaro, L. Hepatocellular carcinoma. A worldwide problem and the major risk factors. Dig. Dis. Sci. 1991, 36, 962–972. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Hepatocellular carcinoma. New Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Perz, J.F.; Armstrong, G.L.; Farrington, L.A.; Hutin, Y.J.; Bell, B.P. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006, 45, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Noureddin, M.; Rinella, M.E. Nonalcoholic Fatty liver disease, diabetes, obesity, and hepatocellular carcinoma. Clin. Liver Dis. 2015, 19, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Bodzin, A.S.; Busuttil, R.W. Hepatocellular carcinoma: Advances in diagnosis, management, and long term outcome. World J. Hepatol. 2015, 7, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- European Association For The Study of The Liver; European Organisation For Research and Treatment of Cancer. EASL-EORTC clinical practice guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2012, 56, 908–943. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. New Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Farazi, P.A.; DePinho, R.A. Hepatocellular carcinoma pathogenesis: From genes to environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef] [PubMed]
- Moeini, A.; Cornella, H.; Villanueva, A. Emerging signaling pathways in hepatocellular carcinoma. Liver Cancer 2012, 1, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Zender, L.; Villanueva, A.; Tovar, V.; Sia, D.; Chiang, D.Y.; Llovet, J.M. Cancer gene discovery in hepatocellular carcinoma. J. Hepatol. 2010, 52, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Kang, Y.K.; Mulcahy, M.; Polite, B.N.; Lim, H.Y.; Walters, I.; Baudelet, C.; Manekas, D.; Park, J.W. Phase II, open-label study of brivanib as second-line therapy in patients with advanced hepatocellular carcinoma. Clin. Cancer Res. 2012, 18, 2090–2098. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.J.; Qin, S.; Park, J.W.; Poon, R.T.; Raoul, J.L.; Philip, P.A.; Hsu, C.H.; Hu, T.H.; Heo, J.; Xu, J.; et al. Brivanib versus sorafenib as first-line therapy in patients with unresectable, advanced hepatocellular carcinoma: Results from the randomized phase III BRISK-FL study. J. Clin. Oncol. 2013, 31, 3517–3524. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Decaens, T.; Raoul, J.L.; Boucher, E.; Kudo, M.; Chang, C.; Kang, Y.K.; Assenat, E.; Lim, H.Y.; Boige, V.; et al. Brivanib in patients with advanced hepatocellular carcinoma who were intolerant to sorafenib or for whom sorafenib failed: Results from the randomized phase III BRISK-PS study. J. Clin. Oncol. 2013, 31, 3509–3516. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.T.S.; Thongprasert, S.; Lim, H.Y.; Sukeepaisarnjaroen, W.; Yang, T.-S.; Wu, C.-C.; Chao, Y.; Chan, S.L.; Kudo, M.; Ikeda, M.; et al. Phase II study of front-line dovitinib (TKI258) versus sorafenib in patients (Pts) with advanced hepatocellular carcinoma (HCC). Gastrointestinal Cancers Symposium (15–17 January 2015). J. Clin. Oncol. 2015, 33 (Suppl. S3), 237. [Google Scholar]
- McCormack, P.L. Nintedanib: First global approval. Drugs 2015, 75, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.M.Y.; Ma, Y.T.; Peck-Radosavljevic, M.; Ross, P.J.; Graham, J.S.; Fartoux, L.; Deptala, A.; Wenz, A.; Hocke, J.; Loembe, A.-B.; et al. Randomized phase II trial comparing the efficacy and safety of nintedanib versus sorafenib in patients with advanced hepatocellular carcinoma (HCC). 2015 Gastrointestinal Cancers Symposium. J. Clin. Oncol. 2015, 33 (Suppl. S3), 238. [Google Scholar]
