
Clinical Development of c-MET Inhibition in Hepatocellular Carcinoma

Abstract
:1. Introduction
2. c-MET Pathway and Relevance in HCC
2.1. The HGF/c-MET Pathway
2.2. Abnormalities in HGF/c-MET Signalling Pathways in Cancer
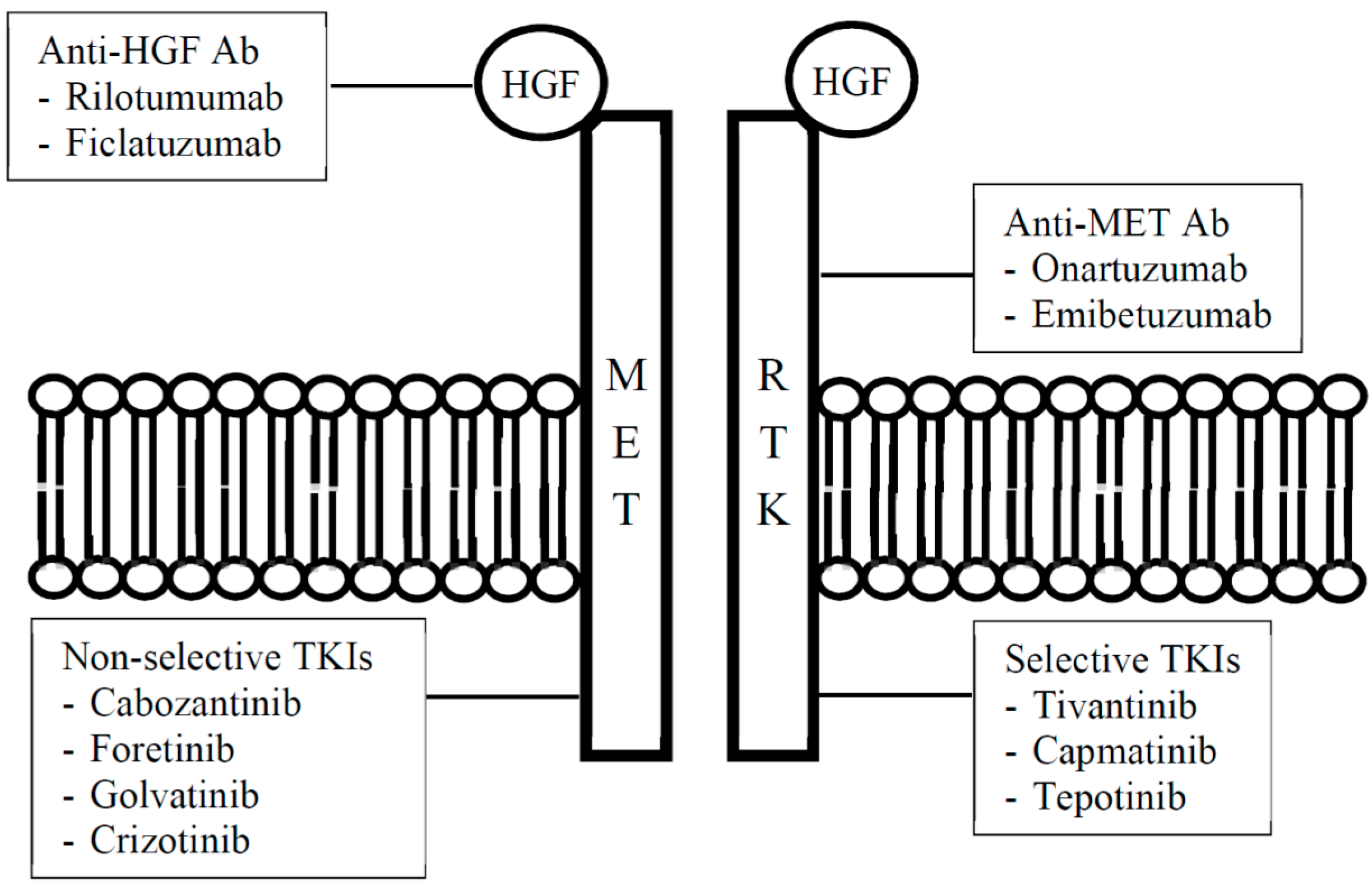
3. Overview of HGF/c-MET Pathway Inhibitors

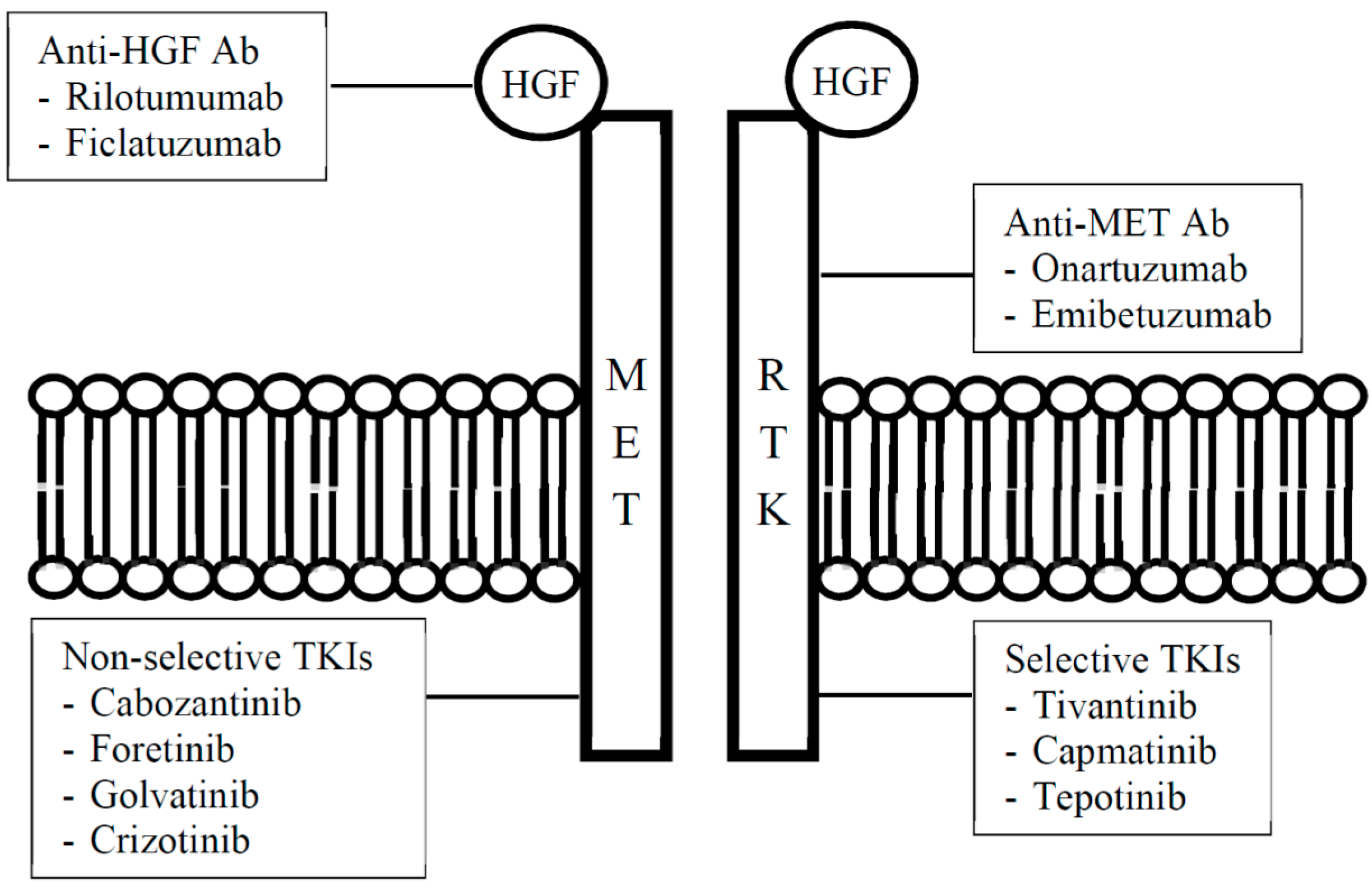{kind=link}
| Type of Inhibitor | Drug | |
|---|---|---|
| Name | Synonym(s) | |
| Antibody | ||
| Anti-HGF | Rilotumumab | AMG 102 |
| Ficlatuzumab | AV-299 | |
| Anti-c-MET | Ornartuzumab | MetMAb |
| Emibetuzumab | LY2875358 | |
| Small molecule inhibitor | ||
| Selective | Tivantinib | ARQ 197 |
| Capmatinib | INC280 (formerly INCB028060) | |
| Tepotinib | MSC2156119J, EMD 1214063 | |
| Non-selective | Cabozantinib | XL184 |
| Foretinib | GSK1363089 (formerly XL880) | |
| Golvatinib | E7050 | |
| Crizotinib | PF-2341066 | |
4. Preclinical Studies of c-MET Inhibitors in HCC
5. Clinical Studies of c-MET Inhibitors in HCC
| Drugs | Phase | Patient Selection | Trial Status | ClinicalTrials.gov Identifier |
|---|---|---|---|---|
| Tivantinib (ARQ 197) | ||||
| Monotherapy | I | Advanced solid tumours † | Recruiting | NCT02150733 |
| With bevacizumab | I | Advanced solid tumours | Active, not recruiting | NCT01749384 |
| With temsirolimus | I | Advanced solid tumours | Recruiting | NCT01625156 |
| With topotecan | I | Advanced solid tumours | Active, not recruiting | NCT01654965 |
| Tivantinib vs. Placebo | III | MET-high HCC | Recruiting | NCT02029157 |
| Tivantinib vs. Placebo | III | MET-high HCC | Recruiting | NCT01755767 |
| Cabozantinib (XL 184) | ||||
| Cabozantinib vs. Placebo | III | HCC | Recruiting | NCT01908426 |
| Capmatinib (INC280) | ||||
| Monotherapy | I | Advanced solid tumours | Recruiting | NCT01546428 |
| Monotherapy | I | MET-dysregulated solid tumours | Recruiting | NCT01324479 |
| Monotherapy | II | MET-dysregulated HCC (1st line) | Recruiting | NCT01737827 |
| Tepotinib (MSC2156119J, EMD 1214063) | ||||