- Itoh, N.; Ornitz, D.M. Fibroblast growth factors: From molecular evolution to roles in development, metabolism and disease. J. Biochem. 2011, 149, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- Kir, S.; Kliewer, S.A.; Mangelsdorf, D.J. Roles of FGF19 in liver metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.Y.; Xie, D.M.; Zhu, G.Q.; Huang, G.Q.; Lin, Y.Q.; Wang, L.R.; Shi, K.Q.; Hu, B.; Braddock, M.; Chen, Y.P.; et al. Targeting fibroblast growth factor 19 in liver disease: A potential biomarker and therapeutic target. Expert Opin. Ther. Targets 2015, 19, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.C.; Wang, M.; Blackmore, C.; Desnoyers, L.R. Liver-specific activities of FGF19 require Klotho beta. J. Biol. Chem. 2007, 282, 27277–27284. [Google Scholar] [CrossRef] [PubMed]
- Kan, M.; Wu, X.; Wang, F.; McKeehan, W.L. Specificity for fibroblast growth factors determined by heparan sulfate in a binary complex with the receptor kinase. J. Biol. Chem. 1999, 274, 15947–15952. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, E.; Fu, L.; John, L.; Hultgren, B.; Huang, X.; Renz, M.; Stephan, J.P.; Tsai, S.P.; Powell-Braxton, L.; French, D.; et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology 2002, 143, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Goetz, R.; Beenken, A.; Ibrahimi, O.A.; Kalinina, J.; Olsen, S.K.; Eliseenkova, A.V.; Xu, C.; Neubert, T.A.; Zhang, F.; Linhardt, R.J.; et al. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol. Cell. Biol. 2007, 27, 3417–3428. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Nakagama, H. FGF receptors: Cancer biology and therapeutics. Med. Res. Rev. 2014, 34, 280–300. [Google Scholar] [CrossRef] [PubMed]
- Miura, S.; Mitsuhashi, N.; Shimizu, H.; Kimura, F.; Yoshidome, H.; Otsuka, M.; Kato, A.; Shida, T.; Okamura, D.; Miyazaki, M. Fibroblast growth factor 19 expression correlates with tumor progression and poorer prognosis of hepatocellular carcinoma. BMC Cancer 2012, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Nicholes, K.; Guillet, S.; Tomlinson, E.; Hillan, K.; Wright, B.; Frantz, G.D.; Pham, T.A.; Dillard-Telm, L.; Tsai, S.P.; Stephan, J.P.; et al. A mouse model of hepatocellular carcinoma: Ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am. J. Pathol. 2002, 160, 2295–2307. [Google Scholar] [CrossRef]
- Uriarte, I.; Fernandez-Barrena, M.G.; Monte, M.J.; Latasa, M.U.; Chang, H.C.; Carotti, S.; Vespasiani-Gentilucci, U.; Morini, S.; Vicente, E.; Concepcion, A.R.; et al. Identification of fibroblast growth factor 15 as a novel mediator of liver regeneration and its application in the prevention of post-resection liver failure in mice. Gut 2013, 62, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Uriarte, I.; Latasa, M.U.; Carotti, S.; Fernandez-Barrena, M.G.; Garcia-Irigoyen, O.; Elizalde, M.; Urtasun, R.; Vespasiani-Gentilucci, U.; Morini, S.; de Mingo, A.; et al. Ileal FGF15 contributes to fibrosis-associated hepatocellular carcinoma development. Int. J. Cancer J. Int. Cancer 2015, 136, 2469–2475. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, J.; Ahn, S.; Lee, J.J.; Song, D.H.; Park, C.K. Expression of fibroblast growth factor 19 is associated with recurrence and poor prognosis of hepatocellular carcinoma. Dig. Dis. Sci. 2013, 58, 1916–1922. [Google Scholar] [CrossRef] [PubMed]
- Gauglhofer, C.; Paur, J.; Schrottmaier, W.C.; Wingelhofer, B.; Huber, D.; Naegelen, I.; Pirker, C.; Mohr, T.; Heinzle, C.; Holzmann, K.; et al. Fibroblast growth factor receptor 4: A putative key driver for the aggressive phenotype of hepatocellular carcinoma. Carcinogenesis 2014, 35, 2331–2338. [Google Scholar] [CrossRef] [PubMed]