| Monotherapy | I | Advanced solid tumours | Active, not recruiting | NCT01014936 |
| Monotherapy | Ib/II | MET + HCC (1st line) ‡ | Recruiting | NCT01988493 |
| Monotherapy | Ib/II | MET + HCC | Recruiting | NCT02115373 |
| Golvatinib (E7050) | ||||
| With sorafenib | Ib/II | HCC (1st line) § | Active, not recruiting | NCT01271504 |
| Onartuzumab (MetMAb) | ||||
| Monotherapy | I | Advanced solid tumours | Recruiting | NCT02031731 |
| Emibetuzumab (LY2875358) | ||||
| Monotherapy | I | Advanced solid tumours | Active, not recruiting | NCT01287546 |
| With Ramucirumab | Ib/II | Advanced solid tumours | Recruiting | NCT02082210 |
5.1. Tivantinib (ARQ 197)
5.1.1. Phase I Studies
5.1.2. Phase II Study
| Trial | Study Design | Patient Selection | Toxicity Outcomes | Efficacy Outcomes | Dose |
|---|---|---|---|---|---|
| Phase I studies | |||||
| Rosen et al.,(2011) [35] | Dose-escalation study | Advanced solid tumours (N = 79) | Most common AE: fatigue (14%), nausea (14%), vomiting (10%), anaemia (8%), diarrhoea (6%) | Three patients (3.8%) achieved PR; 40 patients (50.6%) maintained SD for a median of 19.9 weeks | MTD not reached R2PD: 360 mg BD |
| DLT: leucopaenia, neutropaenia, thrombocytopaenia, vomiting, dehydration in 2 patients treated with 360 mg BD | |||||
| Yap et al., (2011) [36] | Dose-escalation study | Advanced solid tumours (N = 51) | Most common AE (>10%): grade 1/2 fatigue (16%), nausea (14%), vomiting (12%) | Best response of SD ≥ 4 months in 14 patients (27%) | MTD/R2PD: 360 mg BD |
| Santoro et al., (2013) [41] | Phase Ib study | HCC (N = 21), including Child-Pugh A (N = 17) or B (N = 4) liver cirrhosis | No drug-related worsening of liver function | Best response of SD in nine patients (43%) | RP2D: 360 mg BD |
| Grade ≥ 3 drug-related AEs in 11 patients (52%), including neutropaenia in eight patients (38%) | |||||
| Grade 5 neutropaenic septic shock (N = 1) | |||||
| Four cardiac events were considered possibly or probably related to study drug | |||||
| Phase II study | |||||
| Santoro et al., (2013) [15] | Placebo-controlled randomised phase II study; crossover allowed at radiologic PD (N = 23) | Advanced HCC (N = 107) failing or intolerant of first-line systemic therapy with sorafenib or sunitinib | Most common AE: asthenia (42%), loss of appetite (27%), neutropaenia (21%), fatigue (12%) | Increased TTP for the ITT population (6.9 vs. 6.0 weeks). Greatest clinical benefit for MET-high patients: TTP (11.7 vs. 6.1 weeks), PFS and OS (7.2 vs. 3.8 months) | 240 mg BD |
5.1.3. Phase III Studies
5.2. Cabozantinib (XL 184)
5.2.1. Phase I Study
5.2.2. Phase II Study
| Trial | Study Design | Patient Selection | Toxicity Outcomes | Efficacy Outcomes | Dose |
|---|---|---|---|---|---|
| Phase I study | |||||
| Kuzrock et al., (2011) [48] | Dose escalation study | Advanced solid tumours (N = 85) | DLT: HFS, mucositis, transaminitis | In one patient with HCC whose disease was measurable, SD for at least three months | MTD: 175 mg OD |
| Phase II study | |||||
| Cohn et al., (2012) [50] | Randomised discontinuation study | HCC (N = 41) | Most common grade ≥ 3 AE: HFS (15%), diarrhoea (9%), thrombocytopaenia (9%) | DCR at 12 weeks: 71% (Asian subgroup: 77%) | 100 mg OD |
5.2.3. Phase III Study
5.3. Capmatinib (INC280, Formerly INCB028060)
5.3.1. Phase I Study
5.3.2. Phase II Studies
5.4. Tepotinib (MSC2156119J, EMD 1214063)
5.4.1. Phase I Study
5.4.2. Phase II Studies
5.5. Foretinib (GSK1363089, Formerly XL880)
5.5.1. Phase I Study
5.5.2. Phase I/II Study
5.6. Golvatinib (E7050)
5.6.1. Phase I Studies
5.6.2. Phase Ib/II Study
5.7. Onartuzumab (MetMAb)
Phase I Studies
5.8. Emibetuzumab (LY2875358)
5.8.1. Phase I Study
5.8.2. Phase Ib/2 Study
6. Biomarkers for c-MET-Targeted Therapies
7. Conclusions
Author Contributions
Conflicts of Interest
References
- The global and regional burden of cancer. In World Cancer Report; Steward, B.W.; Wild, C.P. (Eds.) IARC Press: Lyon, France, 2014; pp. 16–53.