- French, D.M.; Lin, B.C.; Wang, M.; Adams, C.; Shek, T.; Hotzel, K.; Bolon, B.; Ferrando, R.; Blackmore, C.; Schroeder, K.; et al. Targeting FGFR4 inhibits hepatocellular carcinoma in preclinical mouse models. PLoS ONE 2012, 7, e36713. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.K.; Pok, S.; Streit, S.; Ruhe, J.E.; Hart, S.; Lim, K.S.; Loo, H.L.; Aung, M.O.; Lim, S.G.; Ullrich, A. Fibroblast growth factor receptor 4 regulates proliferation, anti-apoptosis and alpha-fetoprotein secretion during hepatocellular carcinoma progression and represents a potential target for therapeutic intervention. J. Hepatol. 2009, 50, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Da Costa Andrade, V.C.; Parise, O., Jr.; Hors, C.P.; de Melo Martins, P.C.; Silva, A.P.; Garicochea, B. The fibroblast growth factor receptor 4 (FGFR4) Arg388 allele correlates with survival in head and neck squamous cell carcinoma. Exp. Mol. Pathol. 2007, 82, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Streit, S.; Bange, J.; Fichtner, A.; Ihrler, S.; Issing, W.; Ullrich, A. Involvement of the FGFR4 Arg388 allele in head and neck squamous cell carcinoma. Int. J. Cancer J. Int. Cancer 2004, 111, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Stockton, D.W.; Ittmann, M. The fibroblast growth factor receptor-4 Arg388 allele is associated with prostate cancer initiation and progression. Clin. Cancer Res. 2004, 10, 6169–6178. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Xie, B.; Zhu, Q.; Xia, Q.; Jiang, S.; Cao, R.; Shi, L.; Qi, D.; Li, X.; Cai, L. FGFR4 and TGF-beta1 expression in hepatocellular carcinoma: Correlation with clinicopathological features and prognosis. Int. J. Med. Sci. 2013, 10, 1868–1875. [Google Scholar] [CrossRef] [PubMed]
- Poh, W.; Wong, W.; Ong, H.; Aung, M.O.; Lim, S.G.; Chua, B.T.; Ho, H.K. Klotho-beta overexpression as a novel target for suppressing proliferation and fibroblast growth factor receptor-4 signaling in hepatocellular carcinoma. Mol. Cancer 2012, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Hoover, H.; Li, J.; Marchese, J.; Rothwell, C.; Borawoski, J.; Jeffery, D.A.; Gaither, L.A.; Finkel, N. Quantitative Proteomic Verification of Membrane Proteins as Potential Therapeutic Targets Located in the 11q13 Amplicon in Cancers. J. Proteome Res. 2015, 14, 3670–3679. [Google Scholar] [CrossRef] [PubMed]
- Sawey, E.T.; Chanrion, M.; Cai, C.; Wu, G.; Zhang, J.; Zender, L.; Zender, L.; Zhao, A.; Busuttil, R.W.; Yee, H.; Stein, L.; et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by Oncogenomic screening. Cancer Cell 2011, 19, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.M.; Jang, S.J.; Shim, J.H.; Kim, D.; Hong, S.M.; Sung, C.O.; Baek, D.; Haq, F.; Ansari, A.A.; Lee, S.Y.; et al. Genomic portrait of resectable hepatocellular carcinomas: Implications of RB1 and FGF19 aberrations for patient stratification. Hepatology 2014, 60, 1972–1982. [Google Scholar] [CrossRef] [PubMed]
- Touat, M.; Ileana, E.; Postel-Vinay, S.; Andre, F.; Soria, J.C. Targeting FGFR Signaling in Cancer. Clin. Cancer Res. 2015, 21, 2684–2694. [Google Scholar] [CrossRef] [PubMed]
- Guagnano, V.; Kauffmann, A.; Wohrle, S.; Stamm, C.; Ito, M.; Barys, L.; Pornon, A.; Yao, Y.; Li, F.; Zhang, Y.; et al. FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discov. 2012, 2, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Gavine, P.R.; Mooney, L.; Kilgour, E.; Thomas, A.P.; Al-Kadhimi, K.; Beck, S.; Rooney, C.; Coleman, T.; Baker, D.; Mellor, M.J.; et al. AZD4547: An orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res. 2012, 72, 2045–2056. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Wang, J.; Tanizaki, J.; Huang, Z.; Aref, A.R.; Rusan, M.; Zhu, S.J.; Zhang, Y.; Ercan, D.; Liao, R.G.; et al. Development of covalent inhibitors that can overcome resistance to first-generation FGFR kinase inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, E4869–E4877. [Google Scholar] [CrossRef] [PubMed]