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [PubMed]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Llovet, J.M.; Decaens, T.; Raoul, J.L.; Boucher, E.; Kudo, M.; Chang, C.; Kang, Y.K.; Assenat, E.; Lim, H.Y.; Boige, V. Brivanib in patients with advanced hepatocellular carcinoma who were intolerant to sorafenib or for whom sorafenib failed: Results from the randomized phase III brisk-ps study. J. Clin. Oncol. 2013, 31, 3509–3516. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Kudo, M.; Assenat, E.; Cattan, S.; Kang, Y.K.; Lim, H.Y.; Poon, R.T.; Blanc, J.F.; Vogel, A.; Chen, C.L. Effect of everolimus on survival in advanced hepatocellular carcinoma after failure of sorafenib: The EVOLVE-1 randomized clinical trial. JAMA 2014, 312, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Park, J.O.; Ryoo, B.Y.; Yen, C.J.; Poon, R.; Pastorelli, D.; Blanc, J.; Chung, H.C.; Baron, A.D.; Pfiffer, T.E. Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 859–870. [Google Scholar] [CrossRef]
- Cooper, C.S.; Park, M.; Blair, D.G.; Tainsky, M.A.; Huebner, K.; Croce, C.M.; Vande Woude, G.F. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984, 311, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.; Kmiecik, T.E.; Vande Woude, G.F.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-MET proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.C.; Maulik, G.; Christensen, J.; Salgia, R. C-MET: Structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003, 22, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Nakagawa, T. The current stage of molecularly targeted drugs targeting HGF/MET. Jpn. J. Clin. Oncol. 2014, 44, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, H.; Rost, S.; Yauch, R.L. Developing biomarkers to predict benefit from HGF-MET pathway inhibitors. J. Pathol. 2014, 232, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Hepatocyte Growth Factor/Scatter Factor (HGF/SF). MET and Cancer References. Available online: http://www.vai.org/Met/Index.aspx (accessed on 26 August 2015).
- Christensen, J.G.; Burrows, J.; Salgia, R. C-MET as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 2005, 225, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Rimassa, L.; Borbath, I.; Daniele, B.; Salvagni, S.; van Laethem, J.L.; van Vlierberghe, H.; Trojan, J.; Kolligs, F.; Weiss, A. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: A randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013, 14, 55–63. [Google Scholar] [CrossRef]
- Rimassa, L.; Abbadessa, G.; Personeni, N.; Porta, C.; Borbath, I.; Daniele, B.; Salvagni, S.; van Laethem, J.L.; van Vlierberghe, H.; Trojan, J. Tivantinib in pretreated hepatocellular carcinoma (HCC): Tumor and plasma biomarker analysis from the randomized controlled phase 2 trial (RCT). In Proceedings of the 9th International Liver Cancer Association Annual Conference, Paris, France, 4 September 2015.
- Kondo, S.; Ojima, H.; Tsuda, H.; Hashimoto, J.; Morizane, C.; Ikeda, M.; Ueno, H.; Tamura, K.; Shimada, K.; Kanai, Y. Clinical impact of c-MET expression and its gene amplification in hepatocellular carcinoma. Int. J. Clin. Oncol. 2013, 18, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Lennerz, J.K.; Kwak, E.L.; Ackerman, A.; Michael, M.; Fox, S.B.; Bergethon, K.; Lauwers, G.Y.; Christensen, J.G.; Wilner, K.D.; Haber, D.A. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J. Clin. Oncol. 2011, 29, 4803–4810. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.H.; Kwak, E.L.; Siwak-Tapp, C.; Dy, J.; Bergethon, K.; Clark, J.W.; Camidge, D.R.; Solomon, B.J.; Maki, R.G.; Bang, Y.J. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification. J. Thorac. Oncol. 2011, 6, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Chi, A.S.; Batchelor, T.T.; Kwak, E.L.; Clark, J.W.; Wang, D.L.; Wilner, K.D.; Louis, D.N.; Iafrate, A.J. Rapid radiographic and clinical improvement after treatment of a MET-amplified recurrent glioblastoma with a MET inhibitor. J. Clin. Oncol. 2012, 2012, e30–e33. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.C.; Sclafani, F.; Cunningham, D. Emerging molecular targets in oncology: Clinical potential of MET/hepatocyte growth-factor inhibitors. Onco Targets Ther. 2014, 12, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Dulak, A.M.; Gubish, C.T.; Stabile, L.P.; Henry, C.; Siegfried, J.M. HGF-independent potentiation of EGFR action by c-MET. Oncogene 2011, 30, 3625–3635. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J. MET amplification leads to gefitinib resistance in lung cancer by activating erbB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Troiani, T.; Martinelli, E.; Napolitano, S.; Vitagliano, D.; Ciuffreda, L.P.; Costantino, S.; Morgillo, F.; Capasso, A.; Sforza, V.; Nappi, A. Increased TGF-α as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signalling in colon cancer cells. Clin. Cancer Res. 2013, 19, 6751–6765. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtort, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef] [PubMed]
- Sulpice, E.; Ding, S.; Muscatelli-Groux, B.; Bergé, M.; Han, Z.C.; Plouet, J.; Tobelem, G.; Merkulova-Rainon, T. Cross-talk between the VEGF-a and HGF signalling pathways in endothelial cells. Biol. Cell 2009, 101, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; de Sousa E Melo, F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Huh, C.G.; Factor, V.M.; Sánchez, A.; Uchida, K.; Conner, E.; Thorgeirsson, S.S. Hepatocyte growth factor/c-MET signalling pathway is required for efficient liver regeneration and repair. Proc. Natl. Acad. Sci. USA 2004, 101, 4477–4482. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Ding, W.; Dang, H.; Jiang, Y.; Rountree, C.B. C-MET represents a potential therapeutic target for personalised treatment in hepatocellular carcinoma. Hepatology 2011, 54, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; You, H.; Dang, H.; LeBlanc, F.; Galicia, V.; Lu, S.C.; Stiles, B.; Rountree, C.B. Epithelial-to-mesenchymal transition of murine liver tumour cells promotes invasion. Hepatology 2010, 52, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Eathiraj, S.; Palma, R.; Volckova, E.; Hirschi, M.; France, D.S.; Ashwell, M.A.; Chan, T.C. Discovery of a novel mode of protein kinase inhibition characterised by the mechanism of inhibition of human c-MET protein autophosphyrlation by ARQ 197. J. Biol. Chem. 2011, 286, 20666–20676. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.; Jeay, S.; Li, Y.; Chen, C.R.; France, D.S.; Ashwell, M.A.; Hill, J.; Moussa, M.M.; Leggett, D.S.; Li, C.J. ARQ 197, a novel and selective inhibitor of the human c-MET receptor tyrosine kinase with antitumour activity. Mol. Cancer Ther. 2010, 9, 1544–1553. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L.S.; Senzer, N.; Mekhail, T.; Ganapathi, R.; Chai, F.; Savage, R.; Waghorne, C.; Abbadessa, G.; Schwartz, B.; Dreicer, R. A phase I dose-escalation study of tivantinib (ARQ 197) in adult patients with metastatic solid tumors. Clin. Cancer Res. 2011, 17, 7754–7764. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Olmos, D.; Brunetto, A.T.; Tunariu, N.; Barriuso, J.; Riisnaes, R.; Pope, L.; Clark, J.; Futreal, A.; Germuska, M. Phase I trial of a selective c-MET inhibitor ARQ 197 incorporating proof of mechanism pharmacodynamic studies. J. Clin. Oncol. 2011, 29, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Murakami, H.; Nishina, T.; Hirashima, T.; Sugio, K.; Muro, K.; Takahashi, T.; Naito, T.; Yasui, H.; Akinaga, S. The effect of CYP2C19 polymorphim on the safety, tolerability, and pharmacokinetics of tivantinib (ARQ 197): Results from a phase I trial in advanced solid tumours. Ann. Oncol. 2013, 24, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Camacho, L.H.; Bendell, J.C.; John-Reid, L.; Campos, L.T.; Jones, S.F.; Kazakin, J.; Savage, R.; Schwartz, B.E.; Abbadessa, G.; Saleh, M.N. Phase Ib dose-escalation trial evaluating c-MET inhibitor ARQ 197 administered in combination with gemcitabine to patients with advanced solid tumours. J. Clin. Oncol. 2010, 28 (Suppl. S15), e13008. [Google Scholar]
- Goldman, J.W.; Laux, I.; Chai, F.; Savage, R.E.; Ferrari, D.; Garmey, E.G.; Just, R.G.; Rosen, L.S. Phase 1 dose-escalation trial evaluation the combination of the selective MET inhibitor tivantinib (ARQ 197) plus erlotinib. Cancer 2012, 118, 5903–5911. [Google Scholar] [CrossRef] [PubMed]
- Puzanov, I.; Sosman, J.; Santoro, A.; Saif, M.W.; Goff, L.; Dy, G.K.; Zucali, P.; Means-Powell, J.A.; Ma, W.W.; Simonelli, M. Phase 1 trial of tivantinib in combination with sorafenib in adult patients with advanced solid tumors. Investig. New Drugs 2015, 33, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Simonelli, M.; Rodriguez-Lope, C.; Zucali, P.; Camacho, L.H.; Granito, A.; Senzer, N.; Rimassa, L.; Abbadessa, G.; Schwartz, B. A phase-1b study of tivantinib (ARQ 197) in adult patients with hepatocellular carcinoma and cirrhosis. Br. J. Cancer 2013, 108, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.R.; Szwaya, J.; Rojnuckarin, A.; Uppalapati, U.; Huang, L.; Nakuci, E.; Cornell-Kennon, S.; Brown, J.; McSweeney, D.; Bruseo, C. Combination studies of tyrosine kinase inhibitors (TKIs): Assessment of potential cytotoxic synergy of ARQ 197 with sorafenib or sunitinib. Cancer Res. 2009, 69, 18–22. [Google Scholar]
- Chai, F.; Abbadessa, G.; Savage, R.; Zahir, H.; Chen, Y.; Lamar, M.; Kazakin, J.; Ferrari, D.; von Roemeling, R.; Schwartz, B. Phase 1 experience of tivantinib in patients with hepatocellular carcinoma (HCC) or biliary tract cancer (BTC). Ann. Oncol. 2012, 23 (Suppl. S9), 245. [Google Scholar]
- Rimassa, L.; Personeni, N.; Simonelli, M.; Santoro, A. Tivantinib: A new promising mesenchymal-epithelial transition factor inhibitor in the treatment of hepatocellular carcinoma. Futur. Oncol. 2013, 9, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Porta, C.; Rimassa, L.; Borbath, I.; Daniele, B.; Finn, R.S.; Raoul, J.L.; Schwartz, L.H.; He, A.R.; Trojan, J.J. Metiv-HCC: A phase III clinical trial evaluating tivantinib (ARQ 197), a MET inhibitor, versus placebo as second-line in patients with MET-high inoperable hepatocellular carcinoma. J. Clin. Oncol. 2013, 31 (Suppl. S15), TPS4159. [Google Scholar]
- Rimassa, L.; Porta, C.; Borbath, I.; Daniele, B.; Finn, R.S.; Raoul, J.L.; Schwartz, L.H.; He, A.R.; Trojan, J.; Peck-Radosavlijevic, M. Tivantinib in MET-high hepatocellular carcinoma patients and the ongoing phase III clinical trial. Hepatic Oncol. 2014, 1, 181–188. [Google Scholar] [CrossRef]
- Xiang, Q.; Chen, W.; Ren, M.; Wang, J.; Zhang, H.; Deng, D.; Zhang, L.; Shang, C.; Chen, Y. Cabozantinib suppresses tumor growth and metastasis in hepatocellular carcinoma by a dual blockade of VEGFR2 and MET. Clin. Cancer Res. 2014, 20, 2959–2970. [Google Scholar] [CrossRef] [PubMed]
- Kurzrock, R.; Sherman, S.I.; Ball, D.W.; Forastiere, A.A.; Cohen, R.B.; Mehra, R.; Pfister, D.G.; Cohen, E.E.; Janisch, L.; Nauling, F. Activity of XL 184 (cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J. Clin. Oncol. 2011, 29, 2660–2666. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.S.; Vogelzang, N.J.; Schoffski, P.; Daud, A.; Spira, A.I.; O’Keeffe, B.A.; Rafferty, T.; Lee, Y.; Berger, R.; Shapiro, G. Activity of cabozantinib (XL 184) in soft tissue and bone: Results of a phase II randomised discontinuation trial (RDT) in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2011, 29 (Suppl. S15). Abstract 3010. [Google Scholar]
- Cohn, A.L.; Kelley, R.K.; Yang, T.S.; Su, W.C.; Verslype, C.; Ramies, D.A.; Lee, Y.; Shen, X.; Cutsem, E.V. Activity of cabozantinib (XL184) in hepatocellular carcinoma patients (pts): Results from a phase II randomized discontinuation trial (RDT). J. Clin. Oncol. 2012, 30 (Suppl. S4). Abstract 261. [Google Scholar]
- Lu, K.V.; Chang, J.P.; Parachoniak, C.A.; Pandika, M.M.; Aghi, M.K.; Meyronet, D.; Isachenko, N.; Fouse, S.D.; Phillips, J.J.; Cheresh, D.A. VEGF inhibits tumour cell invasion and mesenchymal transition through a MET-VEGFR2 complex. Cancer Cell 2012, 22, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Cheng, A.; Meyer, T.; El-Khoueiry, A.B.; Ikeda, M.; Chun, H.G.; Faivre, S.J.; Furuse, J.; Knox, J.J.; Okusaka, T. Phase 3 randomized, double-blind, controlled study of cabozantinib (XL 184) versus placebo in subjects with hepatocellular carcinoma who have received prior sorafenib (CELESTIAL; NCT01908426). J. Clin. Oncol. 2014, 32 (Suppl. S15). Abstract TPS4150. [Google Scholar]
- Liu, X.; Wang, Q.; Yang, G.; Marando, C.; Koblish, H.K.; Hall, L.M.; Fridman, J.S.; Behshad, E.; Wynn, R.; Li, Y. A novel kinase inhibitor INCB28060 blocks c-MET-dependent signalling, neoplastic activities, and crosstalk with EGFR and HER-3. Clin. Cancer Res. 2011, 17, 7127–7138. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.J.; Su, W.C.; Nam, D.H.; Lim, W.T.; Bauer, T.M.; Brana, I.; Poon, R.T.P.; Hong, D.S.; Lin, C.C.; Peng, B. Phase I study of the safety and efficacy of INC280 in patients with advanced MET-dependent solid tumours. J. Clin. Oncol. 2014, 32 (Suppl. S15). Abstract 2520. [Google Scholar]