- Bahleda, R.; Dienstmann, R.; Adamo, B.; Gazzah, A.; Infante, J.R.; Zhong, B.; J. Platero, S.J.; Smit, H.; Perera, T.; Stuyckens, K.; et al. Phase 1 study of JNJ-42756493, a pan-fibroblast growth factor receptor (FGFR) inhibitor, in patients with advanced solid tumors. 2014 ASCO Annual Meeting. J. Clin. Oncol. 2014, 32 (Suppl. S5), 2501. [Google Scholar]
- Zhao, G.; Li, W.Y.; Chen, D.; Henry, J.R.; Li, H.Y.; Chen, Z.; Zia-Ebrahimi, M.; Bloem, L.; Zhai, Y.; Huss, K.; et al. A novel, selective inhibitor of fibroblast growth factor receptors that shows a potent broad spectrum of antitumor activity in several tumor xenograft models. Mol. Cancer Ther. 2011, 10, 2200–2210. [Google Scholar] [CrossRef] [PubMed]
- Mooney, L.S.K.; Shea, K.; Cross, S.; Zhu, C.; Buttar, D.; Pike, K.; Jones, C.; Yang, P.; Simpson, I. Characterisation of AZ709, a potent and selective inhibitor of Fibroblast Growth Factor Receptor 4 (FGFR4). In Proceedings of the 2013 NCRI Cancer Conference, Liverpool, UK, 3–6 November 2013.
- Hagel, M.; Miduturu, C.; Sheets, M.; Rubin, N.; Weng, W.; Stransky, N.; Bifulco, N.; Kim, J.L.; Hodous, B.; Brooijmans, N.; et al. First Selective Small Molecule Inhibitor of FGFR4 for the Treatment of Hepatocellular Carcinomas with an Activated FGFR4 Signaling Pathway. Cancer Discov. 2015, 5, 424–437. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, A.C.E.; Corcoran, E.; Coffey, H.; Prajapati, S.; Hao, M.-H.; Larsen, N.; Satoh, T.; Wu, J.; Bailey, S.; Ichikawa, K.; et al. H3B6527, an ultra-selective and potent FGFR4 inhibitor for FGF19 driven hepatocellular carcinoma. In Proceedings of the 9th ILCA Annual Conference, Paris, France, 4–6 September 2015.
- Desnoyers, L.R.; Pai, R.; Ferrando, R.E.; Hotzel, K.; Le, T.; Ross, J.; Carano, R.; D'Souza, A.; Qing, J.; Mohtashemi, I.; et al. Targeting FGF19 inhibits tumor growth in colon cancer xenograft and FGF19 transgenic hepatocellular carcinoma models. Oncogene 2008, 27, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Pai, R.; French, D.; Ma, N.; Hotzel, K.; Plise, E.; Salphati, L.; Setchell, K.D.; Ware, J.; Lauriault, V.; Schutt, L.; et al. Antibody-mediated inhibition of fibroblast growth factor 19 results in increased bile acids synthesis and ileal malabsorption of bile acids in cynomolgus monkeys. Toxicol. Sci. 2012, 126, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Mellor, H.R. Targeted inhibition of the FGF19-FGFR4 pathway in hepatocellular carcinoma; translational safety considerations. Liver Int. 2014, 34, e1–e9. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Repana, D.; Ross, P. Targeting FGF19/FGFR4 Pathway: A Novel Therapeutic Strategy for Hepatocellular Carcinoma. Diseases 2015, 3, 294-305. https://doi.org/10.3390/diseases3040294
Repana D, Ross P. Targeting FGF19/FGFR4 Pathway: A Novel Therapeutic Strategy for Hepatocellular Carcinoma. Diseases. 2015; 3(4):294-305. https://doi.org/10.3390/diseases3040294
Chicago/Turabian StyleRepana, Dimitra, and Paul Ross. 2015. "Targeting FGF19/FGFR4 Pathway: A Novel Therapeutic Strategy for Hepatocellular Carcinoma" Diseases 3, no. 4: 294-305. https://doi.org/10.3390/diseases3040294
APA StyleRepana, D., & Ross, P. (2015). Targeting FGF19/FGFR4 Pathway: A Novel Therapeutic Strategy for Hepatocellular Carcinoma. Diseases, 3(4), 294-305. https://doi.org/10.3390/diseases3040294