- Bladt, F.; Friese-Hamim, M.; Ihling, C.; Wilm, C.; Blaukat, A. The c-MET inhibitor MSC2156119J effectively inhibits tumor growth in liver cancer models. Cancers 2014, 19, 1736–1752. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Hong, D.S.; Amin, H.M.; Fu, S.; Piha-Paul, S.A.; Janku, F.; Granda, J.G.; Zheng, H.; Klevesath, M.B.; Kohler, K. Results of the first-in-human phase I trial assessing MSC2156119J (EMD 1214063), an oral selective c-MET inhibitor, in patients with advanced solid tumours. J. Clin. Oncol. 2014, 32 (Suppl. S15). Abstract 2521. [Google Scholar]
- Qin, S.; Cheng, A.L.; Lim, H.Y.; Xu, L.; Bladt, F.; Johne, A.; Li, C.; Zheng, H.; Massimini, G. A multicentre, randomised, phase Ib/II trial of the oral c-MET inhibitor MSC2156119J as monotherapy versus sorafenib in Asian patients with MET-positive advanced hepatocellular carcinoma and Child-Pugh A liver function. J. Clin. Oncol. 2014, 32 (Suppl. S15). Abstract TPS4151. [Google Scholar]
- Huynh, H.; Ong, R.; Soo, K.C. Foretinib demonstrates anti-tumour activity and improves overall survival in preclinical models of hepatocellular carcinoma. Angiogenesis 2012, 15, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Eder, J.P.; Shapiro, G.I.; Appleman, L.J.; Zhu, A.X.; Miles, D.; Keer, H.; Cancilla, B.; Chu, F.; Hitchcock-Bryan, S.; Sherman, L. A phase I study of foretinib, a multi-targeted inhibitor of c-MET and vascular endothelial growth factor receptor 2. Clin. Cancer Res. 2010, 16, 3507–3516. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.C.; Sukeepaisarnjaroen, W.; Chao, Y.; Yen, C.J.; Lausoontornsiri, W.; Chen, P.J.; Sanpajit, T.; Lencioni, R.; Camp, A.C.; Cox, D.S. A phase I/II study of foretinib, an oral multikinase inhibitor targeting MET, RON, AXL, TIE-2, and VEGF in advanced hepatocellular carcinoma (HCC). J. Clin. Oncol. 2012, 30 (Suppl. S15). Abstract 4108. [Google Scholar]
- Nakagawa, T.; Tohyama, O.; Yamaguchi, A.; Matsushima, T.; Takahashi, K.; Funasaka, S.; Shirotori, S.; Asada, M.; Obaishi, H. E7050: A dual c-MET and VEGFR-2 tyrosine kinase inhibitor promotes tumour regression and prolongs survival in mouse xenograft models. Cancer Sci. 2010, 101, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Yoshino, T.; Fuse, N.; Bando, H.; Tahara, M.; Ohki, M.; Fujino, M.; Nishioka, Y.; Matsuura, K.; Sawada, T. Phase I dose-finding study of golvatinib (E7050), a c-MET and EPH receptor targeted multi-kinase inhibitor, administered orally bid to patients with advanced solid tumors. J. Clin. Oncol. 2012, 30 (Suppl. S15). Abstract 3079. [Google Scholar]
- Molife, L.R.; Dean, E.J.; Blanco-Codesido, M.; Krebs, M.G.; Brunetto, A.T.; Greystoke, A.P.; Daniele, G.; Lee, L.; Kuznetsov, G.; Myint, K.T. A phase I, dose-escalation study of the multitargeted receptor tyrosine kinase inhibitor, golvatinib, in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 6284–6294. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, B.H.; Bendell, J.C.; Modiano, M.R.; Machiels, J.P.H.; Versola, M.J.; Hodge, J.P.; Sawarna, K.; Tse, N. Phase I/II study of E7050 (golvantinib) in combination with sorafenib in patients (pts) with advanced hepatocellular carcinoma (HCC): Phase I results. J. Clin. Oncol. 2013, 31 (Suppl. S4). Abstract 294. [Google Scholar]
- Merchant, M.; Ma, X.; Maun, H.R.; Zheng, Z.; Peng, J.; Romero, M.; Huang, A.; Yang, N.Y.; Nishimura, M.; Greve, J. Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc. Natl. Acad. Sci. USA 2013, 110, E2987–E2996. [Google Scholar] [CrossRef] [PubMed]
- Mai, E.; Zheng, Z.; Chen, Y.; Peng, J.; Severin, C.; Filvaroff, E.; Romero, M.; Mallet, W.; Kaur, S.; Gelzleichter, T. Nonclinical evaluation of the serum pharmacodynamic biomarkers HGF and shed MET following dosing with the anti-MET monovalent monoclonal antibody onartuzumab. Mol. Cancer Ther. 2014, 13, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Salgia, R.; Patel, P.; Bothos, J.; Yu, W.; Eppler, S.; Hegde, P.; Bai, S.; Kaur, S.; Nijem, I.; Catenacci, D.V. Phase I dose-escalation study of onartuzumab as a single agent and in combination with bevacizumab in patients with advanced solid malignancies. Clin. Cancer Res. 2014, 20, 1666–1675. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Horiike, A.; Nokihara, H.; Horinouchi, H.; Nakamichi, S.; Wakui, H.; Ohyanagi, F.; Kudo, K.; Yanagitani, N.; Takahashi, S. Phase I study of the anti-MET antibody onartuzumab in patients with solid tumors and MET-positive lung cancer. Investig. New Drugs 2015, 33, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zeng, W.; Wortinger, M.A.; Yan, S.B.; Cornwell, P.; Peek, V.L.; Stephens, J.R.; Tetreault, J.W.; Xia, J.; Manro, J.R. LY2875358, a neutralizing and internalizing anti-MET bivalent antibody, inhibits HGF-dependent and HGF-independent MET activation and tumor growth. Clin. Cancer Res. 2014, 20, 6059–6070. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.W.; Rosen, L.S.; Algazi, A.P.; Tumer, P.K.; Wacheck, V.; Tuttle, J.; Wooldrige, J.E.; Banck, M.S. First-in-human dose escalation study of LY2875358 (LY), a bivalent MET antibody, as monotherapy and in combination with erlotinib (E) in patients with advanced cancer. J. Clin. Oncol. 2013, 31 (Suppl. S15). Abstract 8093. [Google Scholar]
- Peters, S.; Adjei, A.A. MET: A promising anticancer therapeutic target. Nat. Rev. Clin. Oncol. 2012, 9, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Sierra, J.R.; Tsao, M.S. C-MET as a potential therapeutic target and biomarker in cancer. Ther. Adv. Med. Oncol. 2011, 3 (Suppl. S1), S21–S35. [Google Scholar] [CrossRef] [PubMed]
- Spigel, D.R.; Ervin, T.J.; Ramlau, R.; Daniel, D.B.; Goldschmidt, J.H.; Blumenschein, G.R.; Krzakowski, M.J.; Robinet, G.; Clement-Duchene, C.; Barlesi, F. Final efficacy results from OAM4558g, a randomised phase II study evaluating MetMAb or placebo in combination with erlotinib in advanced NSCLC. J. Clin. Oncol. 2011, 29 (Suppl. S15). Abstract 7505. [Google Scholar]
- Iveson, T.; Donehower, R.C.; Davidenko, I.; Tjulandin, S.; Deptala, A.; Harrison, M.; Nirni, S.; Lakshmaiah, K.; Thomas, A.; Jiang, Y. Rilotumumab in combination with epirubicin, cisplatin, and capecitabine as first-line treatment for gastric or oesophagogastric junction adenocarcinoma: An open-label, dose de-escalation phase 1b study and a double-blind, randomised phase 2 study. Lancet Oncol. 2014, 15, 1007–1018. [Google Scholar] [PubMed]
- Venepalli, N.K.; Goff, L. Targeting the HGF-cMET axis in hepatocellular carcinoma. Int. J. Hepatol. 2013, 2013, 341636. [Google Scholar] [PubMed]
- Bellon, S.F.; Kaplan-Lefko, P.; Yang, Y.; Zhang, Y.; Moriguchi, J.; Rex, K.; Johnson, C.W.; Rose, P.E.; Long, A.M.; O’Connor, A.B. C-MET inhibitors with novel binding mode show activity against several hereditary papillary renal cell carcinoma-related mutations. J. Biol. Chem. 2008, 283, 2675–2683. [Google Scholar] [CrossRef] [PubMed]
- Berthou, S.; Aebersold, D.M.; Schmidt, L.S.; Stroka, D.; Heigl, C.; Streit, B.; Stalder, D.; Gruber, G.; Liang, C.; Howlett, A.R. The MET kinase inhibitor SU11274 exhibits a selective inhibition pattern toward different receptor mutated variants. Oncogene 2004, 8, 5387–5393. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Wainberg, Z.A.; Catenacci, D.V.; Hochster, H.S.; Ford, J.; Kunz, P.; Lee, F.C.; Kallender, H.; Cecchi, F.; Rabe, D.C. Phase II study evaluating 2 dosing schedules of oral foretinib (GSK1363089), CMET/VEGFRr2 inhibitor, in patients with metastatic gastric cancer. PLoS ONE 2013, 8, e54014. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; von Pawel, J.; Garmey, E.G.; Akerley, W.L.; Brugger, W.; Ferrari, D.; Chen, Y.; Costa, D.B.; Gerber, D.E.; Orlov, S. Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J. Clin. Oncol. 2011, 29, 3307–3315. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.J.X.; Chan, J.J.; Choo, S.P. Clinical Development of c-MET Inhibition in Hepatocellular Carcinoma. Diseases 2015, 3, 306-324. https://doi.org/10.3390/diseases3040306
Lee JJX, Chan JJ, Choo SP. Clinical Development of c-MET Inhibition in Hepatocellular Carcinoma. Diseases. 2015; 3(4):306-324. https://doi.org/10.3390/diseases3040306
Chicago/Turabian StyleLee, Joycelyn J. X., Jack J. Chan, and Su Pin Choo. 2015. "Clinical Development of c-MET Inhibition in Hepatocellular Carcinoma" Diseases 3, no. 4: 306-324. https://doi.org/10.3390/diseases3040306
APA StyleLee, J. J. X., Chan, J. J., & Choo, S. P. (2015). Clinical Development of c-MET Inhibition in Hepatocellular Carcinoma. Diseases, 3(4), 306-324. https://doi.org/10.3390/diseases3040